

Abstract
Rapamycin, a specific inhibitor for mTOR complex 1, is an FDA-approved immunosuppressant for organ transplant. Recent developments have raised the prospect of using rapamycin to treat cancer or diabetes and to delay aging. It is therefore important to assess how rapamycin treatment affects glucose homeostasis. Here, we show that the same rapamycin treatment reported to extend mouse life span significantly impaired glucose homeostasis of aged mice. Moreover, rapamycin treatment of lean C57B/L6 mice reduced glucose-stimulated insulin secretion in vivo and ex vivo as well as the insulin content and beta cell mass of pancreatic islets. Confounding the diminished capacity for insulin release, rapamycin decreased insulin sensitivity. The multitude of rapamycin effects thus all lead to glucose intolerance. As our findings reveal that chronic rapamycin treatment could be diabetogenic, monitoring glucose homeostasis is crucial when using rapamycin as a therapeutic as well as experimental reagent.
Keywords: Rapamycin, mTOR, Glucose intolerance, Insulin, Diabetes
Introduction
Rapamycin, an immunosuppressant with an anti-proliferative ability, inhibits the mammalian target of rapamycin (mTOR) serine/threonine kinase that is related to the phosphatidylinositol 3-kinase (PI3K) family and the cellular integration center for controlling cell growth and proliferation and for sensing nutrients as well as hormonal signals, including insulin released from pancreatic beta cells [1]. Binding of insulin to its tyrosine kinase receptor leads to the phosphorylation of the insulin receptor substrate (IRS) and the activation of the downstream PI3K/Akt kinase pathway as well as the mTOR signaling pathway to mediate insulin actions in liver and other tissues [2]. Rapamycin treatment could therefore have broad impact in glucose homeostasis.
Recent studies suggest that rapamycin and its derivatives may be used as therapeutic agents for treating cancers, diabetes, and Alzheimer’s disease [3, 4]. Moreover, Harrison et al. [5] reported that rapamycin extends the median and maximal life span of both male and female mice and noted that “disease patterns of rapamycin-treated mice did not differ from those of control mice,” thereby raising the prospect of human clinical trials of rapamycin to treat aging and age-related diseases [6, 7]. However, the potential side effects of rapamycin such as glucose intolerance—a significant risk factor for diabetes and cardiovascular diseases [8–11] as well as cognitive impairments [12]—warrant further investigation.
There are conflicting reports of rapamycin effects on glucose homeostasis. On the one hand, hyperinsulinemia and hyperglycemia activates mTOR, raising the possibility that rapamycin inhibition of mTOR may reduce the risk of diabetes [3]. Moreover, activated mTOR causes the phosphorylation of downstream effectors such as S6 kinase (S6K), the ribosomal protein S6, and the eukaryotic initiation factor 4E binding protein (4EBP) [1]. S6K may also phosphorylate IRS to promote IRS deactivation and degradation, thereby causing insulin resistance [13, 14]. Consistent with this notion, S6K1 knockout mice are resistant to diet-induced obesity (DIO) and diabetes [15]. On the other hand, chronic rapamycin treatment increases the risk of diabetes of human kidney transplant recipients [16]. Other preclinical studies have also shown that rapamycin causes diabetes in obese sand rats (Psammomys obesus) [17], lean rats [18], and DIO mice [19].
In this study, we show that aged mice fed with the same mouse chow with encapsulated rapamycin that extends life span [5] became glucose-intolerant. We further show that chronic exposure to rapamycin caused glucose intolerance in mice due to a reduction in beta cell mass and insulin production as well as insulin resistance.
Materials and methods
Animals
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and used protocol approved by the Institutional Animal Care and Use Committee of the University of California San Francisco (approved protocol ID: AN079064-03D). Inbred male C57B/L6 mice were from Jackson Laboratory (Bar Harbor, ME). The aged mice were from National Institute of Aging (Bethesda, MD). Mice (three to five per cage) housed in the animal facility were fed with PicoLab Mouse Diet #5058 (Lab Diet, Brentwood, MO) and subjected to a standard 12-h light/dark cycle. Rapamycin (Apollo Scientific, UK or LC Laboratories, Boston, MA) was dissolved in DMSO (Sigma, St. Louis, MO) and then diluted in PBS before injection. For encapsulated rapamycin diet, rapamycin was micro-encapsulated and incorporated into Purina 5LG6 diet (Southwest Research Institute, San Antonio, TX).
Intraperitoneal glucose tolerance test
Glucose tolerance tests were performed after a 16-h fasting period (6 pm to 10 am) with free access to water. Each mouse received an intraperitoneal injection of glucose (1.5 g/kg body weight). Blood samples were obtained from the tail and the whole blood glucose level determined using an OneTouch Ultra glucometer (LifeScan, Milpitas, CA).
Insulin sensitivity test
Starting at 7 am, mice were fasted for 4 h with free access to water and then injected intraperitoneally with bovine insulin (1 U/kg body weight, Sigma). Blood samples were obtained from the tail and the whole blood glucose level determined using an OneTouch Ultra glucometer.
Plasma insulin and C-peptide measurement
Starting at 7 am, mice were fasted for 4 h with free access to water and then injected intraperitoneally with glucose (1.5 g/kg body weight). Blood samples were first collected into an ethylene glycol tetraacetic acid (EGTA)-coated tube (Sarstedt, Germany) and plasma was isolated by low-speed centrifugation. Plasma insulin concentration was determined using an ultrasensitive insulin and C-peptide ELISA assay (Alpco, Salem, NH). The HOMA-IR index was calculated as (fasting plasma insulin × fasting plasma glucose)/(22.5 × 18) [20].
Insulin secretion from isolated islets
Mice were treated with either rapamycin or vehicle only for 3 weeks (five mice per group). At the end of the third week, mice were euthanized and pancreatic islets were isolated according to published protocol [21]. For each sample, ten islets were pooled together and exposed to low (2.5 mM) glucose for 20 min followed by high (12.5 mM) glucose stimulation for 20 min; all the islets were saved for total insulin and DNA quantification. Insulin level in supernatants was determined using a High Range ELISA insulin plate (Alpco) and normalized to total DNA.
Electrophysiology
Pancreatic slices (100 μm thick) were prepared as described previously [22]. Slices were incubated in the extracellular solution (in millimolars): 126 NaCl, 21.4 NaHCO3, 2.5 glucose, 5 KCl, 1.25 NaH2PO4, 1.2 MgCl2, 2 CaCl2, equilibrated with 95% O2/5% CO2. Beta cells were identified with infrared DIC optics. An Axon200B amplifier (Molecular Devices Corp., Sunnyvale, CA) was used to measure membrane potentials and membrane capacitance in the standard whole-cell patch-clamp configuration. Data were acquired at 5 kHz with Clampex10 software (Molecular Devices Corp.). Data with series resistances higher than 20 MΩ were excluded from further analysis. The intracellular solution contained (in millimolars): 150 KCl, 10 HEPES, 5 Mg2ATP, 1 Na3GTP, 10 sodium phosphocreatine, 0.05 EGTA; pH was adjusted to 7.2 with KOH. All the experiments were performed at room temperature.
Immunostaining
Mice were anesthetized (Avertin, 0.25 g/kg, i.p. injection) and perfused transcardially with 4% paraformaldehyde. Pancreases were removed and post-fixed overnight in 4% paraformaldehyde then cryoprotected overnight in saline containing 30% sucrose. Tissue sections (20 μm) that were 200 μm apart from one another were mounted onto gelatin-coated slides. Sections were washed in blocking medium containing 0.1% Triton X-100 and 5% donkey serum (Jackson Immunoresearch Laboratories, West Grove, PA) and incubated overnight (4°C) with primary antibodies against insulin (guinea pig, 1:100; DAKO) and Ki67 (rabbit, 1:500; Thermo Scientific) followed by Alexa dye-tagged secondary antibodies (donkey, 1:500; Invitrogen, Eugene, OR). The slides were mounted using Fluoromount G mounting medium containing DAPI (Southern Biotech) and the images acquired using a Nikon fluorescent microscope equipped with a CCD camera. TUNEL apoptosis detection kit (Millipore, Billerica, MA) was used to identify apoptotic beta cells.
Western blot analysis
Mice fasted for 4 h were injected intraperitoneally with insulin (1 U/kg body weight). Ten minutes after injection, mice were euthanized and liver tissues were harvested. Western blot analysis was performed as described previously [23]. Phospho-S6 (Ser235/6), Phospho-4EBP (Thr70), Phospho-Akt (Ser473), S6, 4EBP, and Akt antibodies were purchased from Cell Signaling.
Statistical analyses
Statistical analyses were performed with Prism software (Graphpad Software, San Diego, CA) or R (The R Foundation for Statistical Computing, Vienna, Austria) using two-way repeated measures ANOVA with Bonferroni post hoc test, Mann–Whitney U test, or Student’s t test for pairwise comparisons. A p < 0.05 was considered statistically significant. Barnard’s exact test or Fisher’s exact test was used to evaluate the significance of contingency data. Area under the curve (AUC) of intraperitoneal glucose tolerance test (IPGTT) was calculated using the trapezoidal rule method. Dose-dependent curves were constructed using the following equation to determine
Results
Low-dose rapamycin induces glucose intolerance in aged mice
The remarkable finding that encapsulated rapamycin ingestion even commenced from midlife can extend life span in aged mice [5] has raised the prospect for human clinical trials [6, 7]. To examine the effect of this rapamycin treatment on glucose homeostasis, we obtained two strains of aged hybrid mice, BD6F1 and B6D2F1, and fed them Purina 5LG6 diet with or without encapsulated rapamycin. Remarkably, these aged mice developed glucose intolerance within 4 weeks on rapamycin diet (p < 0.05, Fig. 1a, b). Using a criterion of glucose intolerance as a blood glucose level above 10 mM (180 mg/dl) at 120 min post-intraperitoneal glucose injection, we found that a significant fraction of mice on rapamycin diet showed glucose intolerance (p < 0.05, Fig. 1c). This rapamycin-induced impairment of glucose homeostasis was associated with hypoinsulinemia: mice on rapamycin diet had plasma insulin levels lower than control values both before and after glucose injection (Fig. 1d), indicative of impaired beta cell function since the rapamycin diet did not cause insulin resistance in these aged mice (Fig. 1e).
Fig. 1.
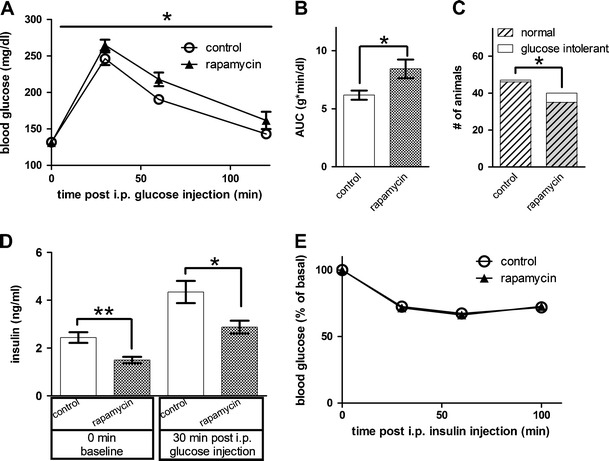
Aged mice fed with diet containing rapamycin showed impaired glucose homeostasis. a Rapamycin-induced glucose intolerance in aged mice (p < 0.05, two-way repeated measures ANOVA, n = 47 and 40 for control and rapamycin groups, respectively). b Rapamycin-fed aged mice had a significantly higher AUC than control group (p < 0.05, Mann–Whitney U test). c More aged mice in the rapamycin-fed group showed glucose intolerance than in control group (p < 0.05, Barnard’s exact test). d Rapamycin significantly reduced plasma insulin level (p < 0.01 for 0 min and p < 0.05 for 30 min after glucose injection, Mann–Whitney U test, n = 35 and 29 for control and rapamycin groups, respectively). e Rapamycin diet did not alter insulin sensitivity in aged mice (p > 0.05, two-way repeated measures ANOVA, n = 50 and 45 for control and rapamycin group, respectively)
Chronic rapamycin treatment causes hyperglycemia and hypoinsulinemia in young mice
Having found that low-dosage rapamycin caused glucose intolerance in a subset of aged mice with a hybrid genetic background, we decided to explore the possible pathophysiologic mechanism by studying C57B/L6 mice, the most widely used inbred mouse line to reduce possible variability due to genetic background. We administrated rapamycin by intraperitoneal injection to have better dosage control and reduce the variability due to differences in diet ingestion. Similar to the aged hybrid mice, 1-year-old C57B/6 mice developed glucose intolerance when injected with rapamycin intraperitoneally (Fig. 2a). Next, we asked whether this rapamycin-induced glucose intolerance is age-dependent, given that the turnover rate of pancreatic beta cells in aged mice is slower [24]. We found that 3 weeks of intraperitoneal administration of rapamycin also caused glucose intolerance in young (2 months old) mice (Fig. 2b). Interestingly, both young and old mice regained glucose sensitivity 2 weeks after termination of the rapamycin regimen (Fig. 2a–c). The sensitivity to rapamycin was comparable between young and old mice, with EC50 of 0.28 and 0.26 mg/kg for 2-months-old and 1-year-old mice, respectively (Fig. 2d). We found that intraperitoneal rapamycin injection for 3 weeks did not affect the bodyweight of both young and old mice (Fig. 2e, f). Since similar rapamycin-induced glucose intolerance was observed in mice of different ages and strains, we focused on young healthy male C57B/6 mice to study the underlying physiological and biochemical mechanism. We chose the paradigm of daily intraperitoneal injection for 3 weeks of either DMSO-only as control or 0.5 mg/kg rapamycin since this dose is within the effective therapeutic concentration for immunosuppression in mammals [25] and is effective in inducing glucose intolerance in mice (Fig. 2d).
Fig. 2.

Intraperitoneal rapamycin injection caused glucose intolerance in both young and old mice. a Old mice (1 year old) treated with different rapamycin dosages. Each group of mice (n = 10 for each dose) received a particular dose of rapamycin (from left to right): DMSO vehicle control, 0.05, 0.5, and 5 mg/kg; for each graph, the IPGTT curves before (baseline, solid black line), 3 weeks after rapamycin treatment (treatment, red line), and 2 weeks after stopping rapamycin treatment (recovery, dash black line) were plotted together. b Young (2 months old) mice (n = 10 for each dose) had comparable responses to intraperitoneal injection of rapamycin for the same range of doses and regimen. c Representative time course of young mice treated with rapamycin (0.5 mg/kg). After 1 week of rapamycin treatment, mice became glucose-intolerant (p < 0.001, two-way ANOVA with Bonferroni post hoc test), and it took about 2 weeks to restore the glucose sensitivity after termination of the rapamycin treatment that lasted for 3 weeks (n = 10 for each group). d Rapamycin dose dependence curves of old (open circle) and young (solid circle) mice. The EC50 were 0.28 and 0.26 mg/kg for young (2 months old) and old (1 year old) mice, respectively. Three weeks of 0.5 mg/kg rapamycin injection did not affect mouse bodyweight from young (e) or old (f) mice. Bodyweights were normalized to the starting value prior to rapamycin treatment
Having found that rapamycin diet caused glucose intolerance by reducing plasma insulin level (Fig. 1d), we further showed that intraperitoneal rapamycin treatment also caused a significant reduction of plasma insulin level (Fig. 3a). Moreover, the plasma insulin level of rapamycin-treated mice failed to rise following intraperitoneal glucose administration (p < 0.05, Fig. 3a). As reduced plasma insulin may arise from either decreased insulin secretion from the pancreatic beta cells or increased hepatic insulin clearance, we measured the plasma level of C-peptide, which accompanies insulin release with longer half-life in the plasma due to low hepatic clearance. We found that glucose injection also failed to raise C-peptide levels in the same plasma samples from rapamycin-treated mice (p < 0.05, Fig. 3b) without altering the plasma insulin/C-peptide ratio (Fig. 3c), suggesting that insulin secretion from pancreatic beta cells is impaired in rapamycin-treated mice. Indeed, although islets isolated from both DMSO- and rapamycin-treated mice still responded to high (12.5 mM) glucose stimulation (p < 0.05), islets from rapamycin-treated mice exhibited much less glucose-induced insulin release (p < 0.05, Fig. 3e), which was associated with a reduction in the total insulin content in islets (p < 0.05, Fig. 3f), suggesting that the suppression of insulin production by chronic rapamycin treatment led to impaired glucose homeostasis.
Fig. 3.
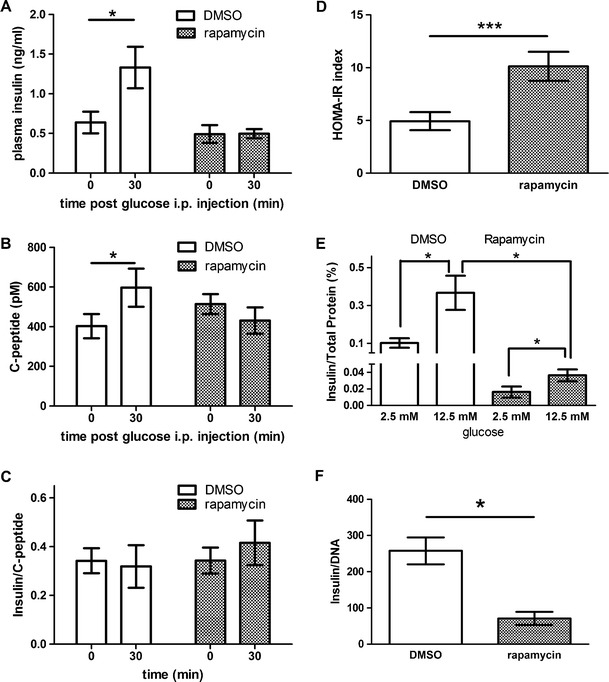
Rapamycin reduced glucose-stimulated insulin secretion in vivo and ex vivo. a Plasma insulin level was assessed before (0 min) and 30 min (30 min) after intraperitoneal glucose injection (1.5 g glucose/kg). Rapamycin blunted glucose-induced insulin secretion (30 min) in vivo (p < 0.05, paired t test). Each group had six to ten animals. b Analyses of the same plasma samples revealed that rapamycin also blunted glucose-induced C-peptide secretion (30 min) in vivo (p < 0.05, paired t test). c Rapamycin treatment did not alter the plasma insulin/C-peptide ratio with or without glucose stimulation. d Rapamycin treatment increased HOMA-IR index (p < 0.05, Mann–Whitney U test). e Pancreatic islets isolated from rapamycin-treated mice showed a decreased insulin secretion upon glucose stimulation. Insulin level was normalized to total protein extract from the isolated islets (p < 0.05, paired t test). f Three weeks of rapamycin treatment reduced total insulin content in islets (p < 0.05, unpaired t test). For experiments using isolated islets (d–f), each data point was for five to seven sets of ten islets pooled from five mice
Glucose induces insulin secretion by closing KATP channels to depolarize the beta cell and induce action potential firing, which in turn causes calcium influx to trigger exocytosis [26]. To ask whether chronic rapamycin treatment impairs the stimulus–secretion coupling for glucose-induced insulin release, we performed patch-clamp recording from beta cells in pancreatic slices. Since beta cells form a syncytium within the islet of Langerhans, whole-cell recording from the intact islet preparation can monitor the membrane potential changes of the entire islet that maintains electrical coupling through gap junctions [22, 27]. Like beta cells from control mice (Fig. 4a), beta cells from rapamycin-treated mice responded to glucose stimulation with depolarization and action potential firing (Fig. 4b). We also measured the increase of membrane capacitance following fifty 3-Hz depolarization pulses from −70 to 0 mV for 100 ms, a stimulation mimicking action potential firing induced by glucose stimulation [22, 26]. This membrane capacitance measurement revealed that the depolarization pulses caused similar increases in the cell surface area of beta cells from rapamycin-treated mice and control mice (Fig. 4d). Thus, rapamycin did not alter the glucose stimulus–excitation coupling or the exocytotic machinery of beta cells.
Fig. 4.
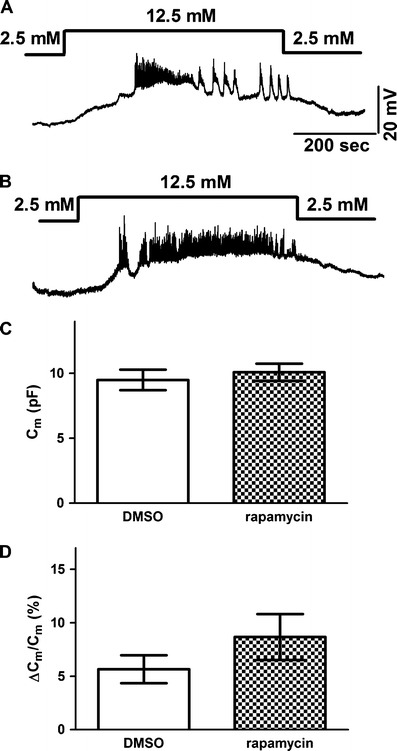
Rapamycin does not alter glucose stimulus–secretion coupling in beta cells. DMSO-treated (a) and rapamycin-treated (b) mice had beta cells that responded similarly to a rise of glucose in the extracellular solution from 2.5 to 12.5 mM with membrane depolarization and action potential firing. c Beta cells from DMSO- or rapamycin-treated mice had similar membrane capacitance, indicating that the cell size was comparable (p > 0.05, unpaired t test). d A train of 100-ms pulses from −70 to 0 mV delivered at 3 Hz for 50 s caused similar capacitance increases in beta cells from DMSO- and rapamycin-treated mice (p > 0.05, unpaired t test). Each group had six to eight cells from three animals
Rapamycin reduces beta cell mass in pancreatic islets by decreasing beta cell proliferation
As a previous study has shown that rapamycin reduces beta cell mass in the islets of obese sand rats [17], we examined the beta cell mass in these lean C57B/L6 mice and found that the pancreatic islets from rapamycin-treated mice were significantly reduced in size (p < 0.05, Fig. 5a–c) while the beta cell density remains unchanged (Fig. 5d). Moreover, using membrane capacitance as a surrogate measure for cell size, we found that the surface area of beta cells from rapamycin-treated mice was normal (Fig. 4c). A reduction of beta cell mass could be caused by reducing beta cell proliferation, increasing beta cell apoptosis, or both [17, 28]. We found that the pancreatic islets from rapamycin-treated mice had a reduction of Ki67-positive cells and insulin-positive cells (0.1% for the DMSO group and 0.03% for the rapamycin group, p < 0.05, Fisher’s exact test; Fig. 6), but TUNEL staining of a cell apoptotic marker did not reveal any difference (data not shown). This result indicates that rapamycin treatment reduced islet size by inhibiting beta cell proliferation rather than increasing beta cell apoptosis.
Fig. 5.
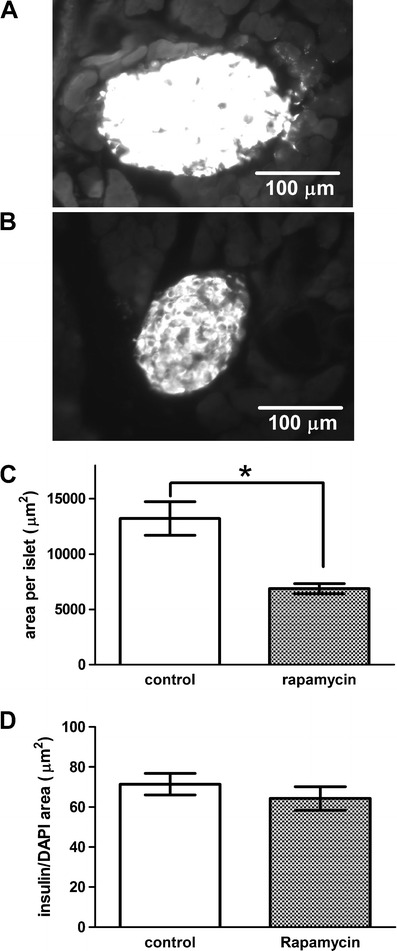
Rapamycin reduced islet size. Representative images of pancreatic sections from DMSO-treated (a) or rapamycin-treated (b) mice, with beta cell mass outlined by insulin-positive cells. Rapamycin significantly reduced islet size (c) (p < 0.05 unpaired t test), but not the average insulin-positive cell area that was determined by measuring the area of insulin staining for each islet normalized by the total cell number measured by DAPI counts (d)
Fig. 6.
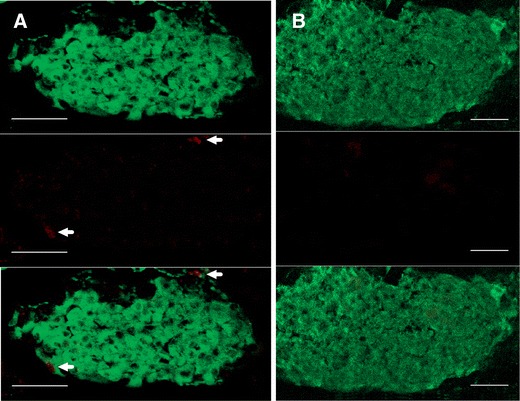
Rapamycin reduced beta cell proliferation. Immunostaining of pancreatic islets from control mice (a) or mice treated with rapamycin for 3 weeks (b). Beta cells were identified by insulin (green, top panel) and proliferating cells were marked by Ki67, a cell proliferation marker (red, middle panel). Bottom panels showed merged images of both insulin and Ki67. Proliferating beta cells can be seen in control islets (white arrows) (a), but not in islets from rapamycin-treated mice (b). Scale bars, 50 μm
Rapamycin reduces insulin sensitivity
Given that mTOR is a key molecule in the insulin signaling pathway [2, 29], rapamycin inhibition of mTORC1 may cause not only hypoinsulinemia but also other effects such as reduced insulin sensitivity, as observed in sand rats [17]. Although the low dosage of encapsulated rapamycin in the diet did not alter insulin sensitivity of the aged mice, daily injection of lean C57B/L6 mice with rapamycin at 0.5 mg/kg for 3 weeks increased the homeostatic model assessment of insulin resistance (HOMA-IR) index (p < 0.005, Fig. 3d), indicating that chronic rapamycin treatment could cause insulin resistance. Indeed, in the intraperitoneal insulin tolerance test, rapamycin-treated mice showed a moderate but significant reduction of insulin sensitivity (p < 0.05, Fig. 7a).
Fig. 7.
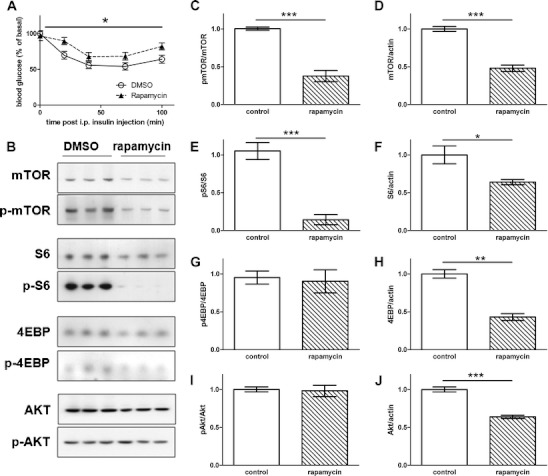
Rapamycin reduced insulin signaling and caused insulin resistance in lean mice. a After a 4-h fasting followed with 1 U/kg bovine insulin injection, the blood glucose measured at the designated time points from these mice revealed a reduction of insulin sensitivity in rapamycin-treated mice (p < 0.05, two-way ANOVA repeat measurement). Blood glucose levels were normalized to the initial value (t = 0 min). Each group had 12–16 mice. b Representative Western blot of mTOR, S6, and Akt in liver samples from mice treated for 3 weeks with either rapamycin or DMSO. c Phospho-mTOR/total mTOR (p < 0.005, unpaired t test). d Total mTOR/total actin (p < 0.005, unpaired t test). e Phospho-S6/total S6 (p < 0.005, unpaired t test). f Total S6/total actin (p < 0.05, unpaired t test). g Phospho-4EBP/total 4EBP (p > 0.05, unpaired t test). h Total 4EBP/total actin (p < 0.01, unpaired t test). i Phospho-Akt/total Akt (p > 0.05, unpaired t test). j Total Akt/total actin (p < 0.005, unpaired t test). n = 3 for each group
To determine how rapamycin treatment might reduce insulin sensitivity, we examined liver protein extracts from rapamycin-treated mice and control mice (Fig. 7b). Chronic rapamycin treatment reduced the extent of phosphorylated mTOR (Ser2448) and phosphorylated S6 (Ser235/6, p < 0.005) but not phosphorylated 4EBP (Thr70) after normalization with the respective protein level (Fig. 7c, e, g). We also found lower levels of mTOR (p < 0.005, Fig. 7d), S6 (p < 0.05, Fig. 7f), and 4EBP (p < 0.01, Fig. 7h) proteins in the liver from rapamycin-treated mice. Whereas there was no significant alteration of the extent of phosphorylated Akt (Ser473) after normalization with the total Akt protein level (Fig. 7i), rapamycin treatment did cause a significant reduction of the Akt protein level (p < 0.005, Fig. 7j) so that the amount of phosphorylated Akt was actually lower in the liver from rapamycin-treated mice. This reduction of mTOR signaling as well as Akt activity could compromise insulin signaling [30].
Discussion
We found that the low-dose rapamycin treatment that increases life span in aged mice [5] caused glucose intolerance by reducing plasma insulin levels before and after glucose stimulation. In lean C57B/L6 mice, a higher dose of rapamycin treatment not only reduced plasma insulin and plasma C-peptide levels but also caused insulin resistance accompanied with compromised hepatic mTOR and insulin signaling. We further characterized the impairment in insulin secretion of pancreatic islets from rapamycin-treated mice, showing that it likely arose from a reduction in beta cell mass and insulin content.
Previous studies report that chronic rapamycin treatment raises plasma insulin of DIO mice [19] and of rats displaying impaired hepatic insulin clearance [18]. In our study, we show that chronic rapamycin treatment of young as well as old mice causes glucose intolerance in a dose-dependent and reversible manner (Fig. 2). Under standard rapamycin regimen (0.5 mg/kg), it took about 2 weeks to fully develop glucose intolerance, and glucose tolerance was restored 2 weeks after stopping the rapamycin regimen (Fig. 2c). Given that the beta cell proliferation rate is very slow, we suspect that a reduction of insulin content upon rapamycin treatment may contribute significantly to a faster onset and offset of glucose intolerance (Fig. 3e, f). Previous studies have shown that insulin turnover rate is about 48–72 h in rodents [31]. Since both the insulin secretory machinery and the excitation–secretion coupling are not affected, rapamycin may slow down the de novo synthesis of insulin without affecting preexisting insulin in the secretory granules. This way, it may take a few days, if not weeks, to deplete the preexisting insulin pool even if rapamycin halts the synthesis of insulin. Also, we suspect that during the first few days after stopping rapamycin treatment, the residual rapamycin in the system might still be effective in suppressing insulin production and inhibiting beta cell proliferation so that the recovery is delayed until rapamycin level is reduced below its pharmacological effective concentration. Indeed, in vivo pharmacokinetic analysis has revealed that rapamycin has a relatively long half-life (5–12 h), larger volume of distribution, and extensive tissue binding [32].
Rapamycin treatment of lean C57B/L6 mice reduces the glucose-stimulated insulin secretion in both in vivo and ex vivo conditions (Fig. 3). Our electrophysiological recordings revealed no alterations in glucose stimulus–excitation coupling and the exocytotic machinery of beta cells from rapamycin-treated mice (Fig. 4). Therefore, the impaired insulin secretion is likely due to a reduction of the total insulin content by 70% (Fig. 3f) and of the average beta cell mass by 50% in rapamycin-treated mice (Fig. 5). Since both the insulin-positive cell area (Fig. 5e) and the membrane capacitance of beta cells were normal in islets from rapamycin-treated mice, these mice most likely have fewer beta cells. Indeed, we found a reduction in dividing cells as well as insulin-containing cells in islets from rapamycin-treated mice (Fig. 6), consistent with previous studies [17, 28]. Nir et al. [33] have demonstrated the regenerative capacity of mouse pancreatic beta cells after diabetogenic injury, and they have also shown that immunosuppressants such as rapamycin and tarcolimus used in the Edmonton protocol, a pancreatic islet transplantation procedure for treating type I diabetes in patients, prevents beta cell proliferation.
Our study has important clinical implications, given that long-term rapamycin (sirolimus) exposure is required to prolong life span or treat illnesses such as Alzheimer’s disease, organ transplant, and cancer [3–5] and given the consideration of rapamycin in human clinical trials [6, 7]. Our findings raise the concern that chronic low-dose rapamycin regimen has the potential to disrupt glucose homeostasis, thereby elevating the risk for hyperglycemia, which could lead to diabetes. A reduction in circulating insulin causing glucose intolerance could be a double-edged sword: lowering the insulin level impairs glucose homeostasis, extending life span, but also likely compromising the quality of life.
How can the conflicting reports of rapamycin effects be reconciled? On the one hand, overactivation of the mTOR-S6K pathway may be diabetogenic in mammals. For example, in a short-term in vivo study on human subjects, rapamycin ameliorates glucose intolerance caused by nutrient abundance of amino acids [34]. Additionally, deleting S6K1 alleviates insulin resistance and improves beta cell survival under conditions of chronic hyperglycemia [15], and rapamycin injection prevents the onset of insulin dependent diabetes in non-obese diabetic mice [35]. Conversely, other studies have suggested that rapamycin is diabetogenic: a retrospective study has shown that kidney transplant patients taking rapamycin have a significantly higher risk of developing diabetes [16]; rapamycin also induces diabetes in DIO sand rats [17] and mice [19]. Our study has further uncovered the diabetogenic effects of rapamycin in young and old lean inbred C57B/L6 mice. One way to reconcile these conflicting findings is to consider that the effects of rapamycin are different depending on the duration of the treatment: short-term rapamycin treatment seems to alleviate the metabolic symptoms [34], whereas chronic rapamycin treatment reduces insulin secretion and decreases insulin sensitivity; hence, chronic rapamycin treatment is diabetogenic and causes deterioration of metabolic symptoms in mice of different ages and strains (Figs. 1 and 2).
In summary, our study shows that chronic rapamycin is diabetogenic in mice. Chronic inhibition of mTORC1 with rapamycin impairs insulin secretion and causes insulin resistance. It would be prudent to monitor glucose homeostasis in patients treated with rapamycin or other mTOR inhibitors to minimize the risk of developing metabolic syndromes.
Acknowledgements
We thank members of the Xu and Vaisse laboratories (Diabetes Center, UCSF) for discussions. We thank Greg Zsot for excellent technical support (Diabetes Center, UCSF). This work was supported by NIH grant R37MH065334 and the American Diabetes Association Mentor-Based Fellowship 7-06-MN-29 (to S.B.Y.). Y.N.J. and L.Y.J. are investigators of the Howard Hughes Medical Institute.
Disclosure of potential conflict of interests
The authors declare no conflict of interests related to this study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Wullschleger S, Loewith R, Oppliger W, Hall MN. Molecular organization of target of rapamycin complex 2. J Biol Chem. 2005;280:30697–30704. doi: 10.1074/jbc.M505553200. [DOI] [PubMed] [Google Scholar]
- 2.Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. 2009;296:E581–E591. doi: 10.1152/ajpendo.90437.2008. [DOI] [PubMed] [Google Scholar]
- 3.Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamerman D. Can biogerontologists and geriatricians unite to apply aging science to health care in the decade ahead? J Gerontol A Biol Sci Med Sci. 2010;65:1193–1197. doi: 10.1093/gerona/glq117. [DOI] [PubMed] [Google Scholar]
- 7.McCormick MA, Tsai SY, Kennedy BK. TOR and ageing: a complex pathway for a complex process. Philos Trans R Soc Lond B Biol Sci. 2011;366:17–27. doi: 10.1098/rstb.2010.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alberti KG. Impaired glucose tolerance: what are the clinical implications? Diabetes Res Clin Pract. 1998;40(Suppl):S3–S8. doi: 10.1016/s0168-8227(98)00035-7. [DOI] [PubMed] [Google Scholar]
- 9.Baron AD. Impaired glucose tolerance as a disease. Am J Cardiol. 2001;88:16H–19H. doi: 10.1016/S0002-9149(01)01832-X. [DOI] [PubMed] [Google Scholar]
- 10.Nijpels G, Popp-Snijders C, Kostense PJ, Bouter LM, Heine RJ. Cardiovascular risk factors prior to the development of non-insulin-dependent diabetes mellitus in persons with impaired glucose tolerance: the Hoorn Study. J Clin Epidemiol. 1997;50:1003–1009. doi: 10.1016/S0895-4356(97)00119-4. [DOI] [PubMed] [Google Scholar]
- 11.Meigs JB, Nathan DM, D’Agostino RB, Sr, Wilson PW. Fasting and postchallenge glycemia and cardiovascular disease risk: the Framingham Offspring Study. Diabetes Care. 2002;25:1845–1850. doi: 10.2337/diacare.25.10.1845. [DOI] [PubMed] [Google Scholar]
- 12.Lamport DJ, Lawton CL, Mansfield MW, Dye L. Impairments in glucose tolerance can have a negative impact on cognitive function: a systematic research review. Neurosci Biobehav Rev. 2009;33:394–413. doi: 10.1016/j.neubiorev.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 14.Lee DF, Kuo HP, Chen CT, Wei Y, Chou CK, Hung JY, Yen CJ, Hung MC. IKKbeta suppression of TSC1 function links the mTOR pathway with insulin resistance. Int J Mol Med. 2008;22:633–638. doi: 10.3892/ijmm_00000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Johnston O, Rose CL, Webster AC, Gill JS. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol. 2008;19:1411–1418. doi: 10.1681/ASN.2007111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, Berthault MF, Magnan C, Cerasi E, Kaiser N, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57:945–957. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- 18.Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, Marette A. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59:1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang GR, Wu YY, Chiu YS, Chen WY, Liao JW, Hsu HM, Chao TH, Hung SW, Mao FC. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol. 2009;105:188–198. doi: 10.1111/j.1742-7843.2009.00427.x. [DOI] [PubMed] [Google Scholar]
- 20.Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295:E1323–E1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- 21.Sawada T, Matsumoto I, Nakano M, Kirchhof N, Sutherland DE, Hering BJ. Improved islet yield and function with ductal injection of University of Wisconsin solution before pancreas preservation. Transplantation. 2003;75:1965–1969. doi: 10.1097/01.TP.0000068871.09469.E0. [DOI] [PubMed] [Google Scholar]
- 22.Speier S, Rupnik M. A novel approach to in situ characterization of pancreatic beta-cells. Pflugers Arch. 2003;446:553–558. doi: 10.1007/s00424-003-1097-9. [DOI] [PubMed] [Google Scholar]
- 23.Lee HY, Yea K, Kim J, Lee BD, Chae YC, Kim HS, Lee DW, Kim SH, Cho JH, Jin CJ, et al. Epidermal growth factor increases insulin secretion and lowers blood glucose in diabetic mice. J Cell Mol Med. 2008;12:1593–1604. doi: 10.1111/j.1582-4934.2007.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 25.Saunders RN, Metcalfe MS, Nicholson ML. Rapamycin in transplantation: a review of the evidence. Kidney Int. 2001;59:3–16. doi: 10.1046/j.1523-1755.2001.00460.x. [DOI] [PubMed] [Google Scholar]
- 26.Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 27.Rozzo A, Meneghel-Rozzo T, Delakorda SL, Yang SB, Rupnik M. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Ann N Y Acad Sci. 2009;1152:53–62. doi: 10.1111/j.1749-6632.2008.04003.x. [DOI] [PubMed] [Google Scholar]
- 28.Zahr E, Molano RD, Pileggi A, Ichii H, San Jose S, Bocca N, An W, Gonzalez-Quintana J, Fraker C, Ricordi C, et al. Rapamycin impairs beta-cell proliferation in vivo. Transplant Proc. 2008;40:436–437. doi: 10.1016/j.transproceed.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007;13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagle SR. Studies on biosynthesis and catabolism of insulin. Biochim Biophys Acta. 1965;107:524–530. doi: 10.1016/0304-4165(65)90196-0. [DOI] [PubMed] [Google Scholar]
- 32.Wood M, Bierer B. Rapamycin: biological and therapeutic effects, binding by immunophilins and molecular targets of action. Perspect Drug Discov Des. 1994;2:163–184. doi: 10.1007/BF02171742. [DOI] [Google Scholar]
- 33.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. 2007;117:2553–2561. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Furnsinn C, Promintzer M, Anderwald C, et al. The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007;56:1600–1607. doi: 10.2337/db06-1016. [DOI] [PubMed] [Google Scholar]
- 35.Baeder WL, Sredy J, Sehgal SN, Chang JY, Adams LM. Rapamycin prevents the onset of insulin-dependent diabetes mellitus (IDDM) in NOD mice. Clin Exp Immunol. 1992;89:174–178. doi: 10.1111/j.1365-2249.1992.tb06928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]