

Abstract
During the last 10 years, a series of exciting observations has led to a new theory of pathophysiology using insights from evolutionary biology and neuroendocrine immunology to understand the sequelae of chronic inflammatory disease. According to this theory, disease sequelae can be explained based on redirection of energy-rich fuels from storage organs to the activated immune system. These disease sequelae are highly diverse and include the following: sickness behavior, anorexia, malnutrition, muscle wasting–cachexia, cachectic obesity, insulin resistance with hyperinsulinemia, dyslipidemia, increase of adipose tissue near inflamed tissue, alterations of steroid hormone axes, elevated sympathetic tone and local sympathetic nerve fiber loss, decreased parasympathetic tone, hypertension, inflammation-related anemia, and osteopenia. Since these disease sequelae can be found in many animal models of chronic inflammatory diseases with mammals (e.g., monkeys, mice, rats, rabbits, etc.), the evolutionary time line goes back at least 70 million years. While the initial version of this theory could explain prominent sequelae of chronic inflammatory disease, it did not however address two features important in the pathogenesis of immune-mediated diseases: the time point when an acute inflammatory disease becomes chronic, and the appearance of hypertension in chronic inflammation. To address these aspects more specifically, a new version of the theory has been developed. This version defines more precisely the moment of transition from acute inflammatory disease to chronic inflammatory disease as a time in which energy stores become empty (complete energy consumption). Depending on the amount of stored energy, this time point can be calculated to be 19–43 days. Second, the revised theory addresses the mechanisms of essential hypertension since, on the basis of water loss, acute inflammatory diseases can stimulate water retention using a positively selected water retention system (identical to the energy provision system). In chronic smoldering inflammation, however, there is no increased water loss. In contrast, there is increased water generation in inflamed tissue and inflammatory cells, and the activation of the water retention system persists. This combination leads to a net increase of the systemic fluid volume, which is hypothesized to be the basis of essential hypertension (prevalence in adults 22–32%).
Keywords: Hormone, Autoimmunity, System biology
Introduction
Despite the enormous progress in genetic and genomic studies, including extensive analysis of the genome of several species, as few as 5–10% of discovered gene functions are known and many more remain to be discovered. To help unravel this complexity, some investigators have developed conceptual frameworks to organize the ever increasing mass of data and to facilitate incorporation of new information. Most of us accept the idea that insights into networks are crucial to elucidate the pathogenesis of chronic diseases (e.g., ref. [1]). Indeed, the following network approaches have been successfully applied for different levels of biomedical science: (1) statistical bioinformatics (correlations of gene expression, metabolite levels, and protein concentrations without any causal relationships), (2) intracellular molecular networks as exemplified by the pathways identified in classical biochemistry [2], (3) paracrine cytokine and immune cell networks in immunological systems [3], and (4) psychoneuroendocrine immune networks that operate at higher integrative physiological levels [4].
Although evolutionary biology and medicine have strong explanatory power to understand disease- and age-related problems [5–7], until recently, these disciplines have not been significantly incorporated into the network theories. The presence of the neuroendocrine and immune systems that are instrumental in this report is documented in all mammals but also in more remote vertebrates such as the fowl, rainbow trout, or shark. Thus, the evolutionary time line of the neuroendocrine immune network that is basis of the present theory goes back at least 400 million years (distance to the suggested common ancestor).
During the last 10 years, the author has developed a framework to understand important steps in disease pathogenesis on the basis of insights from the fields of evolutionary biology relevant to the role of neuroendocrine immune energy regulation in homeostasis. The theory explains many seemingly unrelated disease sequelae in chronic inflammatory diseases [8–10]. The published concept is shortly recapitulated here (in form of a minireview), and two novel amendments to the theory are presented.
The framework of evolutionary biology and neuroendocrine immune energy regulation in pathophysiology of chronic inflammatory diseases
To meet the challenges of infection and other environmental stress, an activated immune system has an urgent need for energy-rich substrates that must be allocated from internal and external energy stores (glycogen, proteins, triglycerides, or free fatty acids) [9]. While an activated immune system requires substantial energy in a quiescent state, this requirement rises in an active phase of inflammation. Table 1 lists energy expenditure under various conditions of different systems and organs.
Table 1.
| System/organ | Energy expenditure per day (kJ/day) |
|---|---|
| Total body basal MR | 7,000 |
| Total body MR with usual activity | 10,000 |
| Total body MR of a Tour de France bicyclist | 30,000a |
| Total body MR during minor surgery | 11,000 |
| Total body MR with multiple bone fractures | up to 13,000 |
| Total body MR with sepsis | 15,000 |
| Total body MR with extensive burns | 20,000 |
| Total body daily uptake (absorptive capacity in the gut) | 20,000 |
| Immune system MR under normal conditions | 1,600b |
| Immune system MR moderately activated | 2,100b |
| Central nervous system MR | 2,000 |
| Muscle MR at rest | 2,500 |
| Muscle MR activated | 2,000–10,000 and more |
| Liverc MR | 1,600 |
| Kidneys MR | 600 |
| Gastrointestinal tractc MR | 1,000 |
| Abdominal organs (together)c MR | 3,000–3,700 |
| Lungc MR | 400 |
| Heart MR | 1,100 (and more when activated) |
| Thoracic organs (together)c MR | 1,600–2,400 |
10,000 kJ = 2,388 kcal
MR metabolic rate
aSuch a high energy expenditure can be maintained only for a very short period of time
bSee derivation of energy need in ref. [9]. Leukocytes use all types of fuels, but the main source is glucose and glutamine making roughly 70% of the fuels needed [54–56]
cEnergy need is difficult to estimate independent of the immune system in these organs
Since uptake of energy-rich substrates has an upper threshold of 20,000 kJ per day (the absorptive capacity in the gut) [11, 12], it is important for the organism to partition energy-rich substrates according to fine-tuned mechanisms of energy regulation. Studies in a number of systems have demonstrated that circadian rhythms of neuronal, endocrine, and immune systems can regulate energy metabolism and partition fuels to daytime and nighttime consumers [9, 13–15]. Since most aspects in a living body occur with constraints on energy availability, regulation of energy storage and provision occupies a very high position in the hierarchy of homoeostatic neuroendocrine immune control. Energy regulation operates not only in the cell but also in coordinating centers of the brain and in endocrine organs that integrate organismal function. Important questions concern the origin of these regulatory mechanisms and the extent to which they have been subject to positive selection in the context of chronic inflammation.
It has been hypothesized that genes responsible for energy regulation have been positively selected under conditions without chronic inflammation [8, 10]. Chronic diseases and chronic inflammatory diseases in particular can exert a high negative selection pressure in evolution. Chronic inflammatory diseases can lead to loss of reproduction potential because affected individuals are at strong disadvantage. Such individuals can be excluded or impaired in the competition for food; they can also have low social status in the group and limited choice or availability of sexual partners. In addition, intense, persistent inflammation can inhibit the hypothalamic–pituitary–gonadal axis, leading to impairment of fertility as demonstrated in studies on the effect of chronic inflammatory diseases on fertility despite good medical control [16–20].
In some situations, however, the presence of a chronic inflammatory disease may not exert any selective pressure despite its potential severity. Many chronic inflammatory diseases only become manifested in patients at older ages. Because of the short life expectancy in the past, our ancestors have not suffered from the chronic inflammatory diseases that we know today. Their genes have been positively selected for different aspects such as reproduction, muscular work, defense against infections, metabolism, and similar important activities. Furthermore, in the past history, there may have been no time for natural selection. This situation would occur if various chronic inflammatory diseases did not exist until 100 to 200 years ago. Indeed, there is a suggestion that rheumatoid arthritis and Crohn’s disease, for example, are relatively new diseases.
It has been hypothesized that the genes relevant to the etiology of today’s chronic inflammatory diseases have been positively selected for their effects on normal host defense as well as the host response to serious, albeit non-life-threatening, inflammatory episodes in contrast to chronic inflammatory diseases (Table 2) [8]. These issues remain under investigation. Nevertheless, despite uncertainty about their origin, many genes have been linked to an increased risk or more severe forms of chronic inflammatory diseases including polymorphisms for human leukocyte antigens (HLA) and polymorphisms for other elements of the immune system detected by genome-wide association studies. However, these genes were most probably evolutionarily conserved because they supported positive selection at younger ages. This concept was developed in the 1950s in the context of aging research and the theory was called “antagonistic pleiotropy” [7].
Table 2.
Non-life-threatening episodes of inflammation that can occur normally and form the basis of selection and conservation during evolution
| Immune response due to infection |
| Control of inner and outer body surfaces |
| Reactions with foreign bodies |
| Wound healing |
| Immunosurveillance in tissue |
| Implantation of stems cells into injured tissue |
| Implantation of a blastocyst into the uterine epithelium |
| Immune phenomena facilitating semiallogenic pregnancy |
| Replacement of cells and tissue (physiological regeneration and degeneration) |
| Apoptosis |
An example of antagonistic pleiotropy illustrates these ideas. HLA DR4 (DRB1*04) is a well-known risk factor for rheumatoid arthritis and other chronic autoimmune diseases, but HLA DR4 (DRB1*04) also has a strong negative association with dengue hemorrhagic fever [21]. HLA DR4 (DRB1*04) homozygous individuals are 12 times less likely to develop dengue hemorrhagic fever [21]. Thus, it seems likely that HLA DR4 (DRB1*04) was positively selected to overcome dengue hemorrhagic fever but not to stimulate rheumatoid arthritis and other autoimmune diseases in later life. Further examples of antagonistic pleiotropy are demonstrated in Table 3. Antagonistic pleiotropy can explain why genes positively selected at younger ages can have disease-supporting roles in later life independent of the selection process.
Table 3.
Examples of antagonistic pleiotropy for genes that increase risk or severity of chronic inflammatory diseases
| Genes | Chronic inflammatory disease | Pleiotropic meaning outside of chronic inflammatory diseases (with selection advantage) | Refs. |
|---|---|---|---|
| HLA DR4 (DRB1*04) | Rheumatoid arthritis and other autoimmune diseases | Decrease of risk of dengue hemorrhagic fever (defense against infectious agents) | [21] |
| HLA B27 | Ankylosing spondylitis and other axial forms of spondyloarthritis | Decrease of viral infection (defense against infectious agents) | [57, 58] |
| PTPN22 1858 C>T* | Many autoimmune diseases | Higher body mass index, higher waist-to-hip ratio in women (storage of energy-rich fuels) | [59] |
| CTLA4 49 A>G | Many autoimmune diseases | Better defense against hepatitis B virus and Helicobacter pylori (defense against infectious agents) | [60, 61] |
| NOD2/CARD15 | Crohn’s disease | Hypertension (activation of the sympathetic nervous system) | [62] |
*PTPN22 1858 C>T is associated with many autoimmune diseases but is also linked to a higher risk of infection. This seems to contradict the theory of antagonistic pleiotropy. Until today, nobody has focused on a possible selection advantage in the context of reproduction. It might well be that this mutation is related to a decreased T cell - dependent rejection of the semiallogenic fetus. This would support reproduction. The PTPN22 1858 C>T mutation is linked to increased risk of endometriosis which already demonstrates a role in the context of reproduction [63]
In summary, it seems plausible that specific genes have not been selected or conserved in evolution because of their putative role in chronic inflammatory diseases, either positive or negative. Rather, genes, signaling pathways, and networks operative in symptomatic chronic inflammatory diseases were likely adapted from normal physiology or serious, albeit non-life-threatening, inflammatory episodes; the outcomes of such transient events, for which recovery is possible, would not have blocked the gene transmission to the progeny (Table 2). The same situation is true for neuroendocrine immune regulation of energy storage and energy allocation to consumers.
On the basis of these considerations, typical disease-related sequelae that are very common in chronic inflammatory diseases have been linked to transient inflammatory episodes [9, 10]. Here, these sequelae have an important function in supporting the transiently activated immune system and tissue inflammation (Table 4). The disease sequelae noted in Table 4 have usually been considered as independent phenomena in the absence of a unifying model to explain both their prominence and their coexistence. As a common denominator, the redirection of energy-rich fuels from energy stores to the activated immune system can explain systemic disease sequelae [9]. This theory can also explain the observed disturbances of the neuroendocrine immune regulation in chronic inflammatory diseases [9].
Table 4.
Sequelae of chronic inflammatory diseases in the light of altered energy regulation
| Disease sequelae | Pathophysiological elements in chronic inflammation leading to energy allocation to an activated immune system |
|---|---|
| Depressive symptoms/fatigue | Cytokine (e.g., IL-1β)-driven sickness behavior and fatigue which increase time at rest (muscles and brain in an inactive state) |
| Anorexia | Consequence of sickness–behavior and fatigue |
| Malnutritiona | Consequence of anorexia and sickness behavior |
| Muscle wasting–cachexia | Protein breakdown in muscles as a consequence of anorexia, sickness behavior and androgen deficit |
| Cachectic obesity | Protein breakdown in muscles as a consequence of anorexia and sickness behavior (protein breakdown > fat breakdown) |
| Insulin (IGF-1) resistance (with hyperinsulinemia) | Cytokine (e.g., TNF)-induced insulin signaling defects in the liver, muscle, and fat tissue but not in immune cells. Immune cells need insulin so that high insulin levels support the activity of the immune system (similar for IGF-1) |
| Dyslipidemiab | Cytokine-driven acute phase reaction of lipid metabolism leading to higher delivery of cholesterol and other lipids to macrophages |
| Increase of adipose tissue in the proximity of inflammatory lesions | Presence of adipose tissue surrounding lymph nodes and in the proximity of inflammatory lesions reflects a local store of energy-rich fuels (increased local estrogens might be important to drive local accumulation of adipose tissue). Adipokines play a proinflammatory role |
| Alterations of steroid hormone axes | Cytokine/leptin-driven hypoandrogenemia supports muscle breakdown and protein delivery for gluconeogenesis and support of an activated immune system (alanine, glutamine). Cortisol-to-androgen preponderance in chronic inflammation is catabolic |
| Elevated sympathetic tone and local sympathetic nerve fiber loss | Cytokine-driven increase of SNS activity increases gluconeogenesis and lipolysis. The parallel loss of sympathetic nerve fibers in inflamed tissue supports local inflammation [64]. It also stimulates lipolysis in the surrounding adipose tissue because sympathetic nerve fibers are increased there [65] |
| Hypertension | Cytokine-driven activation of the water retention system due to systemic water loss during inflammation |
| Decreased parasympathetic tone | Cytokine-driven decrease of the PSNS activity supports allocation of energy-rich fuels to an activated immune system |
| Inflammation-related anemia | Cytokine-driven anemia is linked to reduced energy expenditure for erythropoiesis, increased time at rest, and insulin resistance (see above), all of which support energy allocation to the immune system |
| Osteopenia | High calcium and phosphorus are mandatory for energy-consuming reactions. Driven by cytokines and PTH-related peptide during inflammation. In addition, an activated SNS and HPA axis stimulate bone resorption |
References that demonstrate the respective disease sequelae and the pathophysiological explanation can be found in detail in reference [9]
HPA axis hypothalamic–pituitary–adrenal axis, IGF insulin-like growth factor, IL interleukin, PTH parathyroid hormone, SNS sympathetic nervous system, TNF tumor necrosis factor
aHypovitaminosis D and others, deficiency in zinc, iron, copper, magnesium, and similar
bDyslipidemia in chronic inflammation reflects low levels of HDL cholesterol and/or apolipoprotein A–I and appearance of an “inflammatory HDL subfraction” with increased serum amyloid A and ceruloplasmin
In this conceptualization, the induction of systemic responses results from an appeal for energy-rich substrates, called the energy appeal reaction [9]. In this scenario, the energy appeal reaction has been positively selected for transient inflammatory episodes. Respective genes and networks provided a survival value if used for a short period of time (transient inflammatory process). However, prolonged operation of these adaptive programs such as in chronic inflammatory diseases of today can in itself become pathogenic. Thus, one can add the “systemic response” to the list of classical etiologic factors of chronic inflammatory diseases (Table 5) [10]. The “systemic response” in itself can be disease perpetuating.
Table 5.
The etiological factors in chronic inflammatory diseases
| Genetic susceptibility (gene polymorphisms; polygenic) |
| Complex environmental priming (microbes, toxins, drugs, injuries, radiation, cultural background, and geography) |
| Immune response (exaggerated and continuous immune response against harmless self or foreign antigen) |
| Tissue destruction (continuous wound response without proper healing but fibrotic scarring) |
| Systemic response (support of the immune and wound response by redirection of energy-rich fuels leading to unwanted disease sequelae) |
Refinement of the existing theory
From acute to chronic inflammatory disease—where is the boundary?
An acute infectious disease can be self-limiting and involve an innate immune response of 2 to 3 days; the subsequent phase of the adaptive immune response can last approximately 3 to 4 weeks. Similarly, many transient inflammatory episodes include a healing phase that involves adaptive immune responses (Table 2). The typical germinal center reaction of affinity maturation of B cells occurs for 3 to 4 weeks from the beginning to end [22, 23]. The accompanying proliferative T cell response undergoes a similar time course [24]. The response of the adaptive immune system with a maximum of proliferation until days 12 to 14 and subsequent contraction until days 21 to 28 is surprisingly constant in various acute infectious diseases [24]. Upon generation of immunological memory, the entire process can be markedly shortened, and this was a selection advantage [24]. An important question concerns the dynamics of this response: why do the increase and decrease of the adaptive immune response not last longer?
Acute self-limiting infectious diseases can be very energy consuming although, in the presence of disease-induced sickness behavior and related anorexia (Table 4) [9, 25], intake of energy-rich substrates can be significantly inhibited. To explain these seemingly disparate observations, it can be proposed that sickness behavior represents an element of an adaptive program that has been positively selected for transient immune and inflammatory reactions to limit energy utilization for such activities as foraging and courtship behavior [8, 26, 27]. In such a situation, the immune response can not last forever because energy stores run empty.
Furthermore, sickness behavior can restrain activity and, for example, confine the affected individual to a safe place to keep away predators [8, 26, 27]. In considering energy stores available during these responses, it is important to note that storage occurs primarily in fat tissue (12 kg of triglycerides in the body of a contemporary person, 500,000 kJ), as well as in the liver (150 g glycogen, 2.500 kJ) or muscles (300 g glycogen, 5,000 kJ; 6–7 kg muscle protein, 50,000 kJ) [11, 12].
Under conditions of sickness behavior and anorexia without uptake of energy-rich substrates but an increased sickness-related metabolic rate (Tables 1 and 6), the total amount of stored energy would only last for 19 to 43 days in females and 28 to 41 days in males (Table 6). The number is relatively similar for domestic fowl which have an evolutionary distance to Homo sapiens of 300 million years (Table 6). In other words, an acute consuming infectious disease that uses all energy stores can only last until the stores are empty, say 19 to 43 days. Thus, the evolutionarily positively selected increase and decrease of an adaptive immune response must fit into this prespecified time frame. Due to the physical restrictions of energy storage under natural paleolithic conditions, an acute infectious disease may not last much longer than 3 to 6 weeks. It can be hypothesized that energy considerations can help explain the relatively constant time course of an adaptive immune response in the context of acute infection.
Table 6.
Total consumption time in human evolution
| Species | Date range (Ma, ka) | Body mass (kg) | Sickness-related metabolic ratea (kJ/day) | Stored energy (kJ) | Total consumption time (day) |
|---|---|---|---|---|---|
| Females | |||||
| A. afarensis | 3.9–3.0 Ma | 29 | 7,925 | 152,448 | 19.2 |
| A. africanus | 3.0–2.4 Ma | 30 | 8,061 | 163,943 | 20.3 |
| P. boisei | 2.3–1.4 Ma | 34 | 8,581 | 195,237 | 22.8 |
| P. robustus | 1.9–1.4 Ma | 32 | 8,325 | 177,921 | 21.4 |
| H. habilis | 1.9–1.6 Ma | 32 | 8,325 | 177,921 | 21.4 |
| H. ergaster | 1.9–1.7 Ma | 52 | 10,612 | 404,052 | 38.1 |
| H. erectus | 1.8 Ma–200 ka | 52 | 10,612 | 404,052 | 38.1 |
| H. neanderthalensis | 250 ka–30 ka | 52 | 10,612 | 372,884 | 35.1 |
| H. sapiens | 100 ka–1900 | 50 | 10,406 | 294,034 | 28.3 |
| H. sapiens | Today (USA) | 74 | 12,660 | 545,052 | 43.1 |
| Males | |||||
| A. afarensis | 3.9–3.0 Ma | 45 | 9,872 | 275,502 | 27.9 |
| A. africanus | 3.0–2.4 Ma | 41 | 9,423 | 238,194 | 25.3 |
| P. boisei | 2.3–1.4 Ma | 49 | 10,302 | 303,277 | 29.4 |
| P. robustus | 1.9–1.4 Ma | 40 | 9,308 | 214,241 | 23.0 |
| H. habilis | 1.9–1.6 Ma | 37 | 8,952 | 198,282 | 22.2 |
| H. ergaster | 1.9–1.7 Ma | 66 | 11,956 | 485,500 | 40.6 |
| H. erectus | 1.8 Ma–200 ka | 66 | 11,956 | 485,500 | 40.6 |
| H. neanderthalensis | 250 ka–30 ka | 70 | 12,313 | 509,846 | 41.4 |
| H. sapiens | 100 ka–1900 | 65 | 11,865 | 377,130 | 31.8 |
| H. sapiens | Today (USA) | 86 | 13,648 | 558,908 | 41.0 |
| Animals | |||||
| Domestic pig (adult) | 65 Ma distanceb | 100 | 13,009 | 754,611 | 58.0 |
| Domestic fowl (adult) | 300 Ma distanceb | 3.7 | 1,159 | 21,177 | 18.3 |
Time to total consumption was calculated based on known height and weight of our ancestors [66] using published formula [12, 32]. The last column gives the total consumption time as the ratio of stored energy divided by the sickness-related metabolic rate. Stored energy is a sum of stored triglycerides (roughly 54% of total fat) and usable proteins (roughly 38% of total proteins) using the conversion factor 37.6 kJ/g triglycerides and 16.7 kJ/g protein, respectively [32]. The small amount of glycogen was not entered into the calculation
aThe sickness-related metabolic rate is given as the basal metabolic rate multiplied by a factor of 1.5 (an 150% increase), which was demonstrated to be a good energy expenditure measure for moderate activation of the immune system [9, 67]
bDistance means time in evolution to most recent common ancestor
If an acute inflammatory response lasted longer than the time point at which energy stores were exhausted, the affected person would probably have died due to inanition and starvation, preventing transfer of genes to any offspring. If, however, an immune response lasted for a shorter period, the affected person could survive and be able to transfer favorable genes to offspring. These considerations thus suggest that the time point of total energy consumption marks the upper threshold of an acute disease period for which genes, signaling pathways, and networks can be subject to positive selection. But what happens to the survival of the individual after this physical threshold of total energy consumption?
It can be hypothesized that networks for more persistent inflammatory diseases do not exist because they have not undergone the pressure of positive selection. If a disease is progressive and longstanding (e.g., autoimmunity) and therapies are available to stop lethal emaciation (i.e., immunosuppression), a chronic phase can develop. For the chronic phase of such immune-mediated diseases, the organism would lack positively selected genes, signaling pathways, and networks to be able to stop the chronic disease in a coordinated fashion. Thus, sickness behavior and energy disturbance would remain and impair the function of the individual. It becomes a perpetuating factor (Table 5).
The framework of evolutionary biology and neuroendocrine immune energy regulation thus defines the point of transition from acute disease to chronic disease as the time point of complete energy consumption (19–43 days). In this regard, the point can represent a range in time because the consumption curve depends on stored energy reserves (Table 6).
Inflammation, energy demand, and water retention—an evolutionary link between inflammation and hypertension
Acute inflammatory episodes require abundant energy and are often accompanied by local and systemic water loss (Table 7). Local water loss is particularly important when the inflamed tissue has an exposed surface area such as the skin or to inner surfaces of the gastrointestinal, respiratory, or urogenital tract. For example, water loss through skin wounds depends on the surface area and can amount to 0.35 ml/cm2/day (area, 30 × 30 cm2 = 315 ml/day) [28]. Another example of water loss relates to insensible perspiration. Under normal conditions in adults, insensible perspiration through skin and respiratory tract can reach 0.5 ml/kg/h (example, 80 kg and 24 h = 960 ml/day) [29]. During surgical intervention, which is a model of mild acute inflammatory activation, water loss by insensible perspiration can amount to 1 ml/kg/h (example, 80 kg and 24 h = 1,820 ml/day) [29]. These numbers show that loss of water can be quite high during acute inflammatory episodes.
Table 7.
Water loss during transient inflammatory episodes
| Local loss of water from inflamed tissue |
| Loss from wounds by evaporation |
| Loss from inflamed tissue into the gastrointestinal tract (diarrhea, vomiting) |
| Loss into inflamed upper and lower airways (rhinitis, perspiration, expectoration) |
| Third-space fluid shifts into pleural cavity, peritoneal cavity, and similar |
| Systemic loss of water |
| By perspiration during fever |
| By sweating during fever |
| By triglyceride and glycogen breakdown from fat tissue and liver |
| By gluconeogenesis from lactate (Cori cycle between liver and inflamed tissue) |
| By muscle breakdown in the context of cachexia |
| By complete amino acid breakdown in the urea cycle in the liver |
| By cell proliferation (leukocyte proliferation in lymphoid tissue) |
In addition to physical loss of water via outer and inner surfaces, many metabolic reactions that degrade storage forms of energy-rich substrates need water for hydrolysis (Fig. 1) [30–32]. For example, the breakdown of one molecule of glucose from glycogen needs two molecules of water (Fig. 1, pink area in left upper corner); triglyceride breakdown needs three molecules of water for three molecules of free fatty acids (Fig. 1, pink and yellow area on the left side); and degradation of muscle protein needs particularly many molecules of water (Fig. 1, brown area in right upper corner) [31]. If amino acids are completely degraded within the urea cycle in the liver, one molecule of water is needed for one molecule of amino acid (Fig. 1, pink area in the left upper corner).
Fig. 1.
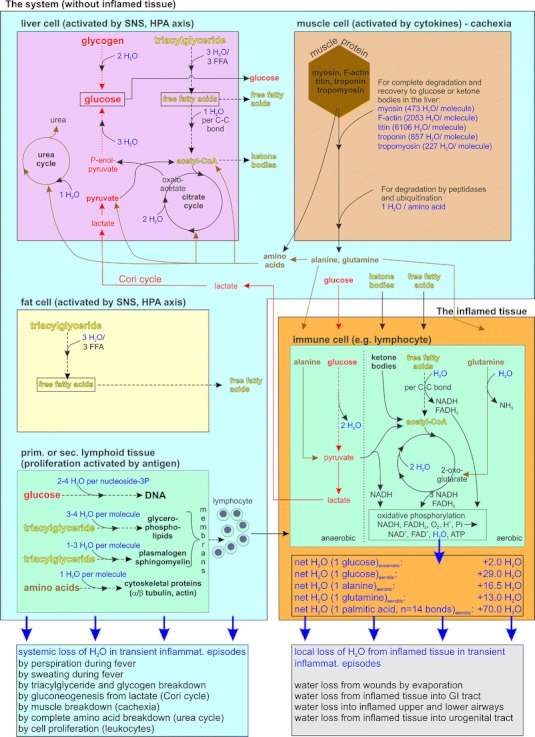
Water fluxes in the system (blue box) and inflamed tissue (orange box). Blue box it is demonstrated that liver cells (pink box) need water for degradation of glycogen, triglycerides, amino acids in the urea cycle, and for gluconeogenesis in the context of the Cori cycle (red arrows in the blue box). Similarly, degradation of muscle proteins in muscle (brown box) and triglycerides in fat tissue (yellow box) withdraw water from the system. An enormous amount of water is needed for proliferation of immune cells in primary and secondary lymphoid organs (green box) due to generation of DNA, membranes, cytoskeletal proteins, and many others. Orange box the inflamed tissue is separated from the system. In inflamed tissue activated cells, mainly immune cells, use provided energy-rich substrates such as glucose, amino acids such as glutamine and alanine, ketone bodies, and free fatty acids to overcome the inflammatory state. In the process of complete degradation of energy-rich substrates to CO2 and H2O, enormous amounts of water are generated (black-edged box in orange box). In an acute inflammatory situation, both, the system and the inflamed tissue, loose water by several pathways (given as boxes below the blue and orange box). C–C bond carbon–carbon bond, FFA free fatty acids, GI tract gastrointestinal tract, HPA axis hypothalamic–pituitary–adrenal axis, Pi inorganic phosphate, SNS sympathetic nervous system. The calculations of water fluxes is derived from biochemistry literature [30–32]
Furthermore, the generation of the Cori cycle between the lactate-producing inflamed tissue and the glucose-producing liver [33], involving gluconeogenesis, needs five molecules of water to reactivate one molecule of lactate (Fig. 1, red lines with arrows) [31]. Finally, the operation of a proper adaptive immune response with proliferation of B cells and T cells and proliferation of innate immune cells (neutrophils, monocytes) in primary and secondary lymphoid organs needs enormous amounts of water (Fig. 1, green area in left lower corner) [31]. The water loss described above is water loss in the system (liver, muscle, fat tissue, lymphoid organs, and others). It is not local water loss in inflamed tissue because here, in contrast, water is produced.
Water is formed when glucose, amino acids, and free fatty acids undergo degradation in the Krebs cycle and during oxidative phosphorylation; these processes occur in the mitochondria of activated cells in inflamed tissue (Fig. 1, orange area). Thus, important differences exist for water loss from the system and the generation of water from locally inflamed tissue. The amount of locally formed water can be large (Fig. 1, orange area), but usually water is lost rapidly in the environment of inflamed tissue via outer and inner surfaces (Table 7).
To overcome systemic water loss during acute inflammatory episodes and events such as trauma, hemorrhage, and burns, a water retention system is activated. The key elements of this system are the sympathetic nervous system (SNS) that activates the renin–angiotensin–aldosterone pathway [34], and the hypothalamic–pituitary–adrenal (HPA) axis with ACTH, aldosterone, and cortisol. ACTH can stimulate aldosterone as well as cortisol, which also has mineralocorticoid activity [35]. It may not be a simple coincidence that the SNS and the HPA axis can redirect energy-rich substrates from energy stores to the activated immune system as well as serve as major water retention systems. In this context, it is important to recall that the acute adaptive response described by Selye in the 1940s was considered a “nonspecific” alarm reaction [36]. Today we can say that Selye’s alarm reaction was not “nonspecific” but rather had important physiologic functions as both, a highly specific energy appeal reaction (as described earlier [9, 10]) as well as a specific water retention reaction.
The SNS and the HPA axis are supported by other alarm hormones such as vasopressin (which is lipolytic and water retentive), growth hormone (glucogenic, lipolytic, and water retentive), and insulin (lipogenic and water retentive). During transient inflammatory episodes, due to sickness behavior and anorexia, lipogenic insulin most probably plays a minor role because insulin secretion activated by the presence of carbohydrates would be downregulated. Thus, while sickness behavior may have antilipogenic consequences by suppressing insulin secretion, it can also be prolipolytic through activation of the SNS and the HPA axis. It leads to redirection of stored energy to the activated immune system.
These ideas can explain other activities of physiological mediators and provide insights in findings that, for example, water retention hormones such as angiotensin II have proinflammatory activities [37]. Angiotensin II uses the important proinflammatory signaling cascade of NF-κB activation similar as TNF [38]. Inhibition of the renin–angiotensin–aldosterone system by angiotensin-converting enzyme inhibitors or angiotensin II receptor antagonists exert anti-inflammatory effects as summarized elsewhere [39]. In the setting of acute inflammatory episodes such as infectious disease, it is important that the energy appeal reaction and the water retention reaction support the proinflammatory process to eliminate the infectious agent. It can be hypothesized that the water retention hormone angiotensin II is immunomodulatory in this respect.
In conclusion, the water retention system shows important similarities to the energy provision system, with operation of the two interacting systems subject to positive selection in evolution to overcome serious, albeit non-life-threatening, transient inflammatory episodes (Table 2). This possibility is supported by the proinflammatory activity of water retention hormones. It is therefore important to consider the role of the water retention system in settings of chronic inflammation such as may occur in aging as well as rheumatic and autoimmune diseases undergoing medical treatment (after entering the chronic phase).
Both the aging process and medically treated chronic inflammatory diseases are accompanied by mild inflammation, a state that one might call low-grade smoldering chronic inflammation [40, 41]. In these settings, circulating cytokine levels can be 5 to 10 times higher than normal (e.g., IL-6 from 2 pg/ml to 10–20 pg/ml), and the overall stimulus might be strong enough to induce a mild systemic water retention and energy appeal reaction (responses shown for low IL-6 serum concentrations in ref. [42, 43]). Since no positively selected programs exist for aging [44] and chronic inflammatory diseases [8, 45], genes, signaling pathways, and networks that are operative in this setting are similar to those that determine acute episodes of inflammation (Table 2).
Under conditions of local inflammation with exposed outer and inner surfaces, circulating cytokines from inflamed tissue activate the energy appeal and water retention reaction. Since the local inflammatory process leads to water loss over local surfaces, locally formed water—by degradation of energy-rich fuels in activated inflammatory cells (Fig. 1, orange area)—can be rapidly lost via the exposed surface (Fig. 2a). It can be hypothesized that, in the presence of local and systemic water loss, the water retention system will not induce hypertension under these acute inflammatory conditions.
Fig. 2.
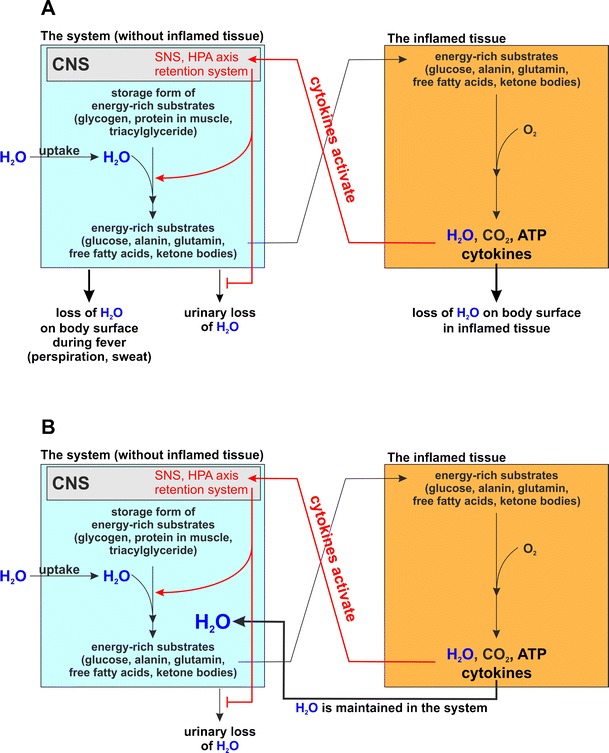
Water fluxes between the system and inflamed tissue with water loss in a transient acute inflammatory episode (a) and without water loss in chronic inflammation (b). a Water fluxes with water loss in acute inflammatory episodes. The inflamed tissue (orange box) releases cytokines or stimulates sensory nerve endings (not shown) in order to induce an energy appeal and water retention reaction in the system (blue box). In the system, urinary water loss is inhibited and water is needed for many important reactions for provision of energy-rich substrates and immune cell proliferation (see Fig. 1). In the acute situation, water is lost from the system and from inflamed tissue via outer and inner surfaces. The water fluxes are in a balance. b Water fluxes without water loss in chronic inflammation. Similarly, as before in panel A the inflamed tissue activates water retention and energy appeal reaction. However, due to low-grade inflammation without fever and due to a lack of inflamed exposed surface areas, water is not lost from the system and from inflamed tissue. Water can recirculate between activated immune cells and the system. In this scenario, it does not matter how the inflamed tissue look like. It might be a separated tissue such as an inflamed joint or an inflamed segment of the aorta. However, it might also be the sum of disseminated activated inflammatory cells in any tissue (orange box). It might also be the sum of disseminated an activated cells in different parts of the body, which might happen during aging when different organs present higher levels of inflammatory activity. The logic behind the water flux between system and inflamed cells remains the same. It is only important that secreted cytokines of these inflammatory cells activate the energy appeal and water retention reaction, and that water loss via inner and outer surfaces is not increased. Chronic inflammation accompanied by essential hypertension is a necessary consequence
The situation of water retention might be very different depending on the setting and operation of different mechanisms of water loss. These mechanisms would include the following events which may be present during disease: local water loss through outer/inner surfaces (Table 7), water loss due to systemic effects such as fever (Table 7), recirculation of formed water from activated inflammatory cells; and cytokine-activated water retention and energy appeal reaction. Depending on the operation and extent of these mechanisms, systemic hypertension could result from the continuous activation of a water retention and energy appeal reaction but without the usual water loss (Fig. 2b).
Indeed, in aging and in chronic inflammatory diseases, both the SNS and the HPA axis are chronically activated [41, 46–48]. In addition, there are obvious signs of volume overload in chronic inflammatory diseases because serum levels of atrial natriuretic factor and NT-brain natriuretic factor are markedly increased in different diseases [49–52]. NT-brain natriuretic factor decreased with TNF neutralization therapy but increased under therapy with volume retention—inducing glucocorticoids [49, 51].
The revised version of the theory predicts that the operation of these mechanisms will not depend on the site of inflammation and whether it occurs in arteries, kidneys, gastrointestinal tract, joints, or at various tissues with inflammatory cells. The key feature relates to the production of cytokines of inflammatory cells, their activation of the energy appeal and water retention reaction, and the extent of water loss via inner and outer surfaces. The magnitude of these changes would determine any detrimental effect on the induction of hypertension. Since prevalence of essential hypertension in adults is 22–32%, these theoretical considerations are highly clinically relevant.
As noted above, the energy appeal reaction and water retention reaction appear to be the result of selection in evolution for their role in transient inflammatory episodes. Induction of energy appeal reaction and water retention reaction provide a survival value if used for a short period of time. However, prolonged operation of these adaptive programs such as in chronic inflammatory diseases of today can in itself become pathogenic because there is no program to counteract continuous water retention and energy appeal reaction during long-term age-related inflammation and in chronic inflammatory diseases.
Conclusions
The study of evolutionary medicine combined with new insights into energy regulation (including the role of neuroendocrine immune axes and water retention systems) can shed new light on the pathophysiology of many age-related and chronic inflammatory diseases. In the past, several notable features of aging and chronic inflammatory diseases were considered as independent phenomena in the absence of a unifying model to explain both their prominence and coexistence. Consistent with emerging data and an overarching model based on the role of evolution in selecting patterns of host response, these features can now be unified in terms of a redirection of energy-rich fuels from energy stores to the activated immune system and activation of the water retention system. This unified picture can help explain sequelae of chronic inflammatory disease in terms of a conceptual framework derived from studies of acute inflammatory diseases.
The induction of systemic responses results from an appeal for energy-rich substrates, called the energy appeal reaction, and for water retention, called the water retention reaction. The revised theory states that energy appeal and water retention reaction have been positively selected for transient inflammatory episodes but prolonged use of these adaptive programs can in itself become pathogenic. Hopefully, recognition of the importance of energy and water fluxes during both acute and chronic inflammation can provide a new perspective of disease pathogenesis and lead to better approaches for diagnosis and treatment.
Acknowledgments
I want to express my sincere appreciation to Prof. Dr. David Pisetsky, Duke University Medical Center, North Carolina, who reviewed an early version of the manuscript and provided helpful editorial comments.
Support
The author has been supported over many years by grants from the DFG (STR 511/5-1, STR 511/9-1, STR 511/10-1, STR 511/11-1, STR 511/14-1, STR 511/15-1, STR 511/16-1, STR 511/17-1, PO 801/1-1, WI 1502/2-1), DFG Research Unit (FOR696, STR 511/9-1, STR 511/20-1, STR 511/21-1, STR 511/23-1, STR 511/25-1, STR 511/26-1, CA 933/1-1), DFG SFB (SFB585 project B8), DFG Mercator (Re 287/35-1), the DAAD (A/96/13772, 323-01-sr, and 423/kcw) and BMBF (Immuno-Pain, Gerontoshield).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Ramsey SA, Gold ES, Aderem A. A systems biology approach to understanding atherosclerosis. EMBO Mol Med. 2010;2:79–89. doi: 10.1002/emmm.201000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karp PD. Pathway databases: a case study in computational symbolic theory. Science. 2001;293:2040–2044. doi: 10.1126/science.1064621. [DOI] [PubMed] [Google Scholar]
- 3.Meyer-Hermann M, Figge MT, Toellner KM. Germinal centres seen through the mathematical eye: B-cell models on the catwalk. Trends Immunol. 2009;30:157–164. doi: 10.1016/j.it.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Besedovsky HO, Del Rey A. Immune-neuro-endocrine interactions. Endocr Rev. 1996;17:64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]
- 5.Nesse RM. How is Darwinian medicine useful? West J Med. 2001;174:358–360. doi: 10.1136/ewjm.174.5.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nesse RM. Is depression an adaptation? Arch Gen Psychiatry. 2000;57:14–20. doi: 10.1001/archpsyc.57.1.14. [DOI] [PubMed] [Google Scholar]
- 7.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411. doi: 10.2307/2406060. [DOI] [Google Scholar]
- 8.Straub RH, Besedovsky HO. Integrated evolutionary, immunological, and neuroendocrine framework for the pathogenesis of chronic disabling inflammatory diseases. FASEB J. 2003;17:2176–2183. doi: 10.1096/fj.03-0433hyp. [DOI] [PubMed] [Google Scholar]
- 9.Straub RH, Cutolo M, Buttgereit F, Pongratz G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med. 2010;267:543–560. doi: 10.1111/j.1365-2796.2010.02218.x. [DOI] [PubMed] [Google Scholar]
- 10.Straub RH. Concepts of evolutionary medicine and energy regulation contribute to the etiology of systemic chronic inflammatory diseases. Brain Behav Immun. 2011;25:1–5. doi: 10.1016/j.bbi.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 12.Blaxter K. Energy metabolism in animals and man. Cambridge: Cambridge University Press; 1989. [Google Scholar]
- 13.Le Magnen J. Lipogenesis, lipolysis and feeding rhythms. Ann Endocrinol (Paris) 1988;49:98–104. [PubMed] [Google Scholar]
- 14.Lange T, Dimitrov S, Fehm HL, Westermann J, Born J. Shift of monocyte function toward cellular immunity during sleep. Arch Intern Med. 2006;166:1695–1700. doi: 10.1001/archinte.166.16.1695. [DOI] [PubMed] [Google Scholar]
- 15.Lange T, Dimitrov S, Bollinger T, Diekelmann S, Born J. Sleep after vaccination boosts immunological memory. J Immunol. 2011;187:283–290. doi: 10.4049/jimmunol.1100015. [DOI] [PubMed] [Google Scholar]
- 16.Ostensen M, Almberg K, Koksvik HS. Sex, reproduction, and gynecological disease in young adults with a history of juvenile chronic arthritis. J Rheumatol. 2000;27:1783–1787. [PubMed] [Google Scholar]
- 17.Silva CA, et al. Hormone profile in juvenile systemic lupus erythematosus with previous or current amenorrhea. Rheumatol Int. 2010;31(8):1037–1043. doi: 10.1007/s00296-010-1389-2. [DOI] [PubMed] [Google Scholar]
- 18.Villiger PM, Caliezi G, Cottin V, Forger F, Senn A, Ostensen M. Effects of TNF antagonists on sperm characteristics in patients with spondyloarthritis. Ann Rheum Dis. 2010;69:1842–1844. doi: 10.1136/ard.2009.127423. [DOI] [PubMed] [Google Scholar]
- 19.Soares PM, Borba EF, Bonfa E, Hallak J, Correa AL, Silva CA. Gonad evaluation in male systemic lupus erythematosus. Arthritis Rheum. 2007;56:2352–2361. doi: 10.1002/art.22660. [DOI] [PubMed] [Google Scholar]
- 20.Suehiro RM, Borba EF, Bonfa E, Okay TS, Cocuzza M, Soares PM, Silva CA. Testicular Sertoli cell function in male systemic lupus erythematosus. Rheumatology (Oxford) 2008;47:1692–1697. doi: 10.1093/rheumatology/ken338. [DOI] [PubMed] [Google Scholar]
- 21.LaFleur C, et al. HLA-DR antigen frequencies in Mexican patients with dengue virus infection: HLA-DR4 as a possible genetic resistance factor for dengue hemorrhagic fever. Hum Immunol. 2002;63:1039–1044. doi: 10.1016/S0198-8859(02)00682-1. [DOI] [PubMed] [Google Scholar]
- 22.Meyer-Hermann ME, Maini PK. Cutting edge: back to “one-way” germinal centers. J Immunol. 2005;174:2489–2493. doi: 10.4049/jimmunol.174.5.2489. [DOI] [PubMed] [Google Scholar]
- 23.Meyer-Hermann M. A mathematical model for the germinal center morphology and affinity maturation. J Theor Biol. 2002;216:273–300. doi: 10.1006/jtbi.2002.2550. [DOI] [PubMed] [Google Scholar]
- 24.Murphy KM, Travers P, Walport M. Janeway’s immunobiology. Oxford: Taylor & Francis; 2011. [Google Scholar]
- 25.Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25:154–159. doi: 10.1016/S0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- 26.Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies JW, Lamke LO, Liljedahl SO. A guide to the rate of non-renal water loss from patients with burns. Br J Plast Surg. 1974;27:325–329. doi: 10.1016/0007-1226(74)90031-9. [DOI] [PubMed] [Google Scholar]
- 29.Jacob M, Chappell D, Hofmann-Kiefer K, Conzen P, Peter K, Rehm M. Determinants of insensible fluid loss. Perspiration, protein shift and endothelial glycocalyx. Anaesthesist. 2007;56:747–758. doi: 10.1007/s00101-007-1235-4. [DOI] [PubMed] [Google Scholar]
- 30.Berg JM, Tymoczko JL, Stryer L. Biochemistry. New York: Freeman; 2002. [Google Scholar]
- 31.Michal G. Biochemical pathways. Heidelberg: Spektrum Akademischer Verlag; 1999. [Google Scholar]
- 32.Rassow J, Hauser K, Netzker R, Deutzmann R. Biochemistry. Stuttgart: Georg Thieme; 2008. [Google Scholar]
- 33.Tredget EE, Yu YM. The metabolic effects of thermal injury. World J Surg. 1992;16:68–79. doi: 10.1007/BF02067117. [DOI] [PubMed] [Google Scholar]
- 34.Bader M, Ganten D. Regulation of renin: new evidence from cultured cells and genetically modified mice. J Mol Med (Berl) 2000;78:130–139. doi: 10.1007/s001090000089. [DOI] [PubMed] [Google Scholar]
- 35.Hattangady N, Olala L, Bollag WB, Rainey WE (2011) Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol. doi:10.1016/j.mce.2011.07.034 [DOI] [PMC free article] [PubMed]
- 36.Selye H. The general adaptation syndrome and the diseases of adaptation. Am J Med. 1951;10:549–555. doi: 10.1016/0002-9343(51)90327-0. [DOI] [PubMed] [Google Scholar]
- 37.Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR, Scholze J. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension. 1999;34:113–117. doi: 10.1161/01.hyp.34.1.113. [DOI] [PubMed] [Google Scholar]
- 38.Phillips MI, Kagiyama S. Angiotensin II as a pro-inflammatory mediator. Curr Opin Investig Drugs. 2002;3:569–577. [PubMed] [Google Scholar]
- 39.Boos CJ, Lip GY. Is hypertension an inflammatory process? Curr Pharm Des. 2006;12:1623–1635. doi: 10.2174/138161206776843313. [DOI] [PubMed] [Google Scholar]
- 40.Pawelec G, Akbar A, Caruso C, Solana R, Grubeck-Loebenstein B, Wikby A. Human immunosenescence: is it infectious? Immunol Rev. 2005;205:257–268. doi: 10.1111/j.0105-2896.2005.00271.x. [DOI] [PubMed] [Google Scholar]
- 41.Straub RH, Cutolo M, Zietz B, Schölmerich J. The process of aging changes the interplay of the immune, endocrine and nervous systems. Mech Ageing Dev. 2001;122:1591–1611. doi: 10.1016/S0047-6374(01)00289-5. [DOI] [PubMed] [Google Scholar]
- 42.Tsigos C, Papanicolaou DA, Defensor R, Mitsiadis CS, Kyrou I, Chrousos GP. Dose effects of recombinant human interleukin-6 on pituitary hormone secretion and energy expenditure. Neuroendocrinology. 1997;66:54–62. doi: 10.1159/000127219. [DOI] [PubMed] [Google Scholar]
- 43.Tsigos C, Papanicolaou DA, Kyrou I, Raptis SA, Chrousos GP. Dose-dependent effects of recombinant human interleukin-6 on the pituitary-testicular axis. J Interferon Cytokine Res. 1999;19:1271–1276. doi: 10.1089/107999099312948. [DOI] [PubMed] [Google Scholar]
- 44.Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 45.Straub RH, Del Rey A, Besedovsky HO. Emerging concepts for the pathogenesis of chronic disabling inflammatory diseases: neuroendocrine-immune interactions and evolutionary biology. In: Ader R, editor. Psychoneuroimmunology. San Diego: Elsevier; 2007. pp. 217–232. [Google Scholar]
- 46.Dekkers JC, Geenen R, Godaert GL, Bijlsma JW, van Doornen LJ. Elevated sympathetic nervous system activity in patients with recently diagnosed rheumatoid arthritis with active disease. Clin Exp Rheumatol. 2004;22:63–70. [PubMed] [Google Scholar]
- 47.Jessop DS, Harbuz MS. A defect in cortisol production in rheumatoid arthritis: why are we still looking? Rheumatology (Oxford) 2005;44:1097–1100. doi: 10.1093/rheumatology/keh644. [DOI] [PubMed] [Google Scholar]
- 48.Straub RH, Paimela L, Peltomaa R, Schölmerich J, Leirisalo-Repo M. Inadequately low serum levels of steroid hormones in relation to IL-6 and TNF in untreated patients with early rheumatoid arthritis and reactive arthritis. Arthritis Rheum. 2002;46:654–662. doi: 10.1002/art.10177. [DOI] [PubMed] [Google Scholar]
- 49.Yasuda M, Yasuda D, Tomooka K, Nobunaga M. Plasma concentration of human atrial natriuretic hormone in patients with connective tissue diseases. Clin Rheumatol. 1993;12:231–235. doi: 10.1007/BF02231533. [DOI] [PubMed] [Google Scholar]
- 50.Straub RH, Hall C, Kramer BK, Elbracht R, Palitzsch KD, Lang B, Schölmerich J. Atrial natriuretic factor and digoxin-like immunoreactive factor in diabetic patients: their interrelation and the influence of the autonomic nervous system. J Clin Endocrinol Metab. 1996;81:3385–3389. doi: 10.1210/jc.81.9.3385. [DOI] [PubMed] [Google Scholar]
- 51.Peters MJ, Welsh P, McInnes IB, Wolbink G, Dijkmans BA, Sattar N, Nurmohamed MT. Tumour necrosis factor alpha blockade reduces circulating N-terminal pro-brain natriuretic peptide levels in patients with active rheumatoid arthritis: results from a prospective cohort study. Ann Rheum Dis. 2010;69:1281–1285. doi: 10.1136/ard.2009.119412. [DOI] [PubMed] [Google Scholar]
- 52.Provan S, Angel K, Semb AG, Atar D, Kvien TK. NT-proBNP predicts mortality in patients with rheumatoid arthritis: results from 10-year follow-up of the EURIDISS study. Ann Rheum Dis. 2010;69:1946–1950. doi: 10.1136/ard.2009.127704. [DOI] [PubMed] [Google Scholar]
- 53.Iemitsu M, Itoh M, Fujimoto T, Tashiro M, Nagatomi R, Ohmori H, Ishii K. Whole-body energy mapping under physical exercise using positron emission tomography. Med Sci Sports Exerc. 2000;32:2067–2070. doi: 10.1097/00005768-200012000-00016. [DOI] [PubMed] [Google Scholar]
- 54.Calder PC. Fuel utilization by cells of the immune system. Proc Nutr Soc. 1995;54:65–82. doi: 10.1079/PNS19950038. [DOI] [PubMed] [Google Scholar]
- 55.Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. 2008;84:949–957. doi: 10.1189/jlb.0108024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172:4661–4665. doi: 10.4049/jimmunol.172.8.4661. [DOI] [PubMed] [Google Scholar]
- 57.McKiernan SM, Hagan R, Curry M, McDonald GS, Kelly A, Nolan N, Walsh A, Hegarty J, Lawlor E, Kelleher D. Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology. 2004;40:108–114. doi: 10.1002/hep.20261. [DOI] [PubMed] [Google Scholar]
- 58.Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pertovaara M, Raitala A, Juonala M, Kahonen M, Lehtimaki T, Viikari JS, Raitakari OT, Hurme M. Autoimmunity and atherosclerosis: functional polymorphism of PTPN22 is associated with phenotypes related to the risk of atherosclerosis. The Cardiovascular Risk in Young Finns Study. Clin Exp Immunol. 2007;147:265–269. doi: 10.1111/j.1365-2249.2006.03278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thio CL, Mosbruger TL, Kaslow RA, Karp CL, Strathdee SA, Vlahov D, O’Brien SJ, Astemborski J, Thomas DL. Cytotoxic T-lymphocyte antigen 4 gene and recovery from hepatitis B virus infection. J Virol. 2004;78:11258–11262. doi: 10.1128/JVI.78.20.11258-11262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raitala A, Karjalainen J, Oja SS, Kosunen TU, Hurme M. Helicobacter pylori-induced indoleamine 2,3-dioxygenase activity in vivo is regulated by TGFB1 and CTLA4 polymorphisms. Mol Immunol. 2007;44:1011–1014. doi: 10.1016/j.molimm.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 62.Gu CC, et al. An investigation of genome-wide associations of hypertension with microsatellite markers in the family blood pressure program (FBPP) Hum Genet. 2007;121:577–590. doi: 10.1007/s00439-007-0349-8. [DOI] [PubMed] [Google Scholar]
- 63.Ammendola M, Bottini N, Pietropolli A, Saccucci P, Gloria-Bottini F. Association between PTPN22 and endometriosis. Fertil Steril. 2008;89:993–994. doi: 10.1016/j.fertnstert.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 64.Miller LE, Jüsten HP, Schölmerich J, Straub RH. The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J. 2000;14:2097–2107. doi: 10.1096/fj.99-1082com. [DOI] [PubMed] [Google Scholar]
- 65.Straub RH, Lowin T, Klatt S, Wolff C, Rauch L (2011) Increased density of sympathetic nerve fibers in metabolically activated fat tissue surrounding human synovium and mouse lymph nodes in arthritis. Arthritis Rheum 63:3234–3242 [DOI] [PubMed]
- 66.Power ML, Schulkin J. The evolution of obesity. Baltimore: The Johns Hopkins University Press; 2009. [Google Scholar]
- 67.Buttgereit F, Burmester GR, Brand MD. Bioenergetics of immune functions: fundamental and therapeutic aspects. Immunol Today. 2000;21:192–199. doi: 10.1016/S0167-5699(00)01593-0. [DOI] [PubMed] [Google Scholar]