

Abstract
Myocardial infarction with ST segment elevation (STE) on electrocardiography (ECG) is a common presentation in emergency rooms across the world. Myocardial injury and necrosis are infrequently the initial presentation in patients with thrombotic thrombocytopenic purpura (TTP). A 48-year-old woman presented with STE myocardial infarction from outside hospital for primary percutaneous coronary intervention. However, her clinical picture was not consistent. Rapid evaluation revealed symptoms associated with microangiopathic hemolytic anemia, thrombocytopenia, acute kidney injury with waxing and waning mental status. A diagnosis of TTP was made with low ADAMST-13 activity. Plasmapheresis was initiated along with intravenous steroid therapy. The patient had a full recovery and went home after full recovery of left ventricular ejection fraction and normal myocardial perfusion studies. Rapid evaluation is needed to identify infrequent causes of STE myocardial infarction. As swift protocols are activated in the emergency room and catheterization laboratories to ensure quality control, it is equally important to integrate all aspects of the patient's clinical and objective data to detect unusual disease entities.
Keywords: Acute coronary syndrome, electrocardiography, microangiopathy, plasmapheresis, ST segment elevation myocardial infarction, thrombotic thrombocytopenic purpura
INTRODUCTION
Patients who present to the emergency department with chest pain require rapid triage, evaluation and management. Myocardial infarction with ST segment elevation (STE) on electrocardiography (ECG) is a common presentation in emergency rooms across the world. Acute coronary syndrome (ACS) marked by STE on the ECG warrants consideration for emergent cardiac catheterization and possible percutaneous coronary intervention (PCI). In fact, there are now core measures and quality metrics in place that grade a hospital's efficiency at caring for such patients.[1] However, the time pressure to optimize such quality metrics may lead to an unintended rush to treatment prior to adequate evaluation.[1]
In this report, we present a rare case where myocardial infarction was seen as a presenting feature of an underlying hematologic disease, thrombotic thrombocytopenic purpura (TTP). This case highlights the importance of a thorough, yet efficient, clinical evaluation in which the history, physical exam, ECG and laboratory data were needed to make the appropriate triage decision and not miss an unusual diagnosis.
CASE REPORT
A 48-year-old woman with no known coronary risk factors was transferred from an outside facility to the cardiac catheterization laboratory of our hospital with the diagnosis of STE myocardial infarction for primary PCI. On arrival to our catheterization laboratory, the ECG from the referring hospital showed sinus tachycardia with normal axis and intervals. There was STE in leads I, II, aVL, V4–6 and reciprocal ST segment depression in lead III [Figure 1]. Laboratory data were not yet available. However, the patient's history of present illness was significant for malaise, fever, chills and lethargy that began 3 days prior to hospitalization. Further questioning established that she had mild generalized abdominal pain and one episode of non-bloody diarrhea. The family also noted that she had been intermittently confused and was talking gibberish. On the morning of admission, she had severe chest pain associated with nausea, vomiting and dyspnea on exertion, which led her to seek medical care. Her medical history was notable for a transient ischemic attack 7 years prior. An extensive thrombophilia work-up at that time was negative. She also had a history of two miscarriages in the past.
Figure 1.
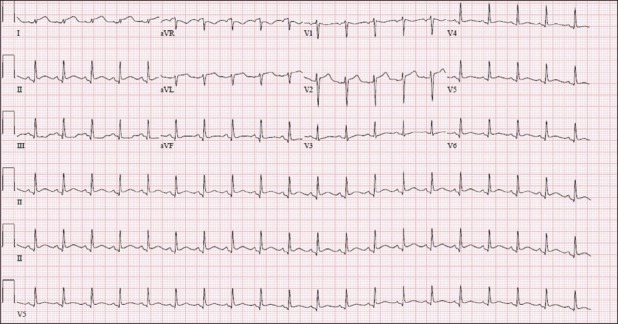
Sinus tachycardia at 121 beats per minute with ST segment elevation in Lead I, II, aVL, V4–6
On examination, she appeared toxic and in respiratory distress. Vital signs revealed a blood pressure of 126/70 mmHg with a heart rate of 121 beats per minute. Her temperature at admission was 34.4°C. The respiratory rate was 30 breaths per minute. Oxygen saturation was 100% on a non-re-breather mask. She was pale, cold and clammy with delayed capillary refill. She had cyanosis in all fingers with mild cyanosis of her tongue and lips. In addition, mottling of her skin and livedo reticularis over the thighs was noted. There were a few purpuric skin lesions observed in her antecubital fossa and upper arms. Her jugular venous pressure was elevated up to the angle of the jaw. Cardiac exam revealed a normal first and second heart sound along with a fourth heart sound. There were no murmurs. Peripheral pulses were not palpable in the feet and were only faintly palpable in the arms. The lungs were clear to auscultation. Abdominal exam was unremarkable. There was no peripheral edema. Neurologically, she was somewhat confused, but the sensory and motor exam was essentially normal. Given that the patient was not having active chest pain, the history was inconsistent with ACS and she appeared more toxic than expected for a lateral wall myocardial infarction; cardiac catheterization was deferred and emergent laboratory studies were obtained.
Initial laboratory studies revealed a white blood cell count of 13.5 × 103/mm3; hematocrit of 24%; mean corpuscular volume of 88.4 fL and platelet count of 6 × 109/L. Her lactate dehydrogenase was elevated at 2820 units/L and haptoglobin was low at less than 10 mg/dL. Coagulation profile showed international normalized ratio of 1.2, prothrombin time of 12.6 seconds, fibrinogen 199 mg/ L and D-dimer 1.27 feu mg/L. Electrolytes were within normal limits; acute kidney injury was noted with blood urea nitrogen 51 mg/ dL and creatinine 1.9 mg/dL. Total bilirubin was markedly elevated at 32 mg% with an indirect bilirubin of 2.1 mg%. Cardiac biomarkers were elevated with creatinine kinase of 487 units/L and MB fraction of 28.8 ng/mL. Troponin-T was 0.86 ng/mL. Urinalysis showed pH of 6.0, 3+ albumin, 3+ hemoglobin, eight WBCs and greater than two RBCs with some amorphous crystals. Peripheral smear showed moderate schistocytes, few spherocytes and low platelet count. Chest radiograph showed no cardiopulmonary abnormalities. Echocardiography demonstrated an ejection fraction of 40–45% with severe hypokinesis of the inferior and basal anteroseptal wall. No significant valvular lesions were noted.
In view of the acute onset of symptoms associated with microangiopathic hemolytic anemia, thrombocytopenia, acute kidney injury and waxing and waning mental status, the presumptive diagnosis of TTP was made. Further coagulopathy testing was negative. A Disintegrin and Metalloproteinase with Thrombospondin Motifs (ADAMST-13) activity was found to be low with presence of ADAMST-13 inhibitors in the plasma.
Plasmapheresis was started immediately. Intravenous steroid therapy was also initiated. She improved clinically with this treatment and normalized her platelets and lactate dehydrogenase, and STE in her ECG resolved [Figure 2]. She was discharged home after a full recovery. Outpatient cardiovascular follow-up demonstrated normalization of the left ventricular ejection fraction by echocardiography and a completely normal nuclear myocardial perfusion stress study. Further testing with invasive catheterization was not pursued as the entire episode was considered to be transient, secondary to metabolic derangement.
Figure 2.
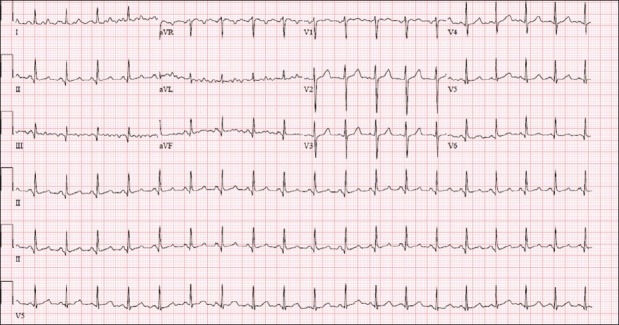
Sinus tachycardia at 113 beats per minute with resolved ST-segment changes
DISCUSSION
TTP is defined as a severe, thrombotic microangiopathy that is characterized primarily by systemic platelet-von Willebrand factor aggregation, organ ischemia, profound thrombocytopenia and fragmentation of erythrocytes.[2] Intravascular coagulation is not considered a prominent feature of the disorder. Clinically, widespread organ dysfunction is usually present. Pathologically, focal areas of necrosis and hemorrhage may be seen in the pancreas, adrenals, heart, brain and kidneys.[3–5] Although myocardial injury and necrosis are observed in a large number of patients with TTP, it is infrequently the initial presentation,[6] and most likely thought to be due to microthrombi from massive platelet aggregation than plaque rupture–thrombosis cascade.[7] Various studies have determined the incidence of myocardial infarction in TTP to range from 15–41%.[6–9] However, the heart is one of the most frequently involved organs at autopsy examination of patients with TTP.[3,10,11] Mortality is considerably higher in patients with TTP who have positive cardiac biomarkers, necessitating closer monitoring in this subgroup.[7,9,12]
Early recognition of myocardial injury in a case of TTP is crucial as it identifies higher risk. However, invasive therapy in the form of cardiac catheterization and PCI may be fraught with complications and is precluded by acute kidney injury and low platelet count.[8] Thrombocytopenia also prevents the use of usual medical management in ACS such as antiplatelet and anticoagulant therapy.[9] Angiotensin converting enzyme inhibitors are also not used because of concomitant renal injury. Beta blockers and HMG CoA reductase inhibitors may be used although their role is questionable.[9] In acute bouts of TTP, such as this case, the treatment of choice is rapid initiation of plasmapheresis. In addition, immunosuppressive therapy including steroid therapy is helpful, especially in the setting of auto-antibodies against ADAMST-13 factor.[13,14] Relapsing cases of TTP have been treated with rituximab, a monoclonal antibody against CD20 on memory B cells with good effect. However, large clinical trials are lacking for this.[13]
In conclusion, STE myocardial infarction is a very common presentation in the emergency rooms. A rapid evaluation is needed to identify infrequent causes of STE myocardial infarction. Although cardiac involvement is common in TTP, as an index event it can be misleading. As swift protocols are activated in the emergency room and catheterization laboratories to ensure quality control, it is equally important to integrate all aspects of patient's clinical and objective data to detect unusual disease entities. Once identified, a multidisciplinary approach is essential for rapid treatment of TTP.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
- 1.Peterman JM, George A, Giugliano GR, Schweiger MJ. Door-to-balloon time: Are we evaluating the wrong metric? J Am Coll Cardiol. 2010;56:158–9. doi: 10.1016/j.jacc.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 2.Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600. doi: 10.1056/NEJMra020528. [DOI] [PubMed] [Google Scholar]
- 3.Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities.A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127:834–9. doi: 10.5858/2003-127-834-TTPAHU. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi V, Robles R, Alberio L, Furlan M, Lämmle B. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: A severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002;100:710–3. doi: 10.1182/blood-2002-02-0344. [DOI] [PubMed] [Google Scholar]
- 5.Sane DC, Streer NP, Owen J. Myocardial necrosis in patients with thrombotic thrombocytopenic purpura: Pathophysiology and rationale for specific therapy. Eur J Haematol. 2009;82:83–92. doi: 10.1111/j.1600-0609.2008.01172.x. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy LJ, Danielson CF, Skipworth EM, Peters SL, Miraglia CC, Antony AC. Myocardial infarction/injury is relatively common at presentation of acute thrombotic thrombocytopenic purpura: The Indiana University experience. Ther Apher. 2002;6:2–4. doi: 10.1046/j.1526-0968.2002.00363.x. [DOI] [PubMed] [Google Scholar]
- 7.Patschan D, Witzke O, Dührsen U, Erbel R, Philipp T, Herget-Rosenthal S. Acute myocardial infarction in thrombotic microangiopathies—clinical characteristics, risk factors and outcome. Nephrol Dial Transplant. 2006;21:1549–54. doi: 10.1093/ndt/gfl127. [DOI] [PubMed] [Google Scholar]
- 8.Wahla AS, Ruiz J, Noureddine N, Upadhya B, Sane DC, Owen J. Myocardial infarction in thrombotic thrombocytopenic purpura: A single-center experience and literature review. Eur J Haematol. 2008;81:311–6. doi: 10.1111/j.1600-0609.2008.01112.x. [DOI] [PubMed] [Google Scholar]
- 9.Gandhi K, Aronow WS, Desai H, Amin H, Sharma M, Lai HM, et al. Cardiovascular manifestations in patients with thrombotic thrombocytopenic purpura: A single-center experience. Clin Cardiol. 2010;33:213–6. doi: 10.1002/clc.20731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amorosi EL, Ultmann JE. Thrombotic thrombocytopenic purpura: Report of 16 cases and review of the literature. Medicine (Baltimore) 1994;45:139–59. [Google Scholar]
- 11.Ridolfi RL, Bell WR. Thrombotic thrombocytopenic purpura.Report of 25 cases and review of the literature. Medicine. 1981;60:413–28. [PubMed] [Google Scholar]
- 12.Lapp H, Shin DI, Kroells W, Boerrigter G, Horlitz M, Schley P, et al. Cardiogenic shock due to thrombotic thrombocytopenic purpura. Z Kardiol. 2004;93:486–92. doi: 10.1007/s00392-004-0077-1. [DOI] [PubMed] [Google Scholar]
- 13.Kremer Hovinga JA, Meyer SC. Current management of thrombotic thrombocytopenic purpura. Curr Opin Hematol. 2008;15:445–50. doi: 10.1097/MOH.0b013e328309ec62. [DOI] [PubMed] [Google Scholar]
- 14.Fontana S, Hovinga JA, Studt JD, Alberio L, Lämmle B, Taleghani BM. Plasma therapy in thrombotic thrombocytopenic purpura: Review of the literature and the Bern experience in a subgroup of patients with severe acquired ADAMTS-13 deficiency. Semin Hematol. 2004;41:48–59. doi: 10.1053/j.seminhematol.2003.10.010. [DOI] [PubMed] [Google Scholar]