

Abstract
Background:
Long QT syndrome (LQTS) is characterized by QT prolongation, syncope and sudden death. This study aims to explore the causes, clinical manifestations and therapeutic outcomes of Jervell and Lange-Nielsen syndrome (JLNS), a rare form of LQTS with congenital sensorineural deafness, in Chinese individuals.
Materials and Methods:
Three JLNS kindreds from the Chinese National LQTS Registry were investigated. Mutational screening of KCNQ1 and KCNE1 genes was performed by polymerase chain reaction and direct DNA sequence analysis. LQTS phenotype and therapeutic outcomes were evaluated for all probands and family members.
Results:
We identified 7 KCNQ1 mutations. c.1032_1117dup (p.Ser373TrpfsX10) and c.1319delT (p.Val440AlafsX26) were novel, causing JLNS in a 16-year-old boy with a QTc (QT interval corrected for heart rate) of 620 ms and recurrent syncope. c.605-2A>G and c.815G>A (p.Gly272Asp) caused JLNS in a 12-year-old girl and her 5-year-old brother, showing QTc of 590 to 600 ms and recurrent syncope. The fourth JLNS case, a 46-year-old man carrying c.1032G>A (p.Ala344Alasp) and c.569G>A (p.Arg190Gln) and with QTc of 460 ms, has been syncope-free since age 30. His 16-year-old daughter carries novel missense mutation c.574C>T (p.Arg192Cys) and c.1032G>A(p.Ala344Alasp) and displayed a severe phenotype of Romano-Ward syndrome (RWS) characterized by a QTc of 530 ms and recurrent syncope with normal hearing. Both the father and daughter also carried c.253G>A (p.Asp85Asn; rs1805128), a rare single nucleotide polymorphism (SNP) on KCNE1. Bizarre T waves were seen in 3/4 JLNS patients. Symptoms were improved and T wave abnormalities became less abnormal after appropriate treatment.
Conclusion:
This study broadens the mutation and phenotype spectrums of JLNS. Compound heterozygous KCNQ1 mutations can result in both JLNS and severe forms of RWS in Chinese individuals.
Keywords: Compound heterozygous mutation, frameshift mutation, Jervell and Lange-Nielsen syndrome, KCNQ1, KCNE1, long QT syndrome, Romano-Ward syndrome, single nucleotide polymorphism
INTRODUCTION
Long QT syndrome (LQTS) is an inherited cardiac disorder characterized by QT interval prolongation, ventricular arrhythmias, syncope and sudden death (SD). Two forms of LQTS have been classified: Autosomal dominant Romano-Ward syndrome (RWS), which presents without hearing deficiency, and the more rarely occurring autosomal recessive Jervell and Lange-Nielsen syndrome (JLNS), which presents with congenital sensorineural deafness.
Physiologically, the slowly activating delayed rectifier K+ current (IKs) is crucial in maintaining the cardiac responses that generate a normal T wave and QT interval. Proteins encoded by the KCNQ1 and KCNE1 genes co-assemble to form IKs potassium channels.[1,2] Homozygous or compound heterozygous mutations of either KCNQ1 or KCNE1 can result in JLNS.[3–6] Single heterozygous mutations found in ≥13 genes, on the other hand, cause RWS and account for the vast majority of LQTS.[7,8]
Severe phenotypes are often seen in patients carrying more than one LQTS-causing mutations. Such a “cumulative effect,” occurring as either homozygous or compound heterozygous mutations, can severely impair or even completely obliterate functional expression of IKs, resulting in severe variants of LQTS. Patients with JLNS often present with early onset of cardiac events, bizarre T waves and marked QTc (QT interval corrected for heart rate) prolongation. Beta-blockers, the first line therapy of LQTS, generally provide limited protection to JLNS patients.[9] Malfunction, or complete loss of function of IKs in the inner ear is the underlying cause of the auditory impairment or sensorineural deafness in JLNS.
Among reported mutations associated with JLNS,[7,10–13] three of 31 cases were found in Chinese individuals and include homozygous mutation T322M[12] and a compound heterozygous mutation T2C/1149insT in KCNQ1.[13] In this study, we report seven additional compound heterozygous mutations in KCNQ1 and one rare SNP on KCNE1, identified in three Chinese JLNS kindreds.
MATERIALS AND METHODS
Study subjects
Three JLNS kindreds, two of Han and one of Miao descent, selected from 160 LQTS families in the Chinese Channelopathy Registry, were enrolled into this study.
Written consents, approved by the Ethics Committee of Peking University People's Hospital, were obtained from participants. Individuals were diagnosed with JLNS based on QT prolongation in the presence of profound sensorineural deafness. Phenotype characteristics were determined by the presence or absence of syncope and precipitating factors, age at first cardiac event, and changes in T wave morphology before and after therapies.
Mutation analysis
Genomic DNA from patients and family members was extracted from whole blood. All samples underwent polymerase chain reaction (PCR) amplification and direct sequencing in accordance to standard protocols (94°C for 3 minutes, followed by 30 cycles of 94°C for 10 seconds, 58°C for 20 seconds, and 72°C for 20 seconds, and a 5 minute extension at 72°C).[4] The sequence of PCR primer pairs was based on reference or re-designed using Primer 3 online (KCNQ1: NM_000218.2; KCNE1: NM_000219.3) to flank all exons and intron-exon junctions (suppl.1). PCR amplification was carried out using standard protocols for all samples with the exception of exon 1c, which was amplified with the GC RICH PCR system (Roche, Rotkreuz, Zug, Switzerland) due to its high GC content. PCR products were purified by vacuum pump Axygen PCR (Microplate, Winooski, USA). Direct sequencing was carried out with BigDye Terminator (Applied Biosystems, Foster City, California, U.S.A) DNA sequencing kit (version 3.1) and 3730XL DNA Analyzer (Applied Biosystems, Foster City, California, U.S.A). DNA samples from 50 healthy Han Chinese volunteers were used as controls. The mutation data were recorded onto a publicly available database LOVD[7] and the accession numbers are: KCNQ1_00582, 00583, 00584, 00585, 00586, 00587, 0589.
Nomenclature of new mutations
LQTS-causing mutations and other variants were denoted using known and accepted nomenclature. The numbering for all mutations started at the ATG initiation codon of the full-length isoform 1 of KCNQ1. The exon numbering was labeled according to Splawski et al.[14] Frameshift mutations resulting from nucleotide insertions or deletions were annotated using the p.Ser6ProfsX2 format. Here, p.Ser6ProfsX2 denotes a frameshift change with Serine 6 as the first affected amino acid, changing into a Proline and the new reading frame ending with stop codon X at position 2 in the shifted reading frame.[15] A substitution of either the first or the last two nucleotides of a particular exon has the capacity to alter proper mRNA splicing, regardless of whether the nucleotide substitution codes for a different amino acid (missense mutation), produces a stop codon (nonsense mutation) or does not alter the open reading frame at all (i.e., a synonymous or silent single nucleotide substitution).
Electrocardiogram (ECG) parameters
The paper speed for all ECGs was 25 mm/s. Each QT interval was defined as the interval between the onset of the Q wave and the end of the T wave, and was based upon the mean of 2–3 consecutive heart beats on leads II, V5 or on any of the 12 leads where the QT interval appeared to be the longest. Heart rate-corrected QT (QTc) was calculated using Bazett's formula (QT/√RR), and a diagnosis of LQTS was considered when QTc was ≥0.47 s for males and ≥0.48 s for females. A QTc in the range of 0.44–0.47 s was considered borderline.[16,17] Bifid T waves were included and U waves were excluded in the QT measurements.
RESULTS
KCNQ1 gene screening in three Chinese JLNS kindreds revealed six disease-causing mutations responsible for JLNS and one novel mutation resulting in RWS. Four mutations previously reported in RWS caused JLNS in our patients when formed as compound mutations [Table 1].
Table 1.
Genotype-phenotype correlation in three kindreds with JLNS
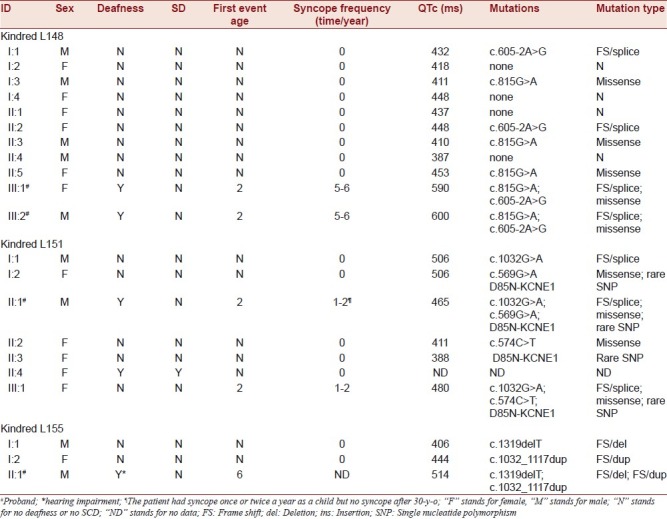
Clinical characteristics and mutation findings in Family L148
Family L148 consisted of two asymptomatic parents and their two profoundly deaf children, a 12-year-old girl and a 5-year-old boy.
The young siblings presented with recurrent syncope starting at age 2, and markedly prolonged QTc intervals (590 ms and 600 ms, respectively) [Figure 1] at time of JLNS diagnosis. No consanguineous marriage was identified in the family and both parents had normal QT intervals. Prior to LQTS therapy, both deaf children experienced approximately 5–6 syncopal episodes per year, mostly triggered by emotional stress or physical exercise, including one reported incidence from the girl during 2008's Wenchuan Earthquake. Considering the severity of their LQTS phenotype, both children were treated with beta-blockers (Propranolol, 2.1–2.5 mg/kg/day) and left cardiac sympathetic denervation (LCSD) in 2009. The girl reported one episode and the boy reported three episodes of syncope at the 2-year follow-up. Due to financial constraints and the possibility of unwarranted side effects, their parents refused installation of an implantable cardioverter defibrillator (ICD).
Figure 1.
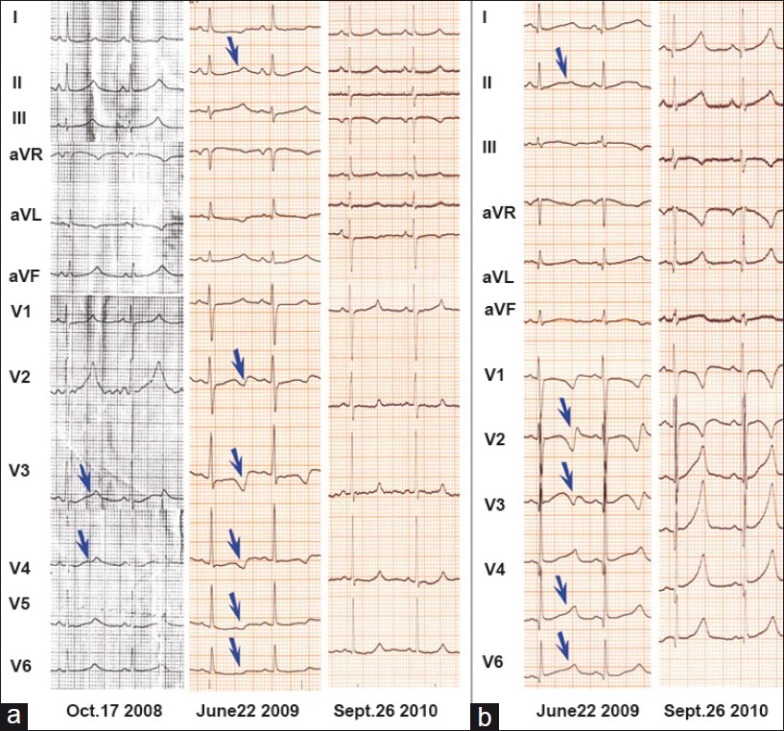
The 12-lead electrocardiogram (ECG) for probands from kindred L148. (a) ECG for L148-01, a 12-year-old girl: The left two ECGs showed very broad and notched or bifid even biphasic T waves on multi-leads (arrows) (QTc 590 ms) before she had received left cardiac sympathetic denervation (LCSD); the right one showed less abnormal ECG 15 months after LCSD (QTc 490 ms). (b) ECG for L148-02, a 5-year-old boy: The left one showed very broad and notched or bifid even biphasic T waves on multi-leads (arrows) (QTc 600 ms) before he had received left cardiac sympathetic denervation (LCSD); the right one showed less abnormal ECG 15 months after LCSD (QTc 490 ms).
Compound heterozygous mutations, c.605-2A>G a splicing mutation and c.815G>A (p.Gly272Asp), a missense mutation of KCNQ1, were identified in these two siblings. Family genotyping revealed that the father carried a heterozygous p.Gly272Asp and the mother carried a heterozygous c.605-2A>G [Figure 2]. These two mutations were absent in 100 control alleles.
Figure 2.
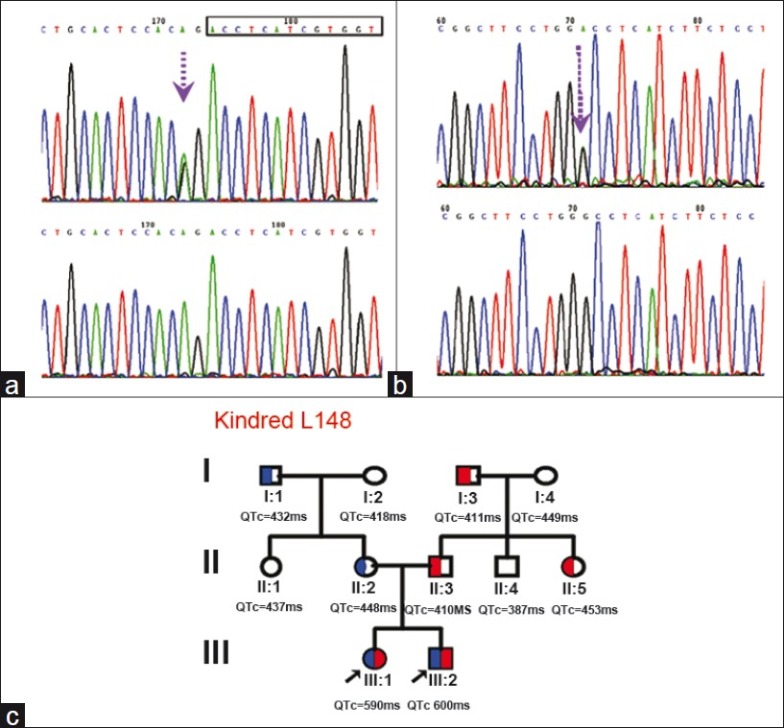
The mutations in kindred L148. (a) shows an A-to-G change at nucleotide position A605-2, which resulted in a splice site alteration c.605-2A>G. The starting point of the coding domain for exon 3 of KCNQ1 is marked in the rectangle. (b) shows a G to A change at position 815, which is a known mutation p.Gly272Asp, previously reported in a Romano-Ward syndrome European patient. (c) shows the family tree for L148, where two mutations were transmitted from both parents. The paternal lineage carries a heterozygous p.Gly272Asp (Red) only, while the maternal lineage carries a heterozygous c.605- 2A>G (Blue).
Clinical characteristics and mutation finding in Family L151
The proband of Miao descent family L151 was a 46-year-old man with congenital deafness, a history of recurrent syncope and borderline QTc prolongation (460 ms). Starting at the age of 2, he experienced syncope 1–2 times per year until the age of 30 and none thereafter without any etiotropic treatment. His 17-year-old daughter, although with normal hearing, was very symptomatic. At age 2, she was diagnosed with epilepsy and treated with Phenobarbital (30–60 mg/day). While on Phenobarbital, she experienced five more seizure-like attacks, triggered by physical or mental stress. At age 12, she was diagnosed with LQTS and treated with Metoprolol 25 mg/day (the equivalent of 0.725 mg/kg of Propranolol). Her QTc on serial ECGs ranged from 480 to 530 ms [Figure 3]. During the third year of beta-blocker treatment, she experienced another syncopal episode, evoked by exercise. Metoprolol dosage was therefore adjusted to 50 mg/day (the equivalent of 1.45 mg/kg of Propranolol) and she has remained event-free for the last 2 years. Family screening revealed that one of her aunts was congenitally deaf and died suddenly at age 33. Both paternal grandparents had QTc of 506 ms, although they had never experienced any cardiac events. There was no consanguineous marriage in the family.
Figure 3.
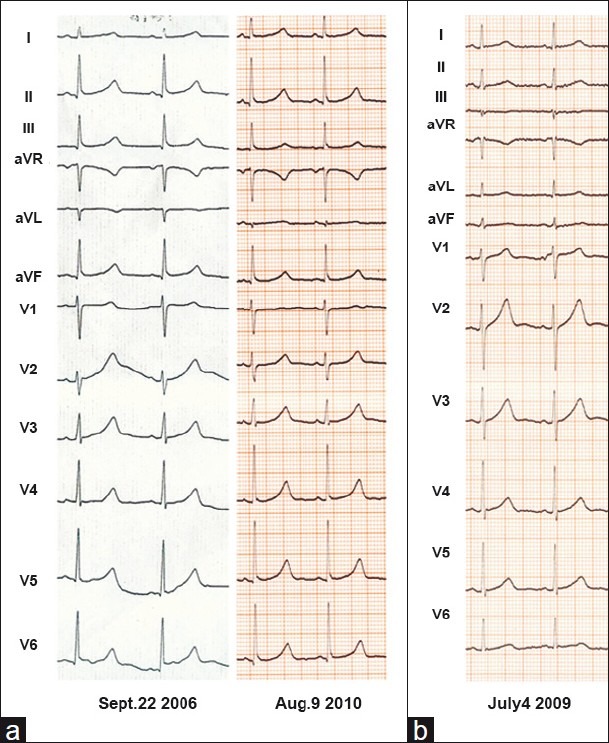
The 12-lead electrocardiogram (ECG) for patients from kindred L151: (a) ECGs for L151-01, now a 17-year-old girl: The left ECG was recorded when she was 12 years old and diagnosed as having Long QT syndrome (QTc 480 ms); the right one was recorded when she was 16 years old and taking metoprolol 0.69 mg/Kg (QTc 530 ms). (b) ECG for the 46-year-old Jervell and Lange-Nielsen syndrome proband: The only abnormality is QT prolongation (QTc 460 ms) for him. He has no syncope after his 30's. No ECG was available for his young ages when he had syncope.
Compound heterozygous mutations c.1032G>A (p.Ala344Alasp) and c.569G>A (p.Arg190Gln) were identified in the proband. His daughter, who displayed a RWS phenotype, inherited c.1032G>A (p.Ala344Alasp) from him and novel missense mutation c.574C>T (p.Arg192Cys) from her mother who was a silent mutation carrier. A rare single nucleotide polymorphism (SNP), c.253G>A (p.Asp85Asn) in KCNE1, was found in the proband, his mother, sister and daughter [Figure 4]. c.253G>A (p.Asp85Asn; rs1805128) and the three pathogenic mutations identified in kindred L151 were absent in 100 control alleles.
Figure 4.
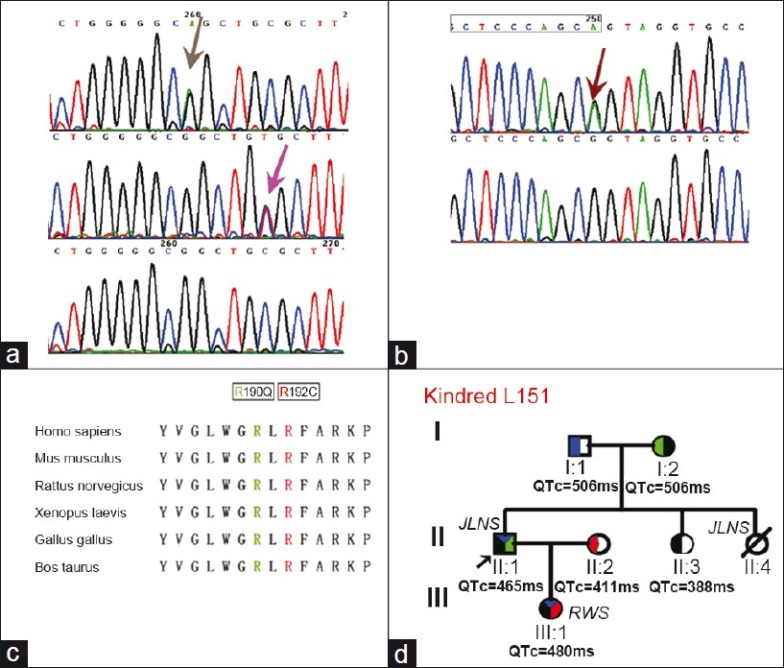
The mutations detected in kindred L151. (a) shows a novel mutation, c.574C>T, discovered in the LQT1 daughter (pink arrow) and a known mutation c.569G>A found in the Jervell and Lange-Nielsen syndrome father (gray arrow). (b) shows a mutation c.1032G>A (arrow) found in both the daughter and father, which induced a splicing error. The ending point of the coding domain for exon 7 of KCNQ1 is marked in the rectangle. (c) shows the conservation across species for c.569G>A and c.574C>T. (d) shows the family tree for L151. Blue represents c.1032G>A, Green c.569G>A, Red c.574C>T, Black D85N-KCNE1 (Sequence not shown).
Clinical characteristics and mutation finding in Family L155
The proband for L155 was a 16-year-old boy with significant hearing loss and required the use of a hearing aid. Between the ages of 6 and 16, he experienced four exercise-induced syncopal episodes. His ECG showed marked sinus bradycardia and a prolonged QTc of 620 ms [Figure 5]. Since being diagnosed with JLNS, he has been treated with Propranolol (2.0 mg/kg) and a pacemaker, and has remained event-free. His parents had normal QT intervals and their marriage was not consanguineous.
Figure 5.
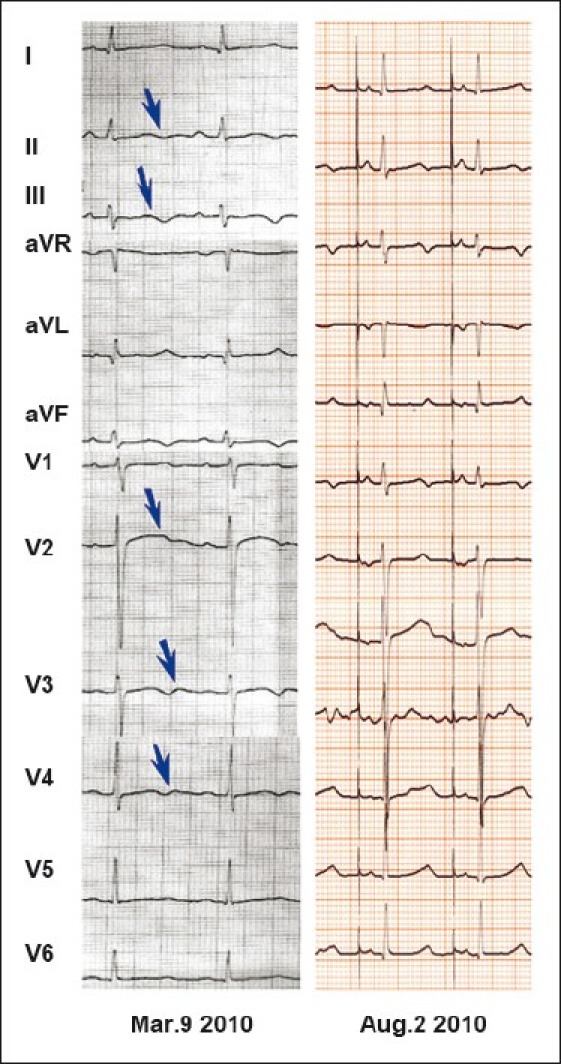
The 12-lead electrocardiogram (ECG) for the proband of L155. The proband is a 16-year-old boy: The left ECG showed very broad and notched or bifid even biphasic T waves on multi-leads (arrows) (QTc 620 ms) before he had received a pacemaker; the right one showed less abnormal ECG 5 months after pacemaker+propranolol 2 mg/Kg (QTc 514 ms).
Two novel complex mutations were found in the proband of kindred L155: c.1032_1117dup (p.Ser373TrpfsX10), a frameshift mutation causing a truncated protein containing a total of 381 amino acids, and c.1319delT (p.Val440AlafsX26), another frameshift mutation resulting in a truncated protein, containing a total of 464 amino acids. His father carries a heterozygous p.Ser373TrpfsX10 only, while the mother carries a heterozygous p.Val440AlafsX26 [Figure 6]. These two mutations were absent in 100 control alleles.
Figure 6.
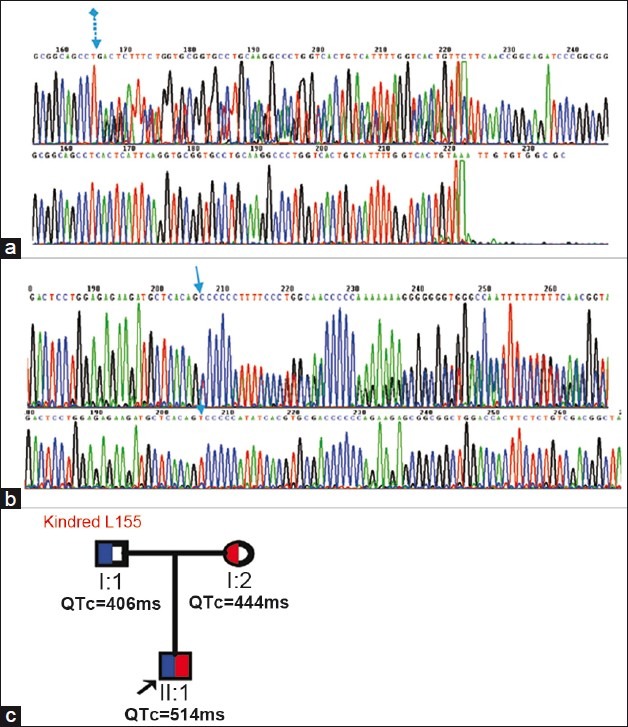
The mutations detected in kindred L155. (a) shows a novel mutation, c.1032_1117dup on exon 8, resulting in a truncated protein containing a total of 381 amino acids. (b) shows another novel deletion mutation, 1319 delT on exon 10, resulting in a truncated protein containing a total of 464 amino acids. The paternal lineage carried a heterozygous p.Ser373TrpfsX10 only, while the maternal lineage carried a heterozygous p.Val440AlafsX26. (c) shows the family tree for L155. The father carried a heterozygous c.1032_1117dup (blue), while the mother a heterozygous 1319 delT (red).
The bizarre T waves in Chinese patients with JLNS
Marked QT prolongation associated with bizarre T waves (broad, bifid, notched or biphasic) was seen on 3/4 JLNS patients [Figures 1 and 5]. These ECG abnormalities were more prominent when patients were symptomatic, and improved after treatment. One individual who did not demonstrate such T wave anomalies was the proband of L151, a 46-year-old male with borderline QTc prolongation (460 ms) who has been event-free for the past 16 years.
DISCUSSION
There have been four reports regarding JLNS-causing mutations in Asian patients thus far,[11–13,18] all found in KCNQ1. Zhang et al.,[12] identified a homozygous mutation T322M in two Chinese siblings; Ohno S et al.,[11] described another homozygous mutation W248F in a Japanese family in 2008. Baek JS et al.,[18] reported compound heterozygous mutation V307sp/S277del in a Korean family in 2010, and Wang RR et al.,[13] described T2C/1149insT in a young Chinese female in 2011. In light of the three additional pairs of compound heterozygous mutations identified in Chinese patients with JLNS in this study, it appears that compound mutations in KCNQ1 may be a primary cause of JLNS among Chinese individuals.
Among the newly identified mutations, c.1032_1117dup and c.1319delT are novel, and c.569G>A, c.605-2A>G, c.815G>A, c.1032G>A are known mutations, previously described in patients with RWS. Interestingly, we found that novel heterozygous mutation c.574C>T evoked a RWS phenotype within a JLNS lineage when joined with c.1032G>A. It should be noted that in Itoh H. et al.'s 2010 study,[19] two out of three of their probands with two KCNQ1 mutations presented without deafness but with severe phenotypes of RWS. Our work further demonstrates that two KCNQ1 mutations can result in a severe variant of RWS in Chinese.
Furthermore, we found that the degree of ECG abnormalities seen in our JLNS patients was associated with the severity of their clinical manifestations. After effective treatments such as beta-blockers, left cardiac sympathetic denervation, or pacemakers, improvement was gauged by reduced syncopal episodes and less abnormal T wave morphologies on ECG readings.
Of the 31 JLNS-causing mutations previously reported [Table 2 and Figure 7], three were splicing, three nonsense, 14 missense, and 11 were frameshift (insertion/deletion) mutations. In the present study, we have identified two additional splice mutations, c.605-2A>G and c.1032G>A combined with c.815G>A and c.569G>A, respectively, in two Chinese families with JLNS. Kapplinger et al.,[20] reported in 2009 that c.605-2A>G was an LQTS-causing mutation, while the c.1032G>A transition was described by Murray et al.,[21] and by six other groups[22–27] as a cause of LQTS. However, each of these previous studies identified single mutations as causes of RWS. Our study on the other hand, demonstrates that when seen as compound mutations, they can also cause JLNS.
Table 2.
JLNS-related mutations
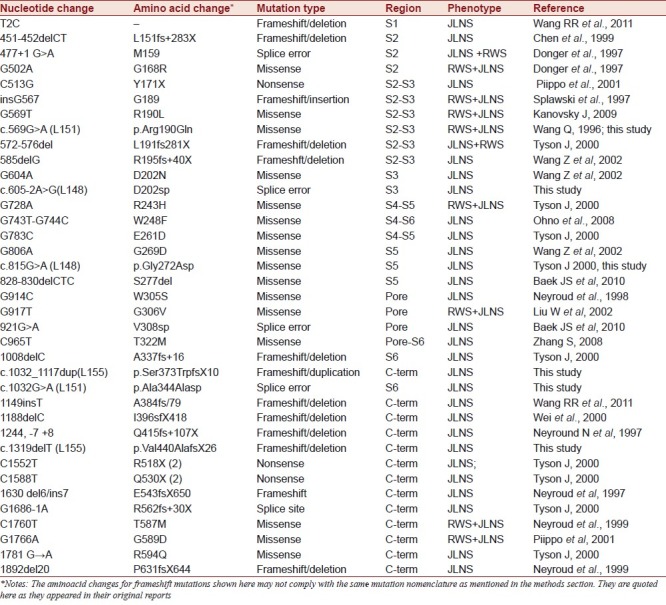
Figure 7.
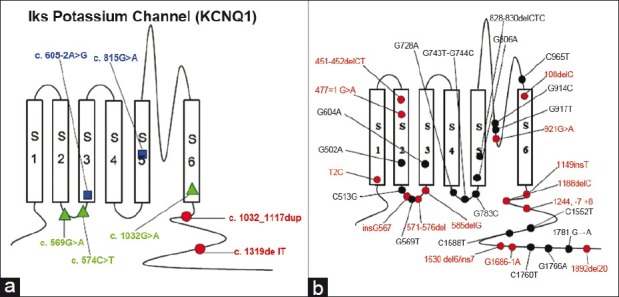
The distribution of mutations within the KCNQ1 channel: (a) Mutations identified in our study: Kindred L148 (blue), kindred L151 (green), kindred L155 (red); (b) Mutations detected previously by other scientists: Frameshift/Deletion/Splice errors are marked in red; Missense/Nonsense are marked in black.
The two novel complex mutations identified in this study, c.1032_1117dup and c.1319delT, expanded the total number of JLNS-causing frameshift mutations in KCNQ1 to 13.
A diverse genotype/phenotype presentation was found in kindred L151 [Figures 3 and 4]. Inherited from each parent, the proband carried compound KCNQ1 mutations and showed a JLNS phenotype. Unlike most JLNS cases, he showed a mild QT interval prolongation (460 ms) and has been event-free for the last 16 years. This finding may imply that with increasing age, cardiac event rates may decrease over time in patients with JLNS, a trend generally seen in most patients with RWS. The proband's parents were silent single-gene mutation carriers and both only presented with markedly prolonged QT intervals. His daughter carried a slightly different set of KCNQ1 compound mutations and presented with a severe RWS phenotype: A significantly prolonged QT interval (530 ms), and recurrent syncope, but without hearing impairment. This observation is consistent with other findings that compound mutations of KCNQ1 do not necessarily lead to JLNS. In 2008, Bhuiyan ZA et al.,[28] described a homozygous intronic mutation (c387-5T>A) that led to an incomplete skipping of exon-2 in KCNQ1 and rescued hearing in an Arabian JLNS patient. This indicated that while a small amount (as little as 10%) of normal KCNQ1 current function was unable to maintain normal cardiac repolarization characteristics, it was enough to effectively maintain hearing function. Furthermore, Kanovsky J et al.,[29] reported a homozygous mutation (R190L) that led to a diagnosis of incomplete JLNS (subclinical hearing impairment) in a 19-year-old female in 2010. Yet, her older sister, who carried the same mutation and presented with similar symptoms, displayed physiologically normal audiometry findings. The mechanism underlying this significant difference could be that some genetic modifiers (yet unknown), when combined with a primary mutation, may exacerbate a patient's phenotype.[30,31]
The allele frequencies of KCNE1-D85N i.e. c.253G>A (p.Asp85Asn), according to Ackerman et al.,[32] are 0.7% in Blacks, 0.7% in Asians, 1.1% in Caucasians, and 0.0% in Hispanics. Additional data from 95 unrelated Han Chinese individuals (50 in the present study and 45 from the International HapMap project[33) indicates that the allele frequency is 0.0% in the Chinese population. A number of studies have demonstrated that D85N can cause loss of function to both IKr (the rapidly activating delayed rectifier K+ channel) and IKs, suggesting that it might function as a disease-causing variant.[34–36] In the present study, both the proband of L151 and his daughter carried a couple of compound mutations on KCNQ1 accompanied by a SNP of D85N on KCNE1, but the daughter maintained normal hearing. It is our hope that the more severe LQTS phenotype seen in the father can help support Lahtinen AM et al.'s[37] proposition that the KCNE1-D85N mutation might preferentially affect males.
The identification of mutations and the investigation of genotype-phenotype relationships of channelopathies have become focal points in the field of genetics and cardiology.[38] Although the molecular genetic mechanisms underlying JLNS remain elusive, important progress has been made. With this study, we have expanded the mutation and phenotype spectrum of JLNS and we have substantiated the important role that compound heterozygous mutations play in the disease. Furthermore, we have documented clinical outcomes and salient ECG irregularities from our patients that with further research may potentially prove to be useful in evaluating the clinical severity of JLNS.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
- 1.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 2.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of KvLQT1 and minK (IsK) proteins to form IKs cardiac potassium channel. Nature. 1996;384:80–3. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 3.Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186–9. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- 4.Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med. 1997;336:1562–7. doi: 10.1056/NEJM199705293362204. [DOI] [PubMed] [Google Scholar]
- 5.Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17:267–68. doi: 10.1038/ng1197-267. [DOI] [PubMed] [Google Scholar]
- 6.Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation. 1998;97:142–46. doi: 10.1161/01.cir.97.2.142. [DOI] [PubMed] [Google Scholar]
- 7.Human variome project in china. [Last accessed on 2011 Dec 25]. Available from: http://www.genomed.org/LOVD/introduction.html .
- 8.Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identification of a Kir3. 4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010;86:872–80. doi: 10.1016/j.ajhg.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. The Jervell and Lange-Nielsen syndrome: Natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783–90. doi: 10.1161/CIRCULATIONAHA.105.592899. [DOI] [PubMed] [Google Scholar]
- 10.Genetic mutations and inherited arrhythmias. [Last accessed on 2011 Dec 25]. Available from: http://www.fsm.it/cardmoc/
- 11.Ohno S, Kubota T, Yoshida H, Tsuji K, Makiyama T, Yamada S, et al. A novel mutation associated with Jervell and Lange-Nielsen syndrome in a Japanese family. Circ J. 2008;72:687–93. doi: 10.1253/circj.72.687. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S, Yin K, Ren X, Wang P, Zhang S, Cheng L, et al. Identification of a novel KCNQ1 mutation associated with both Jervell and Lange-Nielsen and Romano-Ward forms of long QT syndrome in a Chinese family. BMC Med Genet. 2008;9:24. doi: 10.1186/1471-2350-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang RR, Li N, Zhang YH, Wang LL, Teng SY, Pu JL. Novel compound heterozygous mutations T2C and 1149insT in the KCNQ1gene cause Jervell and Lange-Nielsen syndrome. Int J Mol Med. 2011;28:41–6. doi: 10.3892/ijmm.2011.642. [DOI] [PubMed] [Google Scholar]
- 14.Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT. Genomic structure of three long QT syndrome genes: KVLQT1, HERG and KCNE1. Genomics. 1998;51:86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- 15.Ogino S, Gulley ML, den Dunnen JT, Wilson RB. Association for Molecular Pathology Training and Education Committee: Standard mutation nomenclature in molecular diagnostics: Practical and educational challenges. J Mol Diagn. 2007;9:1–6. doi: 10.2353/jmoldx.2007.060081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med. 1992;327:846–52. doi: 10.1056/NEJM199209173271204. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome.An update. Circulation. 1993;88:782–4. doi: 10.1161/01.cir.88.2.782. [DOI] [PubMed] [Google Scholar]
- 18.Baek JS, Bae EJ, Lee SY, Park SS, Kim SY, Jung KN, et al. Jervell and Lange-Nielsen Syndrome: Novel Compound Heterozygous Mutations in the KCNQ1 in a Korean Family. J Korean Med Sci. 2010;25:1522–5. doi: 10.3346/jkms.2010.25.10.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh H, Shimizu W, Hayashi K, Yamagata K, Sakaguchi T, Ohno S, et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: A Japanese multicenter study. Heart Rhythm. 2010;7:1411–8. doi: 10.1016/j.hrthm.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 20.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray A, Donger C, Fenske C, Spillman I, Richard P, Dong YB, et al. Splicing mutations in KCNQ1: A mutation hot spot at codon 344 that produces in frame transcripts. Circulation. 1999;100:1077–84. doi: 10.1161/01.cir.100.10.1077. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Chen Q, Moss AJ, Robinson J, Goytia V, Perry JC, et al. New mutations in the KVLQT1 potassium channel that cause long-QT syndrome. Circulation. 1998;97:1264–9. doi: 10.1161/01.cir.97.13.1264. [DOI] [PubMed] [Google Scholar]
- 23.Kanters JK, Larsen LA, Orholm M, Agner E, Andersen PS, Vuust J, et al. Novel donor splice site mutation in the KVLQT1 gene is associated with long QT syndrome. J Cardiovasc Electrophysiol. 1998;9:620–4. doi: 10.1111/j.1540-8167.1998.tb00944.x. [DOI] [PubMed] [Google Scholar]
- 24.Itoh T, Tanaka T, Nagai R, Kikuchi K, Ogawa S, Okada S, et al. Genomic organization and mutational analysis of KVLQT1, a gene responsible for familial long QT syndrome. Hum Genet. 1998;103:290–4. doi: 10.1007/s004390050819. [DOI] [PubMed] [Google Scholar]
- 25.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–85. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 26.Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation. 2004;110:2119–24. doi: 10.1161/01.CIR.0000144471.98080.CA. [DOI] [PubMed] [Google Scholar]
- 27.Struijk JJ, Kanters JK, Andersen MP, Hardahl T, Graff C, Christiansen M, et al. Classification of the long-QT syndrome based on discriminant analysis of T-wave morphology. Med Biol Eng Comput. 2006;44:543–9. doi: 10.1007/s11517-006-0061-1. [DOI] [PubMed] [Google Scholar]
- 28.Bhuiyan ZA, Momenah TS, Amin AS, Al-Khadra AS, Alders M, Wilde AA, et al. An intonic mutation leading to incomplete skipping of exon-2 in KCNQ1 rescues hearing in Jervell and Lange-Nielsen syndrome. Prog Biophys Mol Biol. 2008;98:319–327. doi: 10.1016/j.pbiomolbio.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Kanovsky J, Novotny T, Kadlecova J, Gaillyova R. A new homozygous mutation of the KCNQ1 gene associated with both Romano-Ward and incomplete Jervell Lange-Nielsen syndromes in two sisters. Heart Rhythm. 2010;7:531–3. doi: 10.1016/j.hrthm.2009.11.034. [DOI] [PubMed] [Google Scholar]
- 30.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–8. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- 31.Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–63. doi: 10.1161/CIRCULATIONAHA.109.879643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic differences in cardiac potassium channel variants: Implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc. 2003;78:1479–87. doi: 10.4065/78.12.1479. [DOI] [PubMed] [Google Scholar]
- 33.International HapMap Project. [Last accessed on 2011 Dec 25]. Available from: http://www.hapmap.org/citinghapmap.html.en .
- 34.Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, et al. D.E.S.I.R. Study Group. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet. 2005;13:1213–22. doi: 10.1038/sj.ejhg.5201489. [DOI] [PubMed] [Google Scholar]
- 35.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: A common cause of severe long-QT syndrome. Circulation. 2004;109:1834–41. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- 36.Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85N, a KCNE1 Polymorphism, Is a Disease-Causing Gene Variant in Long QT Syndrome. J Am Coll Cardiol. 2009;54:812–9. doi: 10.1016/j.jacc.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Lahtinen AM, Marjamaa A, Swan H, Kontula K. KCNE1 D85N polymorphism-a sex-specific modifier in type 1long QT syndrome? BMC Med Genet. 2011;12:11. doi: 10.1186/1471-2350-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou P, Wang J. Genetic testing for channelopathies, more than ten years progress and remaining challenges. J Cardiovasc Dis Res. 2010;1:47–9. doi: 10.4103/0975-3583.64429. [DOI] [PMC free article] [PubMed] [Google Scholar]