

Abstract
The choreographed development of over 200 distinct differentiated cell types from a single zygote is a complex and poorly understood process. Whereas development leads unidirectionally towards more restricted cell fates, recent work in cellular reprogramming has proven that striking conversions of one cellular identity into another can be engineered, promising countless applications in biomedical research and paving the way for modeling disease with patient-derived stem cells. To date, there has been little discussion of which disease models are likely to be most informative. We here review evidence demonstrating that because environmental influences and epigenetic signatures are largely erased during reprogramming, patient-specific models of diseases with strong genetic bases and high penetrance are likely to prove most informative in the near term. However, manipulating in vitro culture conditions may ultimately enable cell-based models to recapitulate gene-environment interactions. Here, we discuss the implications of the new reprogramming paradigm in biomedicine and outline how reprogramming of cell identities is enhancing our understanding of cell differentiation and prospects for cellular therapies and in vivo regeneration.
Plasticity of cellular identity in development and disease
As a zygote cleaves, and through subsequent rounds of cell division develops into a complex organism, cells transition inexorably from one identity to another. Gene expression from a single genome naturally evolves and adapts via a carefully choreographed and directed set of inductive and selective events until lineages become segregated and tissue fates become fixed. This ability of multicellular organisms to create diverse cell types from a single stable genome provides versatility of function, permitting them to adapt and thrive in more varied environments than their single-cell predecessors. While a few complex organisms, such as salamanders, can dedifferentiate their tissues in order to regenerate large portions of their bodies, most multicellular organisms demonstrate very little reversibility of cellular identity after completing embryogenesis. Adult mammals are unable to regenerate organ systems after significant damage or loss, demonstrating that cellular identities in the unaffected tissues are largely stable. Even in the few mammalian organs with high rates of cell turnover, such as the skin, blood system, and gut, the range of possible cell fates is rigidly restricted to those cellular identities comprising the specific tissue.
Evolution has invested heavily in maintaining and restricting cellular identities in mammals. Once a mammalian cell has progressed through its natural developmental and regenerative transitions, its final specialized state is sustained by a loss of self-renewal and inevitable senescence. Mutations in the genetic mechanisms of cellular identity, stability, and senescence predispose cells to the development of malignancy. For example, when granulocyte macrophage precursors acquire self-renewal, these otherwise normal progenitors are transformed into leukemic stem cells (Krivtsov et al., 2006). Pathologic conditions that encourage fluidity of cellular identity can similarly predispose individuals to cancer. Patients with gastroesophageal reflux are a classic example of this phenomenon, where exposure to stomach acid causes affected regions of the esophagus to transform into stomach-like tissue. This tissue metaplasia, while protecting the integrity of the esophagus, also predisposes patients to adenocarcinoma (Lagergren et al., 1999). The in vivo mechanisms by which a differentiated cell transitions to another cell type (metaplasia) or to a more undifferentiated phenotype (dysplasia) are under investigation. Current research suggests that these in vivo alterations of cellular identities are brought about by changes in the epigenome and gene expression of the affected cells, which in turn provide fertile ground for the appearance of mutations that promote malignant transformation (Kang et al., 2003); (Nardone et al., 2007); (Herfs et al., 2009).
Manipulating cellular identity in vitro
The orderly progression of cell differentiation during development has been well described by in vivo studies, but some questions can be addressed more directly in the highly controlled environment of in vitro tissue culture. Human embryonic stem (ES) cells, derived from the inner cell masses of human blastocysts, were first successfully derived less than fifteen years ago by the Thomson group from the University of Wisconsin (Thomson et al., 1998). Pluripotent cells are unique in that they can be grown indefinitely while retaining the ability to differentiate into all three embryonic tissue lineages. Human ES cell derivation has inspired biomedical scientists to exploit stem cells to address questions of human developmental biology, study disease processes in vitro, and even to attempt to replace ailing tissues in human patients. All of these hopes have been pinned on the ability of scientists to engineer specific cellular identities.
This is an ambitious goal. Murine ES cells have been in widespread use for three decades, and yet attempts to generate functional mouse blood cells, pancreatic cells, and highly specialized neurons have so far proven only partially successful. Nonetheless, biologists remain confident that ES cells can be differentiated to specific cell types if culture conditions can be identified that precisely mimic the organizational and signaling events of the developing embryo. This approach necessitates an in-depth understanding of the cellular identity changes that take place in normal development, and requires direct translation of basic developmental biology into painstakingly developed protocols for directed differentiation in vitro. Embryoid bodies have been used as echoes of embryonic organization, and growth factors have been identified that coax pluripotent cells toward one lineage or another. These approaches endeavor to recapitulate the epigenetic changes that occur during embryogenesis to create tissue types analogous to those generated during embryonic development. In the years since the first human ES cell derivation, scientists have crafted differentiation protocols to generate many cell types including motor neurons, retinal pigment epithelium, and hematopoietic precursors from human ES cells (Wichterle et al., 2002); (Klimanskaya et al., 2004); (Ng et al., 2005). These protocols exemplify the reigning paradigm that in vitro manipulations of cellular identity should follow the course of the natural, unidirectional changes that occur during development.
This paradigm was overthrown in 2006 when Takahashi and Yamanaka published the distinctly unnatural conversion of murine fibroblasts into induced pluripotent stem (iPS) cells (Takahashi and Yamanaka, 2006). Not only was their approach not based on mimicking developmental events, but the cellular fate change they engineered went backwards—the implausible reversion of a differentiated, specialized somatic cell to a pluripotent embryonic progenitor. Although conversion of differentiated cells to an embryonic state had previously been accomplished by somatic cell nuclear transfer, that process was and remains to this day inefficient, cumbersome, and poorly understood (Rideout et al., 2001). The Yamanaka reprogramming approach, on the other hand, used a few defined factors to convert a cell to a radically different identity, compelling a bold paradigm shift and introducing the engineering of cell identity as a powerful new strategy for biomedical research and regenerative medicine.
The pluripotency of murine iPS cells has been established in many ways, including gene expression and epigenome profiling, chimera formation, and tetraploid embryo complementation (Okita et al., 2007); (Zhao et al., 2009). Soon after the publication of reprogramming in murine cells, multiple labs confirmed that ectopic expression of defined factors could also generate iPS cells from human tissues (Yu et al., 2007); (Takahashi et al., 2007); (Park et al., 2008b). Both gene expression and epigenetic studies revealed that iPS cells are much more similar to ES cells than they are to their starting cell type (Hawkins et al., 2010; (Doi et al., 2009; Kim et al., 2010; Polo et al., 2010), showing that OSKM transduction indeed alters mammalian cellular identities in the direction opposite to that of development. While rather modest cell fate changes induced by transcription factors between mesodermal or hematopoietic lineages had previously been investigated by Weintraub, Graf and colleagues (Davis et al., 1987); (McNagny and Graf, 2002) engineering cell fate changes as dramatic as reversion of differentiated cells to pluripotency were not envisioned as plausible before Yamanaka’s work.
The original publication of iPS cell reprogramming has inspired researchers to attempt manipulations of cellular identity in new and unexpected directions. Ectopic transcription factor expression is now being investigated as a tool to perform direct conversion, or “transdifferentiation” of one differentiated cell type to another, including β-cells from exocrine cells and neurons and cardiomyocytes from fibroblasts (Zhou et al., 2008); (Vierbuchen et al., 2010); (Ieda et al., 2010); (Efe et al., 2011). Protocols continue to be published for the generation of differentiated cell types from pluripotent stem cells, for more effective generation of stem cells from somatic cells, and for the conversion of one differentiated cell type to another. This diversity of goals represents a radical change in thinking about the categories of identity changes that can be effected in vitro (Figure 1): Adult mammalian cellular identity can be manipulated not just in the direction of stem cell differentiation (a correlate of normal development) but also dedifferentiation (a correlate of dysplasia) and transdifferentiation (a correlate of metaplasia). The recent appreciation for the plasticity of cellular identity has made this area one of the most exciting topics in modern stem cell biology. Takahashi and Yamanaka’s seminal publication has compelled a new and creative open-mindedness about cellular identity, and paved the way for the development of iPS cell-based disease models, drug screens, and cellular therapies. The prospects for cellular therapies are still on the horizon, but use of reprogramming technology for disease modeling and drug testing has already begun. This review provides a discussion of which disease categories are likely to benefit in the near future from reprogramming-based models, and how these models can be used to study gene-environment interactions.
Figure 1. Fluidity of cellular identity, in vivo and in vitro.

Cellular identity is the sum of a cell’s epigenetic landscape. It can become altered through numerous in vivo processes including development, metaplasia, and dysplasia. It can also be manipulated by experimenters in protocols such as directed differentiation, transdifferentiation, and reprogramming.
Reprogramming-based disease models provide a novel platform for disease research
Research into human disease can be performed on platforms as diverse as epidemiology, human genetics, animal modeling, and in vitro cell culture. Each of these approaches provides different kinds of information about the disease under investigation, and each has its own limitations. It is not often that a novel platform for studying disease arises, but in the last few years the advent of patient-specific pluripotent stem cells has inspired researchers to contemplate modeling diseases in a powerful new way. By differentiating patient-specific iPS lines into the cell type responsible for a specific disorder, scientists hope to gain many new research tools. For disorders where etiology is unclear, such as type I diabetes, it is theorized that patient-specific iPS cell models may confirm current theories or inspire new hypotheses about the origins and progression of the disease (Maehr et al., 2009). For diseases in which human-specific cardiac or renal toxicity is a limiting factor in treatment, stem cell-derived models of heart or renal tissues may be used to experimentally measure and reduce drug associated toxicity. Finally, for any disorder whose iPS cell-derived target cells show a measurable disease-specific phenotype, reprogramming-based models can be used as screening tools for development of new drugs that reverse the cellular pathology in vitro, and might therefore carry a greater probability of reversing disease pathology when given to patients.
Because of the inherent pluripotency of the starting cells, their potential applications for cell-autonomous disorders touch virtually every organ system. Researchers studying disorders of hematological, neurological, cardiovascular, metabolic, endocrine, and muscular cell types have already begun the process of creating disease models by reprogramming a disease-specific primary cell sample from patients with cystic fibrosis, Huntington disease, Parkinson disease, sickle cell anemia, dyskeratosis congenita, familial amyotrophic lateral sclerosis and a growing compendium of other conditions recently reviewed by Grskovic et al., 2011 (Park et al., 2008b); (Dimos et al., 2008); (Mali et al., 2008); (Somers et al., 2010); (Ghodsizadeh et al., 2010); (Agarwal et al., 2010). These inexhaustible sources of patient-specific cells are then differentiated towards the lineage affected by the disorder, be it neural, hematopoietic, cardiac, or hepatic. Once the cell type responsible for the disease has been generated, researchers attempt to identify a disease-associated phenotype that manifests characteristics relevant to the disorder. Such cells represent a new research platform for studying both mechanisms and genotype-phenotype interactions of the disease.
For example, congenital long-QT syndrome has been difficult to study in vitro because of the inaccessibility of human cardiomyocytes carrying the causal genetic mutations (Behr et al., 2008). In a recent study by Moretti et al., iPS cells were derived from individuals with monogenic congenital long-QT syndrome type I, differentiated to cardiac lineages, and assayed for characteristic electrophysiological traits (Moretti et al., 2010). The patient-derived cardiomyocytes showed longer-lasting action potentials than the healthy controls, as well as an altered protein localization pattern. These findings allowed the authors to identify a dominant negative mechanism of disease. A paper released shortly afterward described similar studies on another variant of congenital long-QT syndrome, caused by a mutation in a different gene (Itzhaki et al., 2011). This study identified additional disease-associated electrophysiological phenotypes in the patients’ cells, and the authors were able to conduct a limited drug screen to investigate the potency of chemical compounds to ameliorate the disease traits. The existence of these two models will allow for direct comparison of cellular phenotypes between different genotypes of the same disease, and hopefully lead to improved, personalized therapeutic options for patients (Figure 2).
Figure 2. The promise of reprogramming-based disease models.

iPS cell-based models promote research into disease mechanisms, create a framework for screening possible drug compounds, and provide the opportunity to compare different disease genotypes.
Because of these many and varied benefits, reprogramming-based disease models are being rapidly adopted by translational scientists. Dozens have already been published and are the subject of recent review articles (Grskovic et al., 2011; Tiscornia et al., 2011; Unternaehrer and Daley, 2011). However, the concept of iPS cell-based disease modeling is still in its infancy and many challenges remain. Most disease systems still face significant hurdles that need to be overcome before iPS technology can deliver on its promise. The following are specific challenges that each disease field must address.
Directed differentiation and cell culture protocols
For many years, work has been underway to convert pluripotent cells into target cell types. Some highly specialized cell types like motor neurons and cardiomyocytes have been created with great fidelity using protocols that mimic pathways defined through studies of embryo development, while only close facsimiles of other much sought-after cell types, like β-cells, have been created using independent protocols in different laboratories, with phenotypes that differ from actual human target cells (Kroon et al., 2008); (Zhang et al., 2009). Some cell types, such as definitive hematopoietic stem cells (HSCs), have never been successfully derived from pluripotent stem cells purely in vitro. In cases like HSCs, the difficulty rests in large part on the lack of suitable culture conditions to maintain and expand these evanescent cells. Where cell culture conditions remain poorly defined, directed differentiation protocols represent a critical rate-limiting step in iPS cell research.
Definition of “target” cell
Cellular identity is a complex phenotype with many components. For research to proceed on a given iPS cell-derived cell type, the field must first agree that the in vitro product is comparable to its in vivo correlate. This measure of similarity must occur on multiple levels and include analysis of gene expression, chromatin state, and functional assays. Transcriptional activity and methylomes can be evaluated by similar methodologies in all cell types, but appropriate markers of cell identity as well as choice and validation of cell type-specific functional assays must be specifically identified for all target cell types.
Identification of appropriate disease-specific cellular phenotypes
In some cases, target cell types derived from disease-specific iPS cells show clear and predictable phenotypes, such as the electrophysiological abnormalities in the cardiomyocytes described above. For other diseases, the appropriate assay for disease phenotype is unclear or may not show a significant difference between disease and non-disease lines. For example, recent papers have derived iPS lines from patients with idiopathic Parkinson disease (PD) and normal controls (Soldner et al., 2009); (Hargus et al., 2010). The researchers planned to investigate whether PD-iPS cell lines would form dopaminergic neurons at lower frequencies than WT-iPS cells during directed differentiation, as well as how dopaminergic neuron grafts would function in transplantation assays into various animal models of PD. In practice, none of these endpoints showed any difference between the disease and the control-derived iPS cells. The researchers eventually identified only a single outcome measure in the rodent model that showed a difference between the two cell types out of three behavioral tests that were administered after PD was induced. This case underscores the difficulty that befalls the investigator when deciding whether a true phenotypic difference indeed reflects the underlying disease pathophysiology. In another recent paper investigating progeria, the search for a stress-related phenotype in iPS-derived cells led to creative assays such as submersing cells in oil for five hours or exposing them to electrical stimulation for three days (Zhang et al., 2011a). As the search for new disease models expands, cases like this should serve as an instructive reminder that investigators are responsible for demonstrating that their “disease-associated phenotypes” are biologically relevant to the disease at hand.
Identification of diseases amenable to iPS cell-based modeling
To date, there has been little discussion of which disorders are likely to be best informed by iPS cell models. The idiopathic PD example is a cautionary tale that disease modeling with iPS cells will be more informative for some types of disease than for others. Reprogramming may still be too recent a technical advance to reliably predict which diseases can be effectively modeled and which will not, but in the long term it will behoove the field to establish a methodology for analyzing successful and unsuccessful modeling attempts to identify the factors that predict success. Considerations are discussed below for determining whether a particular disease research field is likely to be well served by reprogramming-based models.
Identification of disorders best-suited to ES cell-based disease modeling
Disease-specific ES lines from pre-implantation genetic diagnosis are likely to be better suited for research on certain questions than iPS cell lines. One example of this is Fragile X syndrome (FX), in which the FMR1 gene is inappropriately silenced during development. Because of a failure to reactivate the mutant locus during reprogramming (Urbach et al., 2010), FX-iPS cells do not express the FMR1 gene. Thus while FX-iPS cells may give rise to FMR-deficient neurons (Sheridan et al., 2011), they do not allow studies of the mechanisms by which pathological gene silencing occurs during development.
Reprogramming imperfectly resets the epigenome to an ES cell-like state
Several studies have compared the epigenetic status of iPS cells to that of ES cells and the starting somatic cell type. The global methylation profiles of iPS cells are much closer to ES cells than they are to their tissue of origin (Doi et al., 2009); (Lister et al., 2011), reflecting the phenotypic and functional similarities shared by the pluripotent cells and confirming that the reprogramming process resets the vast majority of the iPS cell epigenome to an ES-like state. However, the epigenomes of the two pluripotent cell types are not identical.
Specific classes of epigenetic marks have been reported to escape reprogramming’s broad epigenetic erasure. While attempting to differentiate iPS cell lines derived from different tissues into various lineages, our laboratory noted that the reprogrammed cells tended to differentiate preferentially into the lineage from which they were originally derived, and in some cases retain residual methylation reflective of the donor cell. These data argue that iPS cells retain epigenetic memory in which a small number of epigenetic marks fail to be reset during reprogramming, apparently at random (Kim et al., 2010);(Kim et al., 2011). Another recent paper identified randomly located methylation differences between iPS and ES cells, and noted that the methylation profiles of the iPS lines became more and more like those of the ES cells upon continued passage (Nishino et al., 2011). Other studies have found that certain chromosomal regions near telomeres and centromeres may be particularly resistant to epigenetic erasure during reprogramming, and that DNA methylation differences in these regions may be maintained even during differentiation (Lister et al., 2011).
Studies on Prader-Willi and Angelman syndromes have also been published on the fate of imprinted genes during reprogramming (Chamberlain et al., 2010); (Yang et al., 2010). To the surprise of investigators, these disease-associated gene imprints were unaltered during iPS cell generation, suggesting that although the majority of developmentally-accrued methylation marks are reset during reprogramming, the process does not revert cellular identity all the way back to a pre-imprinting epigenetic state.
Sources of epigenetic variation in vivo: Environment and stochasticity
Environmental exposures have long been known to introduce changes into the epigenome. Although most environmental events are only transient phenomena, they often have a lasting effect on cellular behavior by causing changes to the epigenome. Cells modify their transcriptional activity in response to chemical exposures, nutrition, or physical stress, and such changes can be an enduring legacy of a significant environmental event (Bell and Spector, 2011) (Waterland and Jirtle, 2004) (McGowan et al., 2009); (Anway et al., 2006).
Stochastic processes also introduce variability to epigenetic organization. The fidelity of inheritance of site-specific maintenance methylation in humans has been estimated at 90 – 98% (Ushijima et al., 2003); (Genereux et al., 2005), but even this modest error rate makes it clear that variation is introduced into the methylome with every cell division at a much higher rate than DNA sequence mutations would ever accumulate. Unlike epigenetic modifications that may occur predictably in response to environmental events, this type of DNA methylation change occurs randomly.
Between environmentally-induced and stochastic epigenetic changes, over time an individual’s epigenome is slowly altered to reflect the events of his or her individual experiences (Wong et al., 2010). Some individuals may also have a genetic predisposition to large or small epigenetic oscillations (Bjornsson et al., 2008; Feinberg and Irizarry, 2010). Given that some or many of these changes will effect gene expression, an individual’s epigenetic signature determines how their cells will react in certain circumstances, and so can predispose to disease or health.
Genetic, environmental, and epigenetic contributions to complex diseases
Despite possessing identical genomes, monozygotic twins are often discordant for complex diseases that strike later in life, such as schizophrenia, type II diabetes, and Parkinson disease. Traditionally, phenotypic variability has been viewed as the sum of genetic variability and environmental variability (Visscher et al., 2008), such that that all differences between monozygotic twins are due to the shared genotype’s reactions to different environments. However, this view commingles the effects of immediate environmental exposures and lasting epigenetic changes, and also ignores epigenetic alterations caused either by stochastic processes or by genetic predisposition to variability. Failure to distinguish between causal environmental exposures and causal epigenetic changes may impair our ability to identify and analyze appropriate phenotypes of in vitro models of disease. Thus when considering disease etiology for modeling, it may be more informative to view cellular disease phenotypes as having three classes of contributing factors, each of which is transmitted differently:
Genetic predisposition. Variations in DNA sequence can increase or decrease the likelihood of a cellular phenotype. DNA sequences are transmitted fully and accurately during cell division.
Environmental exposures (non-epigenetic). Immediate environmental conditions impact cell health and behavior, such as nutritional deficiency, hyperglycemia, or chemical exposure. These conditions are external and not transmittable to daughter cells.
Epigenetic effects. These stable and highly-transmissible changes in gene expression are caused by 1) environmental events; 2) stochastic epigenetic changes; 3) genetic predisposition to variability.
With this in mind, human illness can be viewed as the sum of the genetic, environmental, and epigenetic factors (Petronis, 2006). Attempts to model diseases by iPS cell-derived approaches should take all of these into account. A few rare disorders are caused by a single one of these effects, but many common diseases have the full triad of causes. We will look at examples of these rare, single-factor disorders and then discuss the more common phenomenon of multifactorial disease.
Genetic diseases
Monogenic disorders are those diseases in which a single gene is responsible for the presence, absence, or severity of a particular phenotype. The gene variants responsible for these types of disorders include the CAG repeats of Huntington’s disease, the mutant clotting factors in hemophilia, and the altered ion channels in congenital long QT syndrome. There also may be multigenic conditions in which the combination of specific gene variants causes disease regardless of environmental or epigenetic effects, or where somatic mutation causes the disease. For disorders in which the DNA sequence is responsible for the disease, iPS cells generated from cells with this sequence will bear the causal genotype. Whether or not the responsible locus or loci have been identified, when patient-specific iPS cells are differentiated into relevant cell types, the same genetic defect that caused the patient’s disorder will be present in the differentiated cells.
Environmental diseases
Environmental exposure disorders are those in which an environmental condition directly causes the ailment. In the case of a skin burn caused by exposure to boiling water, 100% of the observed burn phenotype is attributable to the environmental exposure, so there is no epigenetic or genetic cause and the heritability is zero. Comparing iPS cells generated from a patient who has sustained a burn to iPS cells from someone who has not been burned is unlikely to yield information relevant to burn pathophysiology.
Epigenetic diseases
Epigenetic disorders are those in which the phenotype is caused by an epigenetic state of the genome. This may be a wholly aberrant expression pattern of a single gene, as in rare syndromes like Fragile X or Prader-Willi, or the much more common global misregulation in which innumerable environmentally-induced and stochastic epigenetic events affect overall disease predisposition (Feinberg et al., 2010). While the single genes responsible for Fragile X or Prader-Willi are individually regulated during development and so appear to be resistant to epigenetic change during reprogramming, the countless small epigenetic modifications which predispose an individual to complex disease are much more likely to be erased during the reprogramming process. As noted above, epigenetic variation can arise in response to environmental exposures, stochastically, and even in response to specific genotypes. Regardless of the source of the epigenetic change, in a few rare conditions 100% of the observed phenotype is due to epigenetic variation. Phenotypes caused by epigenetic changes include phenomena such as neural tube malformations after global DNA hypomethylation. It has not yet been investigated whether the flawed epigenomes underlying disorders like this one will remain after iPS cell generation, but given the widespread reprogramming of the epigenetic status of the cells, such perpetuation seems unlikely. This issue is discussed in more detail below.
If a disease does not fall cleanly into one of the three categories above, then the phenotypic variation is a sum of the triad of genetic, epigenetic, and environmental variations. An example of such a condition is sunburn after exposure to the sun’s ultraviolet radiation. In this case the genetic makeup of the individual as it relates to skin pigmentation contributes to resistance or predisposition to burning upon exposure to sunlight. In addition, recent ultraviolet exposure that induces genes that produce the photoprotectant melanin may also contribute to burn protection. And finally, intensity and duration of exposure to the damaging radiation will affect the severity of the resulting burn. A reprogramming-based model of sunburn would need to take all three of these factors into account to reproduce a physiologically accurate in vitro model of sunburn.
An interesting subset of complex disorders has both familial and sporadic inheritance patterns but drastically different heritability values. For example, the familial PD variant caused by the α-synuclein A53T mutation has a heritability of nearly 100%, while the diagnosis of idiopathic PD is estimated to show lower heritability of only 30 - 40% (Golbe et al., 1990); (Hamza and Payami, 2010); (Do et al., 2011). Although familial versions of common complex diseases often have distinctive features such as age of onset and severity of symptoms, the underlying cellular pathology often appears consistent between familial and idiopathic versions of disease (e.g., the loss of dopaminergic neurons from the substantia nigra in PD).
Value of stem cell-based disease modeling
Patient-specific disease modeling will be most useful for diseases with large genetic components
During reprogramming of somatic cells to pluripotency, epigenetic histories are erased and standard tissue culture conditions normalize all environmental differences. Of the genetic, environmental, and epigenetic variations that lead to complex disease, the only source of variation likely to be faithfully maintained in iPS cell lines is the genetic variation (Figure 3).
Figure 3. Utility of patient-specific and induced-disease iPS cell-based disease models.

Pink = healthy, green = predisposed to disease. A) Healthy control cells can be cultured, reprogrammed to iPS cells, and then differentiated into a target cell type as a control for disease phenotype assays. B) Cells from patients with monogenic or highly penetrant genetic disease predisposition can be cultured, reprogrammed, and differentiated to the cell type responsible for the disorder. Since the genetic predisposition to disease is present in the final cell, this cell is likely to show disease-specific phenotypes when compared to the healthy control. C) For diseases caused by environmental and/or epigenetic events, some cell types may not have experienced the causative events. Cells without environmental or epigenetic predisposition to disease can be cultured, reprogrammed, and differentiated, but because they never carried a predisposition to disease the resulting cells are unlikely to demonstrate a disease-specific phenotype. Cells affected by a disease-causing environmental or epigenetic event can be cultured, in which environmental conditions are standardized, and reprogrammed, in which the epigenome is reset. After these factors are normalized, these cells no longer carry a predisposition to disease. Thus the iPS lines and differentiated cells resulting from this approach are unlikely to demonstrate disease-specific phenotypes. D) Cells from any source can be exposed to disease-inducing environmental conditions to create a model of an environmentally-induced disorder. This could be either during reprogramming (as shown), during differentiation, or during extended culture. E) Cells from any source can be epigenetically manipulated to mimic epigenetic alterations contributing to disease.
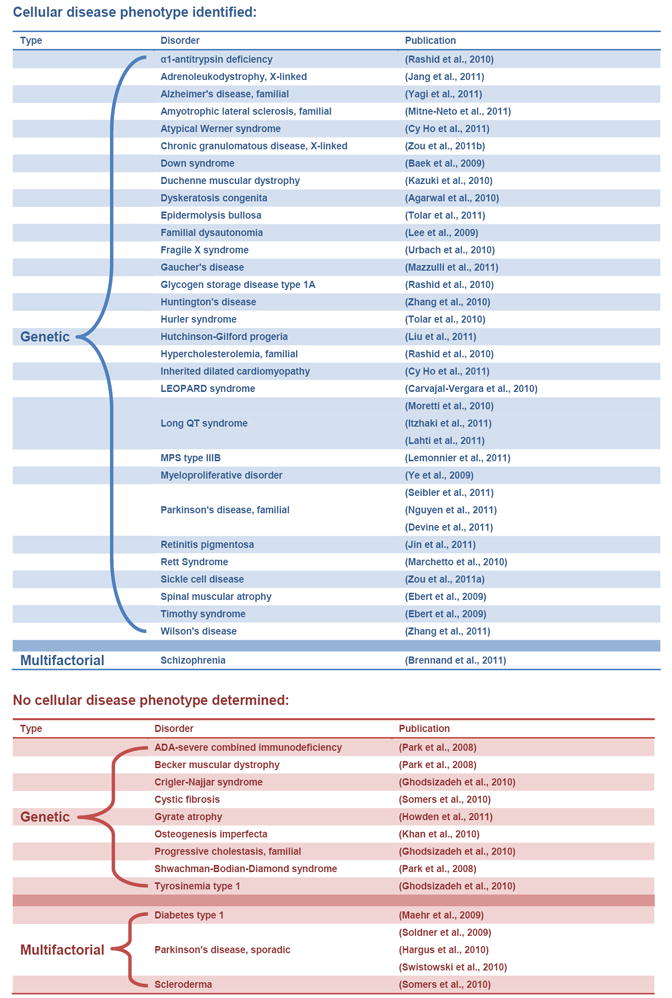
Table 1 lists publications to date which have derived iPS cells from patients and subsequently differentiated them into the target cells affected by the relevant disorder. Some of these papers observed significant differences between the patient-specific lines and control, while others did not. Only one paper has identified a cellular disease phenotype associated with a multifactorial disorder, which was schizophrenia. Schizophrenia is one of the most highly heritable complex disorders, with heritability estimates upwards of 80% (Cardno et al., 1999). All other publications which have shown disease-associated cellular phenotypes have been investigating genetic disorders (Table 1).
Table 1. Patient-specific iPS lines which have been differentiated into a disease-relevant cell type.
Most lines to date which have been differentiated into disease-relevant cell types have been from patients with genetic disease. Identifying a disease-specific phenotype in differentiated cells from patients with multifactorial disease has been less successful than from patients with genetic disease.
|
We predict that the value of patient-specific iPS cell-based disease modeling will be directly proportional to the disease heritability. What then are the prospects for modeling multifactorial diseases with genetic, environmental, and epigenetic components? The field is placing a bet that cells derived from patients who carry a highly-penetrant genetic disease predisposition will provide insight into pathologic mechanisms of both the monogenic and multifactorial versions of the disorder. Is this hope sound? It will be for sporadic forms of disease which have as their molecular underpinnings spontaneously arising mutations in genes that function in the same tissues as the familial forms. These are typically somatic mutations, and consequently are expected to show very similar cellular pathologies in vitro—if the iPS cells happen to capture the somatically acquired disease-causing mutation. In other cases, a patient with sporadic disease may have a genotype that only predisposes to the disease, perhaps in the setting of specific environmental insults, and thus shows incomplete penetrance. This is particularly true for complex diseases, which while not manifesting Mendelian patterns of familial inheritance, nonetheless often show significant heritability. Finally, it is also possible that in some cases the combined environmental and epigenetic influences on the target tissue in an affected patient produces a cellular pathology that mimics that of the genetic version of the disease, but which carries no overt or detectable genetic contributor. In this case, reprogramming will erase all vestiges of the environmental insults that caused the disease, and comparisons of highly heritable familial forms of disease to sporadic cases will prove unrevealing. It is likely that future research will validate each of the above scenarios.
Stem cell-based models for diseases with significant environmental or epigenetic components require experimental induction of the disease
Despite the erasure of environmental and epigenetic causes of disease by reprogramming, pluripotent stem cell-based models may still prove useful for non-genetic diseases. Where the environmental or epigenetic factors responsible for causing disease are well understood, it may be possible to induce them experimentally in culture, even if interactions among distinct cell types are entailed. As an example, by differentiating iPS cells into keratinocytes and melanocytes, which communicate in response to exposure to ultraviolet light (Lin and Fisher, 2007) and could be organized into a facsimile of an epidermis in vitro, the phenomenon of sunburn could be investigated by this experimental platform, as the genetic, epigenetic, and environmental causes are relatively well understood. This leads to a well-controlled triad experimental model, in which as long as two of the three variables are held constant the third can be manipulated to test hypotheses: 1) iPS cell-derived epidermis could be generated from individuals with inherited sunburn predisposition to learn about the genetic factors affecting the process; 2) Various intensities and durations of ultraviolet light could be applied to the epidermis in culture to investigate cellular responses to different severities of environmental exposure; 3) Epidermis in culture could be subjected to repeated ultraviolet exposures to induce gene expression leading to melanin formation, allowing for study of the mechanisms by which the environmental exposure leads to the change in gene expression. This triad model (Figure 4) would provide a much-needed tool to investigate directly the effects of epigenetic manipulations on cellular disease states. It would also be useful for teasing apart genotype by environment or genotype by epigenetic disease effects, because the behavior of cells with various genetic backgrounds could be directly compared.
Figure 4. iPS cell-based triad model of complex disease.

In an experimental system with a reliable cellular disease phenotype readout, constancy of any two variables allows investigation of the other’s effect on the disease phenotype.
Using this technique to approach diseases of poorly-understood etiologies requires a leap of faith that researchers will be able to induce, identify, and exploit a credible cellular pathology. Given that many disorders are late onset, researchers might first discern how to accelerate disease latency from many decades in actual patients to no more than weeks to months in vitro. Moreover, they may be required to re-create the environmental and epigenetic changes induce the cellular phenotype responsible for the disease. A triad model for poorly-understood disorders is an admirable goal which will require much work before it bears fruit.
Future benefits
In addition to the disease models and drug screens currently being developed, the wholesale conversion of one cell type to another has compelled scientists to entertain novel phenomena in cellular and developmental biology, and inspired clinicians to conceive new therapeutic opportunities. Myriad diseases are caused by the lack of a crucial cell or an indispensable protein; the ability to cure disease by replacing the missing component is the promise of stem cell engineering and regenerative medicine.
Two broad approaches could be taken to restore a missing cell type: 1) Creation of iPS cells followed by directed differentiation into the missing cell type, which would then be incorporated into the patient in an anatomically appropriate fashion; or 2) Direct conversion of an extant healthy cell type into the missing or damaged cell type, thus effecting cell regeneration in situ. The first of these possibilities has many elements in common with the approaches currently being taken to create disease models, requiring GMP reprogramming procedures and precise directed differentiation protocols that eliminate the possibility of stem cell-derived tumors. Diseases of different systems each need an approach for attaining lasting integration of the novel cells. This will vary by organ, and may be straightforward for some disorders, like those requiring hematopoietic stem cells, but much more difficult for others, like neurodegenerative conditions requiring specific neuronal connections to be made. The second approach to cell therapy would be to manipulate cellular identity in situ to generate the missing cell type in an appropriate anatomical location. A groundbreaking study from the Melton group investigated in vivo conversion of cell types inside the pancreas (Zhou et al., 2008). They found that infection of pancreatic exocrine cells with viruses expressing developmental pancreatic genes resulted in the in situ formation of cells that looked and behaved like β cells, including an ability to rectify hyperglycemia. Unlike the iPS cell-based approach to cellular therapy, this direct conversion method has no risk of residual pluripotent cells. Instead, the main hurdles are the targeted delivery of the conversion agents to the appropriate areas and the prevention of partially-converted or transformed cells. As with tissue metaplasia in the setting of tissue pathology, manipulation of cellular identity in vivo might carry the worrisome risk of neoplasia.
In addition to the experimental and possibly therapeutic tools generated by manipulating cellular identity in vitro, these techniques may provide novel ways to study diseases of aberrant cell fates, such as metaplasia, neoplasm, and developmental disorders. It has not escaped anyone’s notice that the original Yamanaka reprogramming factors—Oct4, Sox2, KLF4, and Myc—are in distinct contexts potent transforming oncogenes. Indeed, reprogramming mimics the dedifferentiation surmised for some tumors, and insights into the mechanisms of reprogramming are bound to yield new discoveries of mechanisms of oncogenic transformation. Because specific fate changes can now be induced in mammalian cells, researchers have the opportunity to interrogate the mechanisms by which a cell is coerced to change its identity, whether in the natural course of tissue differentiation, the distinctly pathologic course of dedifferentiation, the engineered fates of transdifferentiation, or the most dramatic of fate changes—reprogramming to pluripotency.
Conclusion
The discovery of cellular reprogramming, and the realization that cellular identity is malleable and subject to engineering, has compelled researchers to think in bold ways about novel approaches to research disease mechanisms, drug development, and cell-based treatments for a range of diseases. However, recognizing that many diseases have significant environmental and epigenetic contributions, we anticipate that in the near term patient-specific stem cell-based disease models will be most useful for those disorders with highly penetrant genetic etiologies, and that this first wave of models will teach important lessons about precisely what aspects of pathophysiology can and cannot be gleaned from a disease-in-a-dish, including the impact of epigenetic memory and imperfect reprogramming on disease models. Using pluripotent stem cells to model diseases with only small genetic contributions may prove feasible, but will present more formidable challenges, and will depend on recapitulating environmental and epigenetic influences so that the relevant genetic and non-genetic factors collude to faithfully reproduce disease phenotypes in vitro. Given the pace of current research and the rapid accumulation of publications describing iPS cell-based disease models, the advantages and limitations of this new research platform are bound to come into focus, yielding principles that will guide future research within realistic expectations.
Acknowledgments
GQD is supported by grants from the NIH (RO1-DK70055, RO1-DK59279, UO1-HL100001, and special funds from the ARRA stimulus package- RC2-HL102815), the Roche Foundation for Anemia Research, Alex’s Lemonade Stand, Ellison Medical Foundation, Doris Duke Medical Foundation, and the Harvard Stem Cell Institute. GQD is an affiliate member of the Broad Institute, a recipient of Clinical Scientist Awards in Translational Research from the Burroughs Wellcome Fund and the Leukemia and Lymphoma Society, and an investigator of the Howard Hughes Medical Institute and the Manton Center for Orphan Disease Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal S, Loh YH, McLoughlin EM, Huang JJ, Park IH, Miller JD, Huo HG, Okuka M, dos Reis RM, Loewer S, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–U176. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anway MD, Leathers C, Skinner MK. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology. 2006;147:5515–5523. doi: 10.1210/en.2006-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, Park IH, Yoon SS, Minami T, et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459:1126–1130. doi: 10.1038/nature08062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr ER, Dalageorgou C, Christiansen M, Syrris P, Hughes S, Tome Esteban MT, Rowland E, Jeffery S, McKenna WJ. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 2008;29:1670–1680. doi: 10.1093/eurheartj/ehn219. [DOI] [PubMed] [Google Scholar]
- Bell JT, Spector TD. A twin approach to unraveling epigenetics. Trends in Genetics. 2011;27:116–125. doi: 10.1016/j.tig.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui HM, Yu WQ, Rongione MA, Ekstrom TJ, Harris TB, et al. Intra-individual change over time in DNA methylation with familial clustering. Jama-Journal of the American Medical Association. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. Modelling schizophrenia using human induced pluripotent stem cells. CORD Conference Proceedings. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardno AG, Marshall EJ, Coid B, Macdonald AM, Ribchester TR, Davies NJ, Venturi P, Jones LA, Lewis SW, Sham PC, et al. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch Gen Psychiatry. 1999;56:162–168. doi: 10.1001/archpsyc.56.2.162. [DOI] [PubMed] [Google Scholar]
- Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–U812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cy Ho J, Zhou T, Lai WH, Huang Y, Chan YC, Li X, Ly Wong N, Li Y, Au KW, Guo D, et al. Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging (Albany NY) 2011 doi: 10.18632/aging.100277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB. EXPRESSION OF A SINGLE TRANSFECTED CDNA CONVERTS FIBROBLASTS TO MYOBLASTS. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun. 2011;2:440–440. doi: 10.1038/ncomms1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW, et al. Web-Based Genome-Wide Association Study Identifies Two Novel Loci and a Substantial Genetic Component for Parkinson’s Disease. Plos Genetics. 2011;7 doi: 10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho JS, Loewer S, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature Genetics. 2009;41:1350–U1123. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efe JA, Hilcove S, Kim J, Zhou H, Ouyang K, Wang G, Chen J, Ding S. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nature Cell Biology. 2011;13:215–U261. doi: 10.1038/ncb2164. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Irizarry RA. Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1757–1764. doi: 10.1073/pnas.0906183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Irizarry RA, Fradin D, Aryee MJ, Murakami P, Aspelund T, Eiriksdottir G, Harris TB, Launer L, Gudnason V, et al. Personalized Epigenomic Signatures That Are Stable Over Time and Covary with Body Mass Index. Science Translational Medicine. 2010;2 doi: 10.1126/scitranslmed.3001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux DP, Miner BE, Bergstrom CT, Laird CD. A population-epigenetic model to infer site-specific methylation rates from double-stranded DNA methylation patterns. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5802–5807. doi: 10.1073/pnas.0502036102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodsizadeh A, Taei A, Totonchi M, Seifinejad A, Gourabi H, Pournasr B, Aghdami N, Malekzadeh R, Almadani N, Salekdeh GH et al. Generation of Liver Disease-Specific Induced Pluripotent Stem Cells Along with Efficient Differentiation to Functional Hepatocyte-Like Cells. Stem Cell Reviews and Reports. 2010;6:622–632. doi: 10.1007/s12015-010-9189-3. [DOI] [PubMed] [Google Scholar]
- Golbe LI, Di Iorio G, Bonavita V, Miller DC, Duvoisin RC. A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol. 1990;27:276–282. doi: 10.1002/ana.410270309. [DOI] [PubMed] [Google Scholar]
- Grskovic M, Javaherian A, Strulovici B, Daley GQ. Induced pluripotent stem cells - opportunities for disease modelling and drug discovery. CORD Conference Proceedings. 2011;10:915–929. doi: 10.1038/nrd3577. [DOI] [PubMed] [Google Scholar]
- Hamza TH, Payami H. The heritability of risk and age at onset of Parkinson’s disease after accounting for known genetic risk factors. Journal of Human Genetics. 2010;55:241–243. doi: 10.1038/jhg.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus G, Cooper O, Deleidi M, Levy A, Lee K, Marlow E, Yow A, Soldner F, Hockemeyer D, Hallett PJ, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15921–15926. doi: 10.1073/pnas.1010209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herfs M, Hubert P, Delvenne P. Epithelial metaplasia: adult stem cell reprogramming and (pre)neoplastic transformation mediated by inflammation? TRENDS MOL MED. 2009;15:245–253. doi: 10.1016/j.molmed.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Howden SE, Gore A, Li Z, Fung HL, Nisler BS, Nie J, Chen G, McIntosh BE, Gulbranson DR, Diol NR, et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proc Natl Acad Sci U S A. 2011;108:6537–6542. doi: 10.1073/pnas.1103388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–U113. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- Jang J, Kang HC, Kim HS, Kim JY, Huh YJ, Kim DS, Yoo JE, Lee JA, Lim B, Lee J, et al. Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. CORD Conference Proceedings. 2011;70:402–409. doi: 10.1002/ana.22486. [DOI] [PubMed] [Google Scholar]
- Jin ZB, Okamoto S, Osakada F, Homma K, Assawachananont J, Hirami Y, Iwata T, Takahashi M. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. CORD Conference Proceedings. 2011;6:e17084–e17084. doi: 10.1371/journal.pone.0017084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang GH, Lee HJ, Hwang KS, Lee S, Kim JH, Kim JS. Aberrant CpG island hypermethylation of chronic gastritis, in relation to aging, gender, intestinal metaplasia, and chronic inflammation. American Journal of Pathology. 2003;163:1551–1556. doi: 10.1016/S0002-9440(10)63511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, Hiratsuka M, Takiguchi M, Osaki M, Kajitani N, Hoshiya H, Hiramatsu K, Yoshino T, Kazuki K, Ishihara C, et al. Complete Genetic Correction of iPS Cells From Duchenne Muscular Dystrophy. Molecular Therapy. 2010;18:386–393. doi: 10.1038/mt.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IF, Hirata RK, Wang PR, Li Y, Kho J, Nelson A, Huo YW, Zavaljevski M, Ware C, Russell DW. Engineering of Human Pluripotent Stem Cells by AAV-mediated Gene Targeting. Molecular Therapy. 2010;18:1192–1199. doi: 10.1038/mt.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LIR, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–U260. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P, Hongguang H, Loh YH, Aryee MJ, Lensch MW, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011 doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimanskaya I, Hipp J, Rezai KA, West M, Atala A, Lanza R. Derivation and comparative assessment of retinal pigment epithelium from human embryonic stem cells using transcriptomics. Cloning and Stem Cells. 2004;6:217–245. doi: 10.1089/clo.2004.6.217. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng ZH, Stubbs MC, Wang YZ, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nature Biotechnology. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- Lagergren J, Bergstrom R, Lindgren A, Nyren O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. New England Journal of Medicine. 1999;340:825–831. doi: 10.1056/NEJM199903183401101. [DOI] [PubMed] [Google Scholar]
- Lahti AL, Kujala VJ, Chapman H, Koivisto AP, Pekkanen-Mattila M, Kerkelä E, Hyttinen J, Kontula K, Swan H, Conklin BR, et al. Human disease model for long QT syndrome type 2 using iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2011 doi: 10.1242/dmm.008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier T, Blanchard S, Toli D, Roy E, Bigou S, Froissart R, Rouvet I, Vitry S, Heard JM, Bohl D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. CORD Conference Proceedings. 2011;20:3653–3666. doi: 10.1093/hmg/ddr285. [DOI] [PubMed] [Google Scholar]
- Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. 2007;445:843–850. doi: 10.1038/nature05660. [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–U84. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011 doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehr R, Chen SB, Snitow M, Ludwig T, Yagasaki L, Goland R, Leibel RL, Melton DA. Generation of pluripotent stem cells from patients with type 1 diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15768–15773. doi: 10.1073/pnas.0906894106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Ye ZH, Hommond HH, Yu XB, Lin J, Chen GB, Zou JZ, Cheng LZ. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells. 2008;26:1998–2005. doi: 10.1634/stemcells.2008-0346. [DOI] [PubMed] [Google Scholar]
- Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo GW, Mu YL, Chen G, Gage FH, Muotri AR. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNagny K, Graf T. Making eosinophils through subtle shifts in transcription factor expression. Journal of Experimental Medicine. 2002;195:f43–f47. doi: 10.1084/jem.20020636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitne-Neto M, Machado-Costa M, Marchetto MC, Bengtson MH, Joazeiro CA, Tsuda H, Bellen HJ, Silva HA, Oliveira AS, Lazar M, et al. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. CORD Conference Proceedings. 2011;20:3642–3652. doi: 10.1093/hmg/ddr284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, et al. Patient-Specific Induced Pluripotent Stem-Cell Models for Long-QT Syndrome. New England Journal of Medicine. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- Nardone G, Compare D, De Colibus P, de Nucci G, Rocco A. Helicobacter pylori and epigenetic mechanisms underlying gastric carcinogenesis. Digestive Diseases. 2007;25:225–229. doi: 10.1159/000103890. [DOI] [PubMed] [Google Scholar]
- Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG. Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood. 2005;106:1601–1603. doi: 10.1182/blood-2005-03-0987. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schüle B, Dolmetsch RE, Langston W, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. CORD Conference Proceedings. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, Umezawa A. DNA Methylation Dynamics in Human Induced Pluripotent Stem Cells over Time. Plos Genetics. 2011;7 doi: 10.1371/journal.pgen.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–U311. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008a;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo HG, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008b;451:141–U141. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Petronis A. Epigenetics and twins: three variations on the theme. Trends in Genetics. 2006;22:347–350. doi: 10.1016/j.tig.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li YS, Shioda T, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nature Biotechnology. 2010;28:848–U130. doi: 10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. Journal of Clinical Investigation. 2010;120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout WM, Eggan K, Jaenisch R. Nuclear cloning and epigenetic reprogramming of the genome. Science. 2001;293:1093–1098. doi: 10.1126/science.1063206. [DOI] [PubMed] [Google Scholar]
- Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D. Mitochondrial Parkin Recruitment Is Impaired in Neurons Derived from Mutant PINK1 Induced Pluripotent Stem Cells. J Neurosci. 2011;31:5970–5976. doi: 10.1523/JNEUROSCI.4441-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile x syndrome. PLoS One. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, et al. Parkinson’s Disease Patient-Derived Induced Pluripotent Stem Cells Free of Viral Reprogramming Factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers A, Jean JC, Sommer CA, Omari A, Ford CC, Mills JA, Ying L, Sommer AG, Jean JM, Smith BW, et al. Generation of Transgene-Free Lung Disease-Specific Human Induced Pluripotent Stem Cells Using a Single Excisable Lentiviral Stem Cell Cassette. Stem Cells. 2010;28:1728–1740. doi: 10.1002/stem.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swistowski A, Peng J, Liu QY, Mali P, Rao MS, Cheng LZ, Zeng XM. Efficient Generation of Functional Dopaminergic Neurons from Human Induced Pluripotent Stem Cells Under Defined Conditions. Stem Cells. 2010;28:1893–1904. doi: 10.1002/stem.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Tiscornia G, Vivas EL, Belmonte JC. Diseases in a dish: modeling human genetic disorders using induced pluripotent cells. Nat Med. 2011:1570–1576. doi: 10.1038/nm.2504. [DOI] [PubMed] [Google Scholar]
- Tolar J, Park IH, Xia L, Lees CJ, Peacock B, Webber B, McElmurry RT, Eide CR, Orchard PJ, Kyba M, et al. Hematopoietic differentiation of induced pluripotent stem cells from patients with mucopolysaccharidosis type I (Hurler syndrome) Blood. 2010;117:839–847. doi: 10.1182/blood-2010-05-287607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar J, Xia L, Riddle MJ, Lees CJ, Eide CR, McElmurry RT, Titeux M, Osborn MJ, Lund TC, Hovnanian A, et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. CORD Conference Proceedings. 2011;131:848–856. doi: 10.1038/jid.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unternaehrer JJ, Daley GQ. Induced pluripotent stem cells for modelling human diseases. Philos Trans R Soc Lond B Biol Sci. 2011;366:2274–2285. doi: 10.1098/rstb.2011.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential Modeling of Fragile X Syndrome by Human Embryonic Stem Cells and Induced Pluripotent Stem Cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushijima T, Watanabe N, Okochi E, Kaneda A, Sugimura T, Miyamoto K. Fidelity of the methylation pattern and its variation in the genome. Genome Res. 2003;13:868–874. doi: 10.1101/gr.969603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–U1050. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions. Nat Rev Genet. 2008;9:255–266. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition. 2004;20:63–68. doi: 10.1016/j.nut.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Wong CC, Caspi A, Williams B, Craig IW, Houts R, Ambler A, Moffitt TE, Mill J. A longitudinal study of epigenetic variation in twins. Epigenetics : official journal of the DNA Methylation Society. 2010;5:516–526. doi: 10.4161/epi.5.6.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- Yang JY, Cai J, Zhang Y, Wang XM, Li W, Xu JY, Li F, Guo XP, Deng K, Zhong M, et al. Induced Pluripotent Stem Cells Can Be Used to Model the Genomic Imprinting Disorder Prader-Willi Syndrome. Journal of Biological Chemistry. 2010;285:40303–40311. doi: 10.1074/jbc.M110.183392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Zhan H, Mali P, Dowey S, Williams DM, Jang YY, Dang CV, Spivak JL, Moliterno AR, Cheng L. Human induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JY, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhang DH, Jiang W, Liu M, Sui X, Yin XL, Chen S, Shi Y, Deng HK. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Research. 2009;19:429–438. doi: 10.1038/cr.2009.28. [DOI] [PubMed] [Google Scholar]
- Zhang JQ, Lian QZ, Zhu GL, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang YL, Tse HF, et al. A Human iPSC Model of Hutchinson Gilford Progeria Reveals Vascular Smooth Muscle and Mesenchymal Stem Cell Defects. Cell Stem Cell. 2011a;8:31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Zhang N, An MC, Montoro D, Ellerby LM. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010;2:RRN1193–RRN1193. doi: 10.1371/currents.RRN1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chen S, Li W, Guo X, Zhao P, Xu J, Chen Y, Pan Q, Liu X, Zychlinski D, et al. Rescue of ATP7B function in hepatocyte-like cells from Wilson’s disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin. Hum Mol Genet. 2011b;20:3176–3187. doi: 10.1093/hmg/ddr223. [DOI] [PubMed] [Google Scholar]
- Zhao XY, Li W, Lv Z, Liu L, Tong M, Hai T, Hao J, Guo CL, Ma QW, Wang L, et al. iPS cells produce viable mice through tetraploid complementation. Nature. 2009;461:86–U88. doi: 10.1038/nature08267. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–U630. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011a;118:4599–4608. doi: 10.1182/blood-2011-02-335554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, Dowey SN, Cheng L, Malech HL. Oxidase deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease mediated safe harbor targeting. Blood. 2011b;117:5561–5572. doi: 10.1182/blood-2010-12-328161. [DOI] [PMC free article] [PubMed] [Google Scholar]