

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive and typically fatal lung disease. To gain insight into the pathogenesis of IPF, we reanalyzed our previously published gene expression data profiling IPF lungs. Cytokine receptor-like factor 1 (CRLF1) was among the most highly up-regulated genes in IPF lungs, compared with normal controls. The protein product (CLF-1) and its partner, cardiotrophin-like cytokine (CLC), function as members of the interleukin 6 (IL-6) family of cytokines. Because of earlier work implicating IL-6 family members in IPF pathogenesis, we tested whether CLF-1 expression contributes to inflammation in experimental pulmonary fibrosis. In IPF, we detected CLF-1 expression in both type II alveolar epithelial cells and macrophages. We found that the receptor for CLF-1/CLC signaling, ciliary neurotrophic factor receptor (CNTFR), was expressed only in type II alveolar epithelial cells. Administration of CLF-1/CLC to both uninjured and bleomycin-injured mice led to the pulmonary accumulation of CD4+ T cells. We also found that CLF-1/CLC administration increased inflammation but decreased pulmonary fibrosis. CLF-1/CLC leads to significantly enriched expression of T-cell-derived chemokines and cytokines, including the antifibrotic cytokine interferon-γ. We propose that, in IPF, CLF-1 is a selective stimulus of type II alveolar epithelial cells and may potentially drive an antifibrotic response by augmenting both T-helper-1-driven and T-regulatory-cell-driven inflammatory responses in the lung.
Idiopathic pulmonary fibrosis (IPF) is a progressive disorder characterized by unrelenting accumulation of myofibroblasts and by deposition of collagens in the lower respiratory tract, leading ultimately to breathlessness and respiratory failure. It is a rare disease, with an estimated prevalence of 14 to 43 per 100,000 people, depending on the case definition of IPF.1 The disease is typically discovered late in its course, and the median survival time is close to 3 years from the time of diagnosis.2 Except for lung transplantation, in the United States, there are no approved therapies for IPF.
Although anti-inflammatory or immunologically active agents have proven ineffective in IPF,3–5 studies in cytokine signaling in both IPF and animal models of fibrosis still provide a strong rationale for investigating inflammatory pathways in IPF. The dichotomy between cytokines elaborated by type 1 helper T cells (Th1) and type 2 helper T cells (Th2) has been studied in IPF3,6,7 and in animal models of fibrosis. The Th1 cytokine interferon-γ (IFN-γ) suppresses fibroblast proliferation and collagen synthesis8–10 and tends to attenuate fibrosis in animal models.10–12 In contrast, the Th2 cytokines interleukins 4 and 13 (IL-4 and IL-13) promote fibroblast proliferation13 and have been shown to augment pulmonary fibrosis.14,15 The proinflammatory molecule IL-6 and its family members have also been implicated in the pathogenesis of IPF as a factor promoting the growth and survival of human lung fibroblasts in vitro16–18 and lung injury and fibrosis in animal models.19,20
To study IPF, a disease that is shaped by a complex interplay of genetic,21–23 epigenetic,24–26 and environmental27–29 factors, we and others have used gene expression profiling technologies to identify both individual genes and networks of genes that may contribute to the pathogenesis of IPF.30–33 Because of the roles that growth factor and cytokine signaling pathways likely play in the pathogenesis of both IPF and experimental pulmonary fibrosis, we re-examined our gene expression profiling data comparing IPF lungs and normal controls,33 to identify genes involved in growth factor and cytokine signaling pathways. Our reanalysis revealed significantly increased expression of the secreted receptor cytokine-like factor 1 (CLF-1; also known as cytokine receptor-like factor 1 and encoded by the CRLF1 gene). CLF-1 is secreted as a heterodimer with cardiotrophin-like cytokine (CLC), a member of the IL-6 family of cytokines.34
The functions of CLF-1 in the lung and in pulmonary fibrosis are unknown. From our gene expression data, CLF-1 was the most highly up-regulated member of the IL-6 family. Thus, we reasoned that CLF-1 may play an analogous role to IL-6 in the lung potentiating injury, inflammation, and fibroproliferation. We therefore investigated the expression pattern of CLF-1 in IPF and determined the role of CLF-1 in an experimental model of pulmonary fibrosis.
Materials and Methods
The studies were approved by the Institutional Review Boards and Institutional Animal Care and Use Committees of Columbia University and the University of Pittsburgh.
Reagents
The Sircol collagen assay was from Biocolor (Belfast, UK). Recombinant human CLF-1/CLC, and antibodies directed against leukemia inhibitory factor receptor-β (LIFRβ) and CNTFR were from R&D Systems (Minneapolis, MN). Pentobarbital solution was from the Henry Schein company (Melville, NY). Bleomycin and leukemia inhibitory factor (LIF) were from Sigma-Aldrich (St. Louis, MO). Antibodies against mouse B220 (clone RA3-6B2), CD3 (clone 145-2C11), CD8 (clone YTS105.18), CD68 (clone FA-11), mouse neutrophils (clone 7/4), and human CD68 (clone Ki-M7) were obtained from AbD Serotec (Raleigh, NC). Rat IgG directed against mouse CD4 (clone RM4-5) and mouse Foxp3 (clone NRRF-30) were from eBioscience (San Diego, CA). Rabbit IgG against phosphorylated Stat3 (clone D3A7) and total Stat3 were from Cell Signaling Technology (Danvers, MA). Rabbit antibody against surfactant protein C (SP-C) was obtained from Seven Hills Bioreagents (Cincinnati, OH). Goat anti-rabbit Fab fragments, peroxidase-, rhodamine-, and Cy5-conjugated secondary antibodies, Cy5-conjugated streptavidin, nonimmune rabbit and goat IgG were from Jackson ImmunoResearch (West Grove, PA). Pierce SuperSignal West Pico Chemiluminescent substrate was from Thermo Fisher Scientific (Rockford, IL). Vectastain ABC universal kit, 3,3′-diaminobenzidine, Vector SG substrate, aminoethylcarbazole, and Gill's hematoxylin were from Vector Laboratories (Burlingame, CA). Rabbit IgG against Tbet, Power SYBR Green, high-capacity reverse transcriptase, and a one-step RT-PCR kit were from Life Technologies-Invitrogen (Carlsbad, CA) and Life Technologies-Applied Biosystems (Foster City, CA). Elastase was from Worthington Biochemical (Lakewood, NJ).
Gene Expression Profiling of mRNA From Human Lung Tissue
The details of our gene expression profiling experiment have been reported previously.33 In brief, RNA was extracted from whole-lung homogenates from patients with IPF and from normal control subjects. Hybridization of biotinylated cRNA was performed on a human genome U95Av2 array (Affymetrix, Santa Clara, CA). Data were analyzed with GeneSpring software versions 5.0 (Agilent Technologies, Santa Clara, CA), followed by a supervised analysis using open-source Cluster and TreeView software version 1.60 (http://rana.lbl.gov/EisenSoftware.htm).
RNA Extraction for RT-PCR of Human Tissue
Total RNA was extracted from 30 to 50 μg of snap-frozen lung tissue collected by the Lung Tissue Research Consortium (stored at the University of Colorado, Denver, CO) from 49 normal lung histology control subjects and 50 IPF patients. RNA was processed by tissue homogenization and disruption with an electric homogenizer (PolyTron homogenizer H3660-2A; Kinematica, Lucerne, Switzerland) in 700 μL of QIAzol lysis reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. RNA was further purified using a miRNeasy mini kit (217004; Qiagen) with a QIAcube automated system (9001292; Qiagen). The purity of the RNA was verified spectroscopically, and the quality of the RNA was assessed using an Agilent 2100 bioanalyzer.
Quantitative RT-PCR of mRNA from Human Tissue
Quantitative RT-PCR was performed using the ABI TaqMan system (Life Technologies) to validate the gene expression microarray data. RT-PCR amplifications were generated with a TaqMan RNA-to-CT one-step kit (including master mix containing AmpliTaq Gold DNA polymerase, UTP, dNTPs, and buffers) according to the manufacturer's protocol. The PCR conditions were as follows: 15 minutes at 48°C, 12 minutes at 95°C, and then 40 cycles with 15 seconds at 95°C and 1 minute at 60°C in an ABI 7300 real-time PCR system (Life Technologies). PPIA [peptidylprolyl isomerase A (cyclophilin A)] was used as the housekeeping gene for normalization.
Isolation and Use of Primary Rat Type II Alveolar Epithelial Cells
Primary rat type II alveolar epithelial cells were isolated by elastase-mediated digestion of uninjured lungs from Sprague-Dawley rats (Harlan Sprague-Dawley, Indianapolis, IN), as described previously.35 Cells were plated in Dulbecco's modified Eagle's medium + 10% fetal bovine serum, followed by serum starvation and stimulation with recombinant human (rh) CLF-1/CLC at 25 ng/mL for up to 60 minutes. Cells were lysed in radioimmunoprecipitation assay buffer with 0.1% SDS, protease inhibitors, and sodium orthovanadate. Lysates were processed for immunoblotting for phosphorylated Stat3 (p-Stat3). Immunoblots were then stripped and reprobed for total Stat3. Quantitative densitometry was performed using ImageJ software version 1.4.3.67 (NIH, Bethesda, MD), as described previously.36
Delivery of rhCLF-1/CLC to Mice
Cell culture grade, recombinant human CLF-1/CLC was purchased from R&D Systems (Minneapolis, MN). CLF-1/CLC was resuspended according to the recommendations of the manufacturer in hydrochloric acid 4 mmol/L + 0.1% bovine serum albumin. For delivery to the animal, 2 μg of CLF-1/CLC were then diluted 10-fold in sterile phosphate-buffered saline to achieve a final volume of 30 μL at pH 7.4. The vehicle control was an equivalent dilution of the hydrochloric acid and bovine serum albumin diluent. Each animal received up to 14 doses of CLF-1/CLC or vehicle control. We quantified endotoxin in the delivered reagents by the Limulus amebocyte lysate (LAL) method (GenScript, Piscataway, NJ) and determined that each animal received <2.0 EU endotoxin per experiment. This number is considered low and is similar to that in a published study using intranasal administration of a recombinant protein.19 Mice were anesthetized with isoflurane in an anesthesia chamber, a volume of 30 μL of CLF-1/CLC or vehicle control was slowly beaded onto the nares of the mouse, and the volume was inhaled. For experiments involving bleomycin, CLF-1/CLC or vehicle control was administered for 2 days before bleomycin (see next section), and then treatment was continued for up to 14 days after bleomycin injury. Animals were sacrificed at 7 or 14 days after bleomycin injury.
Intratracheal Injection of CLF-1/CLC and LIF
Animals were anesthetized with ketamine 30 mg/kg and xylazine 6 mg/kg by intraperitoneal injection. After induction of anesthesia, an incision was made in the middle of the neck to expose the trachea. After tracheotomy, a sterile 18-gauge catheter was inserted. A volume of 200 μL of LIF 2.5 μg, PBS, or CLF-1/CLC 5 μg was instilled. Then the animal was ventilated via the catheter by means of a small-animal ventilator (model 683; Harvard Apparatus, Holliston, MA). After 15 minutes, the animal was euthanized and the lungs were inflation-fixed with 10% neutral-buffered formalin. Lung tissue was then processed for immunostaining with rabbit IgG directed against p-Stat3 as described below.
Mouse Model of Bleomycin-Induced Lung Injury and Histopathology
Male C57BL/6 mice (6 to 8 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME). The mice were anesthetized with isoflurane in an anesthesia chamber. Bleomycin at 3 mg/kg (standard dose)36 or 1 mg/kg (low dose) or saline control was administered intranasally in a volume of 30 μL. Mice were sacrificed on days 1, 3, 7, 14, or 21 with pentobarbital, and the lungs were excised for histopathology, immunostaining, isolation of RNA, and determination of collagen content. Lung tissue was flash-frozen in liquid nitrogen or formalin-fixed and paraffin-embedded. Routine H&E and Masson's trichrome staining of tissue sections was performed by the Columbia University or the University of Pittsburgh histology services.
Immunostaining of Human and Rodent Lung Sections
Staining of Human Lung
Deidentified cryosections of human lung from patients with IPF or normal control subjects (histologically normal lung distant from a tumor) were air-dried, fixed in 2% paraformaldehyde, and rehydrated. For IHC, sections were incubated with rabbit IgG directed against CLF-1 (Sigma-Aldrich) or nonimmune rabbit IgG overnight. After incubation with a biotinylated anti-rabbit secondary antibody, color was developed using a Vectastain universal ABC kit and diaminobenzidine (Vector Laboratories). Sections were counterstained with Gill's hematoxylin (Vector Laboratories).
For immunofluorescent staining of lung, two rabbit primary antibodies were used. First, sections were stained using rabbit IgG directed against surfactant protein C (SP-C). Sections were then incubated with goat anti-rabbit Fab fragments, followed by a biotinylated anti-goat secondary antibody. CLF-1 was stained as above. To develop the color, a rhodamine-conjugated donkey anti-rabbit secondary antibody was used to stain CLF-1, and Cy5-conjugated streptavidin was used to stain SP-C. To stain for macrophages and CLF-1, a mouse anti-human CD68 antibody was used in combination with rabbit anti-CLF-1. Color was developed using Alexa Fluor 488-conjugated anti-mouse and rhodamine-conjugated anti-rabbit secondary antibodies (Life Technologies-Invitrogen). Sections were counterstained with DAPI. Images were photographed on a Nikon TE200 microscope equipped with epifluorescence. All images were processed with Adobe Photoshop C5 Extended version 12.04x32 under identical conditions, for presentation.
Staining of Rodent Lung
Conditions for staining rodent lungs are summarized in Table 1. All bright-field images were taken with an Olympus DP25 camera on an Olympus CH2 microscope.
Table 1.
Antibodies and Staining Conditions for Immunohistochemistry
| Lung tissue | Fixation method | Antigen retrieval | Target | Species of primary antibody | Developing agent (color) |
|---|---|---|---|---|---|
| Rat | FFPE | Sodium citrate, 10 mmol/L, pH 6.8 | CNTFR | Goat | AEC (red) |
| Mouse | FFPE | EDTA, 1 mmol/L, pH 8 | Phospho-Stat3 | Rabbit | Ni-DAB (black) |
| Mouse | FFPE | EDTA, 1 mmol/L, pH 8 | SP-C | Rabbit | DAB (brown) |
| Mouse | FFPE | Sodium citrate 10 mmol/L, pH 6.8 | Mouse neutrophils | Rat | DAB (brown) |
| Mouse | FFPE | Sodium citrate, 10 mmol/L, pH 6.8 | T-bet | Rat | DAB (brown) |
| Mouse | FFPE | Sodium citrate, 10 mmol/L, pH 6.8 | FoxP3 | Rat | DAB (brown) |
| Mouse | Cold acetone on cryosection | NA | CD68 | Rat | Ni-DAB (black) |
| Mouse | Cold acetone on cryosection | NA | CD3 | Rat | Ni-DAB (black) |
| Mouse | Cold acetone on cryosection | NA | CD4 | Rat | Ni-DAB (black) |
| Mouse | Cold acetone on cryosection | NA | CD8 | Rat | Ni-DAB (black) |
AEC, aminoethylcarbazole; DAB, diaminobenzidine; FFPE, formalin-fixed, paraffin-embedded; NA, not applicable; Ni, nickel.
To double-stain for p-Stat3 and SP-C, a similar strategy was used to that described above for human lung. Formalin-fixed, paraffin-embedded sections were deparaffinized, rehydrated, and exposed to EDTA antigen retrieval. The first antigen was stained using rabbit polyclonal IgG directed against p-Stat3 (Cell Signaling Technology), followed by incubation with goat anti-rabbit Fab fragments, and then a biotinylated-anti-goat secondary antibody. Color was developed using a Vectastain ABC universal kit and then a Vector SG substrate kit for a blue/gray color in the nucleus (Vector Laboratories). After avidin-biotin blocking (Vector Laboratories), sections were incubated with rabbit anti-SP-C. After incubation with a biotinylated anti-rabbit secondary antibody, color was developed with the Vectastain universal ABC kit and diaminobenzidine.
Quantification of Cells in Mouse Lung Sections
Sections of mouse lungs were stained as described in Table 1. A blinded reader counted the number of positively staining cells in every field on the section under the 40× objective, as we have described previously.36 For each experiment, an average of 113 fields were counted per animal. Data are presented as means + SEM.
Sircol Assay for Acid-Soluble Collagen
After sacrificing the animals, left lungs were excised and snap-frozen in liquid nitrogen, followed by lyophilization. Lungs were homogenized in 0.5 mol/L acetic acid with a protease inhibitor cocktail (Sigma-Aldrich) and mixed gently at 4°C overnight. The homogenate was pelleted, and the supernatant was run across a QIAshredder homogenizer column (Qiagen), as described previously.19 The Sircol assay was performed according to the manufacturer's instructions. Briefly, 100 μL of the lung-acetic acid mixture was added to 1 mL of the Sirius Red reagent and incubated for 60 minutes. The collagen-dye complex was pelleted by centrifugation in a microcentrifuge (Eppendorf, Hamburg, Germany), and the precipitated material was dissolved in 0.5 mol/L NaOH. Optical density at 540 nm (OD540) was recorded using a microplate reader.
Total RNA Isolation from Mouse Lungs
CLF-1/CLC was administered to mice for 7 days after bleomycin injury (3 mg/kg) or to no-injury control mice. The mice were euthanized, and the lungs were harvested for RNA isolation. This RNA was used for RT-PCR and microarray analysis as described below. Mouse lungs were first homogenized (PolyTron; Kinematica) and then processed with the miRNeasy mini kit (Qiagen, Frederick, MD) for total RNA extraction according to the manufacturer's instructions. Total RNA extracted was quantified with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE), and the integrity of the RNA samples was analyzed using an Agilent 2100 bioanalyzer with the accompanying RNA 6000 nano kit. Only RNAs with OD260/OD280 > 1.8 and RIN (RNA integrity number) > 8 were used for microarray experiments.
Quantitative RT-PCR on mRNA from Mouse Lungs
Total RNA was reverse-transcribed using an ABI high-capacity power reverse transcription kit (Life Technologies). Primers specific for GATA3, IL-17, RORc, and PPIA were from Qiagen. Quantitative RT-PCR was performed using SYBR Green chemistry on an ABI 7500 system (Applied Biosystems). Fold change was calculated by the 2−ΔΔCt method, and data were analyzed with ABI 7500 system software version 1.3.1 according to the manufacturer's recommendations.
Microarray Analysis of mRNA from Mouse Lungs
Five hundred nanograms of the total RNA was labeled and amplified with the Agilent Quick Amp labeling kit. The quality of this purified cRNA was assessed using NanoDrop microarray measurement. The fragmented cRNA with specific activity of >9 and a yield of >1.65 μg was hybridized to Agilent 4x44K whole mouse genome microarrays for 17 hours. The chips were washed with Agilent gene expression wash buffers 1 and 2 and scanned with an Agilent scanner.
Array images were processed using the Agilent Feature Extraction protocol version 9.5.3.37 All arrays were cyclic-LOESS normalized using the Bioconductor package (version 2.8) as described previously.38 For statistical analysis, we applied one-way analysis of variance. A false discovery rate of 1% was used as a cutoff for statistical significance in microarray data. Data visualization and clustering were performed using Genomica version 3.040710,39 GeneSpring GX version 11.5.1 and Scoregenes version 1.0,40 and Spotfire Decision Site 9 (TIBCO, Palo Alto, CA). For qRT-PCR, student's t-test was used and significance was defined at P < 0.05. Data were imported into the NIH DAVID bioinformatics database version 6.7 for functional annotation and analysis for gene enrichment.41
Statistical Analysis
Data were analyzed using SAS version 9.2 (SAS Institute, Cary, NC), as well as the R environment version 2.13.1 for statistical analysis42 with confirmation of two-way analysis of variance results using Open BUGS software version 3.2.1 for Bayesian model fitting via Gibbs sampling and the Regression Modeling Strategy package.43 Assay results were natural log-transformed where indicated by analysis of residuals for normality and subjected to one- or two-way analysis of variance. Main effects and interactions between bleomycin and CLF-1 were tested using maximum likelihood.
Results
Gene Expression Profiling Shows Significantly Increased Expression of CLF-1 in IPF Lungs Compared with Normal Controls
To gain insight into the pathogenesis of IPF, we previously performed gene expression profiling of IPF lungs compared with normal controls.33 Considering the top 80 up-regulated genes, we noted that CLF-1 (the CRLF1 protein product) was the only IL-6 family member that was up-regulated in IPF samples. We validated significantly increased expression of CLF-1 in IPF in a separate cohort of patients. CLF-1 mRNA expression was up-regulated 6.5-fold in IPF patients (n = 50), compared with normal control subjects (n = 49) (P < 0.0001; Figure 1A). We also determined whether CLF-1 expression was increased in the lung after bleomycin injury (Figure 1B). Data were natural log-transformed and subjected to one-way analysis of variance. CLF-1 expression increased after administration of bleomycin. Statistically significant increases in CLF-1 expression were detected from day 7 through day 28, compared with the uninjured control (P < 0.05). Thus, we found significantly increased expression of CLF-1 both in humans and in experimental pulmonary fibrosis.
Figure 1.
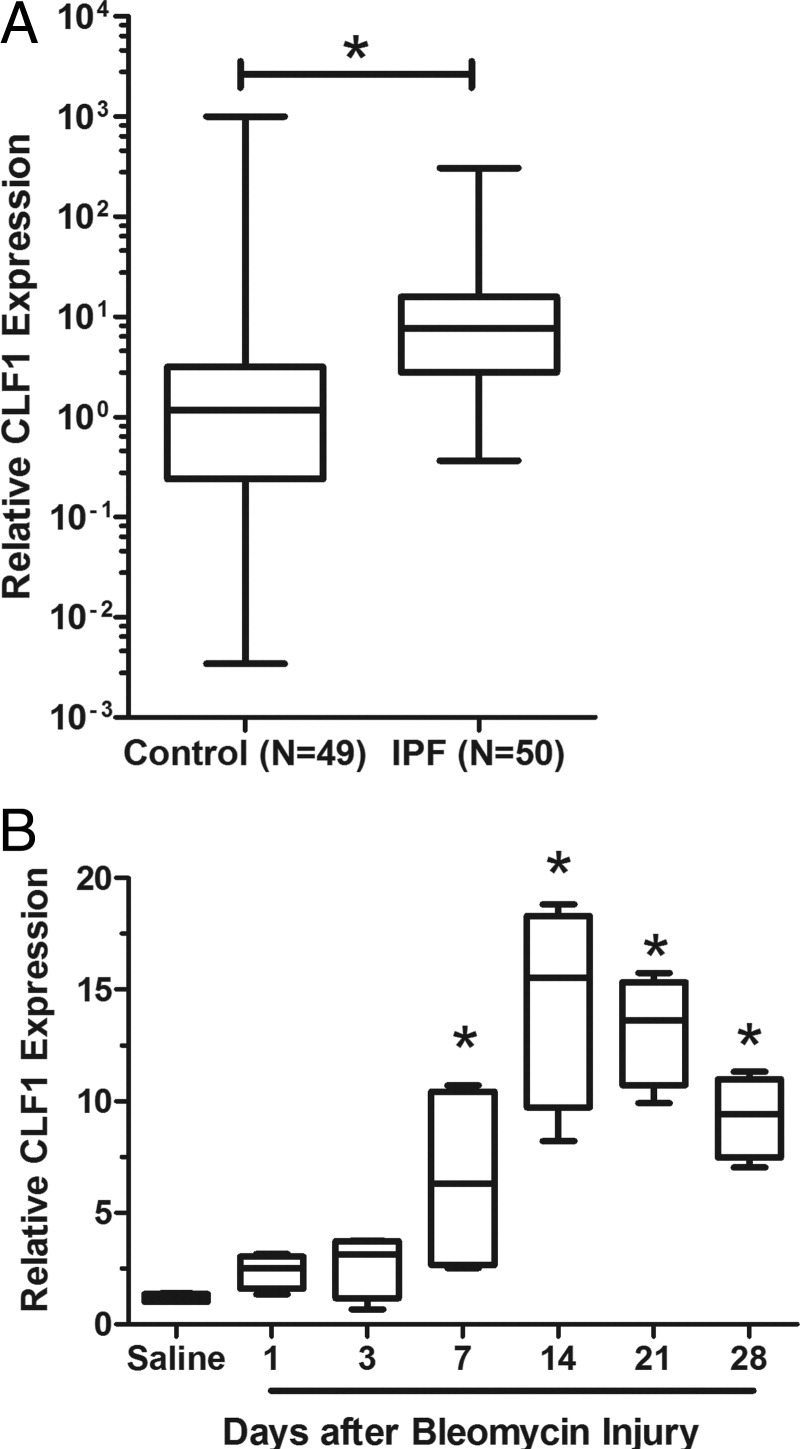
CLF-1 expression is increased in IPF and in bleomycin injury. A: Quantitative RT-PCR of CLF-1 mRNA normalized to peptidyl prolyl isomerase A (PPIA) mRNA on samples obtained from lungs of patients with IPF (n = 50), compared with normal control subjects (n = 49). *P < 0.0001, Wilcoxon rank sum test. B: Quantitative RT-PCR was performed on lungs from mice at various time points after bleomycin injury (3 mg/kg). *P < 0.05 versus saline control, one-way analysis of variance. n = 4 per group. Box plots indicate median, quartiles, and range.
Strong Immunoreactivity for CLF-1 Is Observed in Type II Alveolar Epithelial Cells and Macrophages in IPF Lungs
To determine which cell types express CLF-1 in IPF lungs, we performed IHC for CLF-1 (Figure 2, A–C). Little staining was observed in sections of uninjured lungs from patients. On cryosections of IPF lungs, we observed strong staining for CLF-1 in cells that morphologically appear to be type II alveolar epithelial cells and macrophages (Figure 2, A and B). Staining for CLF-1 appeared to be localized to the alveolar parenchyma. No staining of airway epithelial cells in IPF was noted (Figure 2C). No staining was seen using nonimmune rabbit IgG. To confirm the identity of the positively staining cells in IPF lungs, we performed double-label immunofluorescence for CLF-1 with the type II alveolar epithelial cell marker surfactant protein C (SP-C) (Figure 2, D–G) and the macrophage marker CD68 (Figure 2, H–K). Merged images show colocalization of CLF-1 in SP-C+ and CD68+ cells. These results indicate that CLF-1 expression in the lung was localized to type II alveolar epithelial cells and macrophages.
Figure 2.

CLF-1 expression is limited to type II alveolar epithelial cells and macrophages in IPF lungs. IHC for CLF-1 was performed on cryosections of normal (A) or IPF (B and C) human lungs. Immunostaining was localized to cells that morphologically appear to be type II alveolar epithelial cells (black arrows) and alveolar macrophages (red arrows). C: Staining is absent in airway epithelial cells. Inset: Staining with nonimmune rabbit IgG control. D–K: Immunofluorescence was performed for CLF-1 (D and H, green), the type II alveolar epithelial cell marker SP-C (E, red), and the macrophage marker CD68 (I, red). Nuclei were stained with DAPI (F and J, blue). G and K: Merged samples show colocalization of CLF-1 with SP-C (G) and with CD68 (K). Insets (D–K): Nonimmune controls. Original magnification, ×400 (A–C and inset); ×200 (D–K and insets).
Expression of the Specific CLF-1 Receptor CNTFR Is Localized to Type II Alveolar Epithelial Cells
Having identified the source of CLF-1 expression, we next turned our attention to identifying the cells in the lung that respond to CLF-1 signaling. CLF-1 exists naturally as a secreted heterodimer complexed with cardiotrophin-like cytokine (CLC).34 Like other IL-6 family members, CLF-1/CLC signals through gp130 and leukemia inhibitory factor receptor β (LIFRβ). By IHC, we found that LIFRβ was strongly expressed by multiple cell types in IPF lungs (data not shown). Unlike other IL-6 family members, CLF-1/CLC also requires the participation of a third member of this complex, ciliary neurotrophic factor receptor (CNTFR), to transduce downstream signaling events.44 We then performed IHC for CNTFR using goat IgG specific for rat CNTFR on lung sections from uninjured rats (Figure 3A). We found immunostaining for CNTFR in cells that appear morphologically to be type II alveolar epithelial cells. No staining was detected with nonimmune goat IgG. To determine whether cells in rodent lungs are capable of mediating transmembrane signaling of CLF-1/CLC, we instilled rhCLF-1/CLC, LIF, or vehicle control into the lungs of mice and performed IHC for p-Stat3 (Figure 3, B–D). No immunostaining for p-Stat3 was detected in vehicle-exposed animals. In animals exposed to CLF-1/CLC, immunostaining for p-Stat3 appeared to be localized to SP-C+ cells only; 8.5-fold more p-Stat3+ cells were quantified in CLF-1/CLC-exposed lungs compared with vehicle controls (Figure 3E). The lack of staining for p-Stat3 in other cell types was unlikely to be due to insufficient access of the cytokine to other cell types, because in animals similarly exposed to another cytokine capable of activating the Stat3 pathway, LIF, immunostaining for p-Stat3 was present in many cell types that were either SP-C+ or SP-C−.
Figure 3.

Expression of CNTFR and responsiveness to CLF-1 stimulation is localized to type II alveolar epithelial cells. A: IHC was performed for CNTFR on uninjured rat lung. Immunostaining appears to be localized to type II alveolar epithelial cells. Inset: Staining is absent with nonimmune goat IgG. Animals underwent transtracheal injection of vehicle control (B), LIF (C), or CLF-1/CLC (D). Animals were ventilated for 15 minutes, and the lungs were fixed and processed for IHC for p-Stat3 (black) and SP-C (brown). In vehicle-treated animals (B), no p-Stat3 staining was present. In LIF-treated animals (C), both p-Stat3+/SPC+ (black arrows) and p-Stat3+/SPC− (red arrows) staining was observed. After CLF-1 stimulation (D), p-Stat3+ cells were limited to SP-C+ cells. E: Quantification of p-Stat3+ cells in vehicle-treated and CLF-1-treated animals. *P = 0.0001. n = 4 per group. F: Immunoblotting for p-Stat3 in primary rat type II alveolar epithelial cells up to 60 minutes after CLF-1 stimulation. G: Densitometric analysis of p-Stat3 band intensity normalized to total Stat3, representative of four independent experiments. *P < 0.05 versus unstimulated control, one-way analysis of variance. Original magnification, ×400 (A–D and Inset).
To confirm that type II alveolar epithelial cells are capable of responding to CLF-1 in a cell-autonomous fashion, we incubated primary rat type II alveolar epithelial cells in culture with CLF-1/CLC or vehicle control for 0, 15, 30, or 60 minutes (Figure 3, F and G). By immunoblotting, we detected p-Stat3 in primary rat type II alveolar epithelial cell lysates from CLF-1/CLC stimulated cultures, compared with normal controls. Taken together, these data show that expression of CNTFR, a specific receptor for CLF-1/CLC, is localized to type II alveolar epithelial cells. We also found that type II alveolar epithelial cells are preferentially stimulated by exposure to CLF-1/CLC as measured by phosphorylation of Stat3.
Exposure of Animals to Chronic CLF-1/CLC Administration
Because CLF-1 expression was significantly increased in IPF lungs, we sought to determine whether exposure of animals to CLF-1 at high local concentrations would cause both lung injury and fibrosis or potentiate injury induced by a fibrotic stimulus such as bleomycin. To answer this question, we exposed animals to daily intranasal administration of CLF-1/CLC or vehicle control, with and without bleomycin (Figure 4, A–D). In animals treated with vehicle only, there was a mild inflammatory infiltrate without any architectural distortion. In the CLF-1/CLC-treated animals, there was no architectural distortion, but isolated lymphoid aggregates were observed. In animals treated with CLF-1/CLC or vehicle control daily for 7 days after bleomycin, there was significant inflammation and architectural distortion (Figure 4, C and D). In the CLF-1/CLC-exposed animals injured with bleomycin, there was greatly amplified inflammation and tissue injury. In a second cohort of animals, we continued treatment out to 14 days after bleomycin injury. In the animals exposed to CLF-1/CLC and vehicle alone, all animals survived to 14 days. In the animals injured with bleomycin and treated with vehicle, only one animal died (on day 13). In contrast, in the bleomycin-injured, CLF-1/CLC-treated animals, there was significantly increased mortality beginning on day 7, with most animals having met predefined criteria for euthanasia (data not shown). These data indicate that CLF-1/CLC promotes inflammation, tissue injury, and increased mortality in animals injured with bleomycin at 3 mg/kg.
Figure 4.

Lung histology from animals exposed to CLF-1/CLC. Animals (n = 5 per group) were administered CLF-1/CLC or a vehicle control, 7 days after bleomycin injury (3 mg/kg) or no-injury control. Animals were sacrificed, and the lungs were removed and processed for routine histological staining with H&E. A: Vehicle. B: CLF-1/CLC. C: Bleomycin + vehicle. D: Bleomycin + CLF-1/CLC. Original magnification, ×100.
Quantitative Analysis of Lung Histopathology of Animals Exposed to rhCLF-1/CLC
Because of the marked effects of CLF-1/CLC exposure on lung histology, we performed a quantitative analysis of cell lineages in the lungs from animals exposed to CLF-1/CLC treatment at 7 days after bleomycin injury or uninjured controls (Figure 5, A–E). We performed IHC for markers of type II alveolar epithelial cells, neutrophils, T cells, B cells, and macrophages. Cells were quantified, and the data were natural log-transformed and subjected to two-way analysis of variance as described under Materials and Methods. A significant increase in the number of SP-C+ (Figure 5A) cells was detected after administration of bleomycin (P < 0.0001). An interaction between CLF-1/CLC and bleomycin was present (P < 0.05), but no significant effect of CLF-1/CLC on the number of SP-C+ cells was detected. Significant decreases in the number of neutrophils (Figure 5B) were detected after administration of bleomycin (P < 0.0001). An interaction between CLF-1/CLC and bleomycin was present (P < 0.05), but no significant effect of CLF-1/CLC was detected on the number of neutrophils. Significant increases in the number of CD3+ cells (Figure 5C) were detected in the presence of CLF-1/CLC (P = 0.0005). No interaction between bleomycin and CLF-1/CLC was detected on the number of CD3+ cells. At 7 days after bleomycin administration, the number of B220+ cells (Figure 5D) detected was significantly increased (P = 0.02). No interaction between CLF-1/CLC and bleomycin was detected. At 7 days after bleomycin and CLF-1/CLC administration, a significant increase in the number of CD68+ cells was detected (Figure 5E). No interaction between CLF-1/CLC and bleomycin was detected. When the model was reanalyzed without interaction, the principal effect on the number of CD68+ cells was the result of CLF-1/CLC (P < 0.05), with a borderline contribution from bleomycin (P < 0.08). Taken together, these data show that administration of CLF-1/CLC, both in the presence and in the absence of bleomycin, leads to the accumulation of CD3+ cells in the lung.
Figure 5.

CLF-1/CLC promotes the accumulation of CD3+ and CD68+ cells in mouse lungs. IHC was performed on lungs from animals that were administered CLF-1/CLC or a vehicle control (n = 5 per group) at 7 days after bleomycin injury (3 mg/kg) or no-injury control, using cell-specific markers SP-C for type II alveolar epithelial cells (A), neutrophils (B), CD3 for T cells (C), B220 for B cells (D), and CD68 for macrophages (E). Cell numbers were quantified per high-power field (hpf). *P < 0.05, main effects of CLF-1/CLC; †P < 0.05, main effects of bleomycin. Data were analyzed by two-way analysis of variance (the statistical analysis is further described under Results).
CLF-1/CLC Treatment Increases the Number of CD4+ T Cells in Bleomycin-Injured Lungs
To determine the phenotype of the T cells that accumulated in response to instillation of CLF-1/CLC, we stained lung sections from animals treated with CLF-1/CLC or vehicle control, 7 days after bleomycin injury. We performed IHC for CD3 (Figure 6, A and B) and also for CD4 and CD8, with quantification of the number of positively staining cells. Significantly increased numbers of CD4+ cells (Figure 6C) were detected after administration of bleomycin and CLF-1/CLC. For CD4+ cells, an interaction was detected (P < 0.001), with main effects by bleomycin (P < 0.0001) and CLF-1/CLC (P = 0.05). No significant differences were noted in the number of CD8+ cells between CLF-1/CLC-treated mice and vehicle controls (Figure 6D). After bleomycin injury, CLF-1/CLC treatment nearly tripled the number of both Tbet+ (P < 0.04) and Foxp3+ cells (P < 0.008) in the lung (Figure 6, E and F). For Tbet+ cells, no interaction between bleomycin and CLF-1/CLC was detected using two-way analysis of variance. Performing the two-way analysis of variance without an interaction term showed a significant effect of CLF-1/CLC (P = 0.01). For Foxp3+ cells, no interaction between bleomycin and CLF-1/CLC was detected using two-way analysis of variance. Performing the two-way analysis of variance without an interaction term identified significant effects of both CLF-1/CLC (P = 0.04) and bleomycin (P < 0.001).
Figure 6.

CD4+, Tbet+, and Foxp3+ cells accumulate in the lungs of bleomycin-injured animals treated with CLF-1/CLC. IHC was performed for CD3 in lungs from bleomycin-injured animals treated with vehicle control (A) or CLF-1/CLC (B). IHC was performed for CD4 (n = 5 to 7 per group) (C), CD8 (n = 3 or 4 per group) (D), the Th1 marker Tbet (E), and the Treg marker Foxp3 (F) (n = 3 per group). Quantitative RT-PCR was performed on lung samples for the Th2 marker GATA3 (G), the Th17 marker IL-17 (H), and the Tfh marker RORc (I). Significant differences in gene expression for GATA3 and RORc were detected after bleomycin injury (†P < 0.05), but CLF-1/CLC treatment did not significantly augment expression for GATA3, IL-17, or RORc. *P < 0.05, main effects of CLF-1; †P < 0.05, main effects of bleomycin. Original magnification, ×400 (A and B).
We next performed quantitative RT-PCR on RNA isolated from the lungs of animals exposed to CLF-1/CLC with and without bleomycin to determine the relative expression of markers of other important T-helper cell lineages. A significant effect on gene expression for GATA3 (Figure 6G) was detected after administration of bleomycin (P < 0.01), but no interaction was detected between CLF-1/CLC treatment and bleomycin. No significant effect on gene expression for IL-17 (Figure 6H) was detected after administration of bleomycin both in the presence and in the absence of CLF-1/CLC. A significant effect on gene expression for the Tfh marker RORc (Figure 6I) was detected after administration of bleomycin (P = 0.001), but no interaction was detected between CLF-1/CLC and bleomycin. Therefore, in the context of bleomycin-induced lung injury, CLF-1/CLC induced the accumulation of Th1 and Treg cells.
After Low-Dose Bleomycin, CLF-1/CLC Treatment Decreases Collagen Deposition
The IL-6 family has been implicated in the pathogenesis of IPF as a factor promoting the growth and survival of fibroblasts16–18 and lung injury and fibrosis.19,20 Because we observed that CLF-1/CLC induced increased inflammation in mouse lungs, we investigated whether this is accompanied by a change in the accumulation of collagen, either alone or in the context of bleomycin-induced lung injury and fibrosis. Administration of CLF-1/CLC in animals receiving 3 mg/kg bleomycin produced excessive mortality for quantitative analysis of fibrosis at 14 days after injury. For these experiments, therefore, we used a lower dose of bleomycin (1 mg/kg). Compared with bleomycin-injured, vehicle-treated animals (Figure 7A), treatment with CLF-1/CLC again produced a more highly cellular infiltrate with a greater degree of architectural distortion compared with the vehicle controls (Figure 7B). As a qualitative analysis of collagen deposition, we performed Masson's trichrome staining. Overall, there appeared to be less blue staining in the bleomycin-injured and CLF-1/CLC-treated mice compared with the vehicle controls (Figure 7, C and D). Uninjured animals that received 14 doses of CLF-1/CLC exhibited a similar degree of detected collagen. We quantified the effect of CLF-1/CLC on collagen content in the presence and absence of bleomycin. Bleomycin-injured animals treated with vehicle exhibited a twofold increase in acid-soluble collagen content, compared with uninjured, vehicle-treated controls. In the comparison with vehicle-treated controls, CLF-1/CLC treatment was associated with decreases in collagen content for both injured and uninjured vehicle-treated controls. Data were analyzed by two-way analysis of variance. Significant differences in mean collagen levels were detected (P < 0.0002), but no interaction between CLF-1/CLC and bleomycin was detected (Figure 7E). When the model was reanalyzed without interaction, significant main effects of bleomycin and CLF-1 were detected (P < 0.05). We conclude that CLF-1 treatment decreases collagen content in the lung.
Figure 7.

CLF-1/CLC treatment of bleomycin-injured animals increases inflammation but decreases fibrosis. Animals were pretreated with CLF-1/CLC for 2 days and then were administered bleomycin at 1 mg/kg (low dose). Treatment continued daily for an additional 12 days. Animals were sacrificed, and the lungs were removed and processed for H&E staining (A and C), Masson's trichrome staining (B and D), and determination of collagen content (E). A and B: Vehicle-treated, bleomycin-injured. C and D: CLF-1/CLC-treated, bleomycin-injured. E: At 12 days after injury with low-dose bleomycin or saline controls, left lungs were processed for detection of collagen by Sircol assay. Low-dose bleomycin injury led to significantly increased collagen content, compared with uninjured controls (†P < 0.05), and CLF-1/CLC led to significant reductions in collagen content at baseline and after bleomycin injury (*P = 0.05). Vehicle, n = 6; CLF-1/CLC, n = 5; bleomycin + vehicle, n = 7; bleomycin + CLF-1/CLC, n = 12. Original magnification, ×100.
Gene Expression Profiling of Lungs from Animals Treated with CLF-1/CLC
Because we observed an increase in the number of T cells in the lungs of CLF-1/CLC-treated mice, we hypothesized that these lungs from animals exposed to CLF-1/CLC would exhibit a gene expression profile enriched in inflammatory pathways. To gain further insight into the molecular mechanisms underlying the proinflammatory and antifibrotic phenotype of CLF-1/CLC-treated animals, we performed gene expression profiling of lungs from animals treated with CLF-1/CLC or vehicle control, with and without bleomycin injury. Across the four experimental groups, there were 3875 differentially expressed genes. The top 35 genes induced by administration of CLF-1/CLC are presented in Tables 2 and 3. Of the differentially expressed genes, clustering by K means organized the data set into six distinct clusters (Figure 8). The clustering indicates that the gene expression changes were dominated by bleomycin injury. The genes clustered in cluster 5 (Figure 8), however, showed a stepwise increase in average gene expression after CLF-1/CLC stimulation. All of the clusters were imported into NIH DAVID for functional annotation (Table 4).41 In cluster 5, the most significantly enriched pathway of genes belonged to the immune response (see Supplemental Tables S1–S3 at http://ajp.amjpathol.org.) Cluster 6 was characterized by genes exhibiting decreased expression in response to CLF-1/CLC after bleomycin injury. In this cluster, one of the most significantly enriched pathways involves extracellular matrix proteins (see Supplemental Tables S4 and S5 at http://ajp.amjpathol.org.) These data show that, after administration of CLF-1, there was increased expression of inflammatory genes and decreased expression of several extracellular matrix genes in murine lungs.
Table 2.
Top 35 Probes Induced by CLF-1 versus Vehicle
| Symbol | Gene name | Fold change |
|---|---|---|
| CXCL9 | Chemokine (C-X-C motif) ligand 9 | 27.8 |
| UBD | Ubiquitin D | 22.2 |
| CLCA3P | Chloride channel accessory 3, pseudogene | 16.6 |
| CXCL10 | Chemokine (C-X-C motif) ligand 10 | 12.2 |
| GBP1 | Guanylate binding protein 1, interferon-inducible, 67kDa⁎ | 10.6 |
| LOC667597 | cDNA sequence BC023105 | 9.5 |
| LOC667597 | cDNA sequence BC023105 | 7.5 |
| CCL2 | Chemokine (C-C motif) ligand 2 | 5.6 |
| CXCL11 | Chemokine (C-X-C motif) ligand 11 | 5.5 |
| APOL6 | Apolipoprotein L, 6 | 5.4 |
| CCL7 | Chemokine (C-C motif) ligand 7 | 5.0 |
| MAPK10 | Mitogen-activated protein kinase 10 | 4.8 |
| MYBPC3 | Myosin binding protein C, cardiac | 4.7 |
| IFNG | Interferon, gamma | 4.6 |
| GJA3 | Gap junction protein, alpha 3, 46 kd | 4.6 |
| IFNG | Interferon, gamma | 4.6 |
| ADAMTS19 | ADAM metallopeptidase with thrombospondin type 1 motif, 19 | 4.6 |
| BC061237 | cDNA sequence BC061237 | 4.5 |
| ALPK2 | Alpha-kinase 2 | 4.5 |
| PRAMEF3 | PRAME family member 3 | 4.4 |
| CCL7 | Chemokine (C-C motif) ligand 7 | 4.2 |
| PLN | Phospholamban | 4.2 |
| BC006965 | cDNA sequence BC006965 | 4.0 |
| KCNJ3 | Potassium inwardly rectifying channel, subfamily J, member 3 | 3.9 |
| IGFBPL1 | Insulin-like growth factor binding protein-like 1 | 3.9 |
| CYP1A1 | Cytochrome P450, family 1, subfamily A, polypeptide 1 | 3.9 |
| TTN | Titin | 3.9 |
| BC061237 | cDNA sequence BC061237 | 3.8 |
| FGF12 | Fibroblast growth factor 12 | 3.7 |
| IL12B | Interleukin 12B (natural killer cell stimulatory factor 2, cytotoxic lymphocyte maturation factor 2, p40) | 3.7 |
| FGF12 | Fibroblast growth factor 12 | 3.7 |
| CORIN | Corin, serine peptidase | 3.7 |
| HAMP | Hepcidin antimicrobial peptide | 3.6 |
Includes EG:2633.
Table 3.
Top 35 Probes Induced by Bleomycin + CLF-1 versus Bleomycin + Vehicle
| Symbol | Gene name | Fold change |
|---|---|---|
| CLCA3P | Chloride channel accessory 3, pseudogene | 14.4 |
| CXCL9 | Chemokine (C-X-C motif) ligand 9 | 11.9 |
| CXCL11 | Chemokine (C-X-C motif) ligand 11 | 9.5 |
| IRG1 | Immunoresponsive 1 homolog (mouse) | 7.7 |
| IRG1 | Immunoresponsive 1 homolog (mouse) | 7.6 |
| IFNG | Interferon, gamma | 6.3 |
| IFNG | Interferon, gamma | 5.8 |
| GZMB | Granzyme B (granzyme 2, cytotoxic T-lymphocyte-associated serine esterase 1) | 5.8 |
| UBD | Ubiquitin D | 5.6 |
| GBP4 | Guanylate binding protein 4⁎ | 5.1 |
| CCL1 | Chemokine (C-C motif) ligand 1 | 4.9 |
| VAX2 | Ventral anterior homeobox 2 | 4.8 |
| PDCD1 | Programmed cell death 1 | 4.7 |
| ADAMTS20 | ADAM metallopeptidase with thrombospondin type 1 motif, 20 | 4.2 |
| IL21 | Interleukin 21 | 4.1 |
| IDO1 | Indoleamine 2,3-dioxygenase 1 | 4.1 |
| GBP2 | Guanylate binding protein 2, interferon-inducible | 3.9 |
| CXCL10 | Chemokine (C-X-C motif) ligand 10 | 3.9 |
| CD274 | CD274 molecule | 3.9 |
| IL18BP | Interleukin 18 binding protein | 3.8 |
| TNFSF11 | Tumor necrosis factor (ligand) superfamily, member 11 | 3.8 |
| ARG1 | Arginase, liver | 3.8 |
| CCR8 | Chemokine (C-C motif) receptor 8 | 3.8 |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 | 3.7 |
| DCAF12L2 | DDB1 and CUL4-associated factor 12-like 2 | 3.6 |
| ITLN1 | Intelectin 1 (galactofuranose binding) | 3.5 |
| NUB1 | Negative regulator of ubiquitin-like proteins 1 | 3.5 |
| GBP5 | Guanylate binding protein 5 | 3.4 |
| NKG7 | Natural killer cell group 7 sequence | 3.4 |
| NKG7 | Natural killer cell group 7 sequence | 3.3 |
| IL18BP | Interleukin 18 binding protein | 3.2 |
| IL12RB1 | Interleukin 12 receptor, beta 1 | 3.2 |
| CD4 | CD4 molecule | 3.2 |
| FAM26F | Family with sequence similarity 26, member F | 3.2 |
| CNGB3 | Cyclic nucleotide-gated channel beta 3 | 3.2 |
Includes EG:115361.
Figure 8.

K-means clustering identifies clusters with changes in gene expression induced by CLF-1/CLC administration. CLF-1/CLC or vehicle was administered to mice with and without bleomycin. Gene expression profiling was performed on mouse lungs. K-means clustering was performed using six clusters. Vertical lines represent individual animals in each experimental grouping. Each colored line represents an individual gene. Functional mapping of these gene clusters is presented in Table 4. In clusters 1 to 4, changes in gene expression appear to be dominated by bleomycin injury. In cluster 5, changes in gene expression indicate a stepwise increase in expression after CLF-1/CLC administration, compared with control. The genes in cluster 5 map most significantly (P < 3 × 10−12) to genes coding for inflammatory proteins (Table 4). In cluster 6, genes with increased expression after bleomycin and the vehicle exhibited decreased expression with CLF-1/CLC. The genes in cluster 6 map most significantly to genes coding for extracellular matrix proteins (P < 0.00005).
Table 4.
Functional Mapping of the Clusters
| Term | Count | P value | Fold enrichment |
|---|---|---|---|
| Cluster 1 | |||
| Nucleotide binding | 201 | 1.76 × 10−28 | 2.2 |
| ATP binding | 161 | 3.08 × 10−23 | 2.3 |
| RNA binding | 73 | 1.55 × 10−14 | 2.7 |
| DNA replication | 28 | 4.30 × 10−14 | 5.9 |
| Protein transport | 68 | 3.66 × 10−13 | 2.7 |
| Cell cycle | 60 | 4.89 × 10−10 | 2.4 |
| Kinase | 80 | 1.88 × 10−9 | 2.0 |
| Chaperone | 30 | 3.13 × 10−9 | 3.6 |
| Cytoskeleton | 67 | 2.57 × 10−8 | 2.1 |
| DNA damage | 30 | 1.04 × 10−6 | 2.8 |
| Cluster 2 | |||
| Cell adhesion | 20 | 1.14 × 10−8 | 5.2 |
| Transmembrane protein | 19 | 4.30 × 10−8 | 5.0 |
| Lectin | 12 | 1.10 × 10−6 | 7.0 |
| Immunoglobulin domain | 18 | 2.81 × 10−6 | 4.0 |
| Actin binding | 13 | 3.43 × 10−6 | 5.6 |
| Calmodulin binding | 9 | 1.00 × 10−5 | 8.6 |
| Muscle protein | 7 | 1.10 × 10−5 | 13.7 |
| Receptor | 46 | 4.75 × 10−5 | 1.8 |
| Signal anchor | 14 | 2.58 × 10−4 | 3.4 |
| Lipoprotein | 17 | 3.37 × 10−4 | 2.8 |
| Cluster 3 | |||
| Transcription | 131 | 4.78 × 10−11 | 1.8 |
| Mitochondrion | 74 | 1.04 × 10−10 | 2.3 |
| Differentiation | 52 | 1.62 × 10−10 | 2.7 |
| Transmembrane | 297 | 1.80 × 10−10 | 1.4 |
| Oxidoreductase | 58 | 8.15 × 10−10 | 2.4 |
| Activator | 52 | 9.86 × 10−10 | 2.6 |
| Alternative splicing | 254 | 1.21 × 10−8 | 1.4 |
| Cell membrane | 117 | 6.86 × 10−8 | 1.6 |
| DNA binding | 96 | 1.32 × 10−6 | 1.6 |
| Endoplasmic reticulum | 56 | 1.60 × 10−6 | 2.0 |
| Cluster 4 | |||
| Mitosis | 34 | 1.15 × 10−35 | 23.1 |
| Cell division | 37 | 1.70 × 10−34 | 17.6 |
| Cell membrane | 60 | 2.82 × 10−23 | 4.3 |
| Kinetochore | 15 | 5.01 × 10−17 | 29.9 |
| Extracellular matrix | 20 | 1.08 × 10−14 | 11.4 |
| Glycoprotein | 69 | 5.44 × 10−13 | 2.3 |
| Centromere | 11 | 6.73 × 10−13 | 33.4 |
| Cleavage on pair of basic residues | 19 | 9.73 × 10−13 | 9.7 |
| Signal | 61 | 1.83 × 10−12 | 2.5 |
| Secreted | 39 | 3.68 × 10−11 | 3.3 |
| Cluster 5 | |||
| Cytokine | 16 | 4.89 × 10−13 | 13.9 |
| Immune response | 16 | 8.63 × 10−13 | 13.4 |
| Secreted | 34 | 4.21 × 10−11 | 3.7 |
| Cell membrane | 37 | 7.25 × 10−11 | 3.3 |
| Transmembrane | 65 | 3.21 × 10−9 | 1.9 |
| Inflammatory response | 9 | 3.73 × 10−8 | 17.8 |
| Chemotaxis | 8 | 1.40 × 10−7 | 19.9 |
| Lipoprotein | 18 | 2.24 × 10−7 | 4.7 |
| T cell | 6 | 1.51 × 10−6 | 29.8 |
| Immunoglobulin domain | 14 | 5.57 × 10−6 | 4.9 |
| Cluster 6 | |||
| Extracellular matrix | 8 | 4.57 × 10−9 | 29.2 |
| Acute phase | 4 | 3.23 × 10−6 | 129.4 |
| Cell adhesion | 7 | 5.00 × 10−6 | 14.3 |
| Glycoprotein | 15 | 1.10 × 10−5 | 3.2 |
| Disulfide bond | 12 | 5.59 × 10−5 | 3.8 |
| Amyloid | 3 | 9.50 × 10−5 | 194.1 |
| HDL | 3 | 1.95 × 10−4 | 137.0 |
| Sulfation | 3 | 7.94 × 10−4 | 68.5 |
| Cytokine | 4 | 0.001 | 17.5 |
| Chemotaxis | 3 | 0.002 | 37.6 |
Clusters are depicted in Figure 8.
Discussion
In the present study, our focus was on understanding the biology of CLF-1 in pulmonary fibrosis. We found that CLF-1 expression was significantly increased in IPF lungs, compared with normal controls. We also found that CLF-1 expression in the lung was restricted to type II alveolar epithelial cells and macrophages. CNTFR, the receptor for CLF-1/CLC, was localized mainly to type II alveolar epithelial cells and to a lesser extent in airway epithelial cells. We suggest that this pattern of CNTFR expression mediates the responsiveness of type II alveolar epithelial cells to CLF-1/CLC stimulation, which we found led to phosphorylation of Stat3 in vivo. Administration of CLF-1/CLC to mice induced a dramatic increase in the number of T cells; in the context of bleomycin injury, CLF-1/CLC decreased pulmonary fibrosis.
Like other members of the IL-6 family, CLF-1/CLC signals via the common gp130 pathway and can lead to phosphorylation and activation of Stat3, ERK, and Akt.44,45 Of its known functions, CLF-1/CLC signaling most notably promotes the growth and survival of neurons46 and the differentiation of rat metanephric mesenchyme into mature nephron structures.47 CLF-1 functions as a member of the IL-6 family. Although we did not find any difference in IL-6 expression between IPF and controls (data not shown), several important studies have implicated IL-6 or other family members in the pathogenesis of IPF.16–20 These findings raise the question as to why some IL-6 family members (eg, IL-6 and oncostatin-M) tend to be profibrotic, whereas CLF-1 appears to be antifibrotic, in the context of bleomycin-induced lung injury. Indeed, by stimulating fibroblasts directly, IL-6 is capable of inducing fibroblast proliferation, resistance to apoptosis, and enhanced production of extracellular matrix.41–43,48 We suggest that restricted responsiveness of CLF-1/CLC for type II alveolar epithelial cells to IL-6 and oncostatin-M (in contrast to the more widespread responsiveness of other cell types, including fibroblasts) is a likely explanation for these differences.
CLF-1 expression in the lung has been reported,49 but the role of CLF-1 has been studied mostly in the context of the nervous system. Humans with homozygous mutations in the CLF-1 gene (CRLF1) exhibit Sohar-Crisponi syndrome,50 a rare syndrome characterized by dysmorphic features, muscle contractions, scoliosis, and cold-induced sweating. Many die in infancy with respiratory abnormalities, but the lung disease has not been clearly defined.51 Crlf1−/− and Cntfr−/− mice die on postnatal day 1 because of a suckling defect, but no pulmonary phenotype has been described.52,53
We have shown that delivery of CLF-1/CLC to uninjured lungs leads to the accumulation of CD4+ T cells that organize into aggregates, both in the presence and in the absence of bleomycin injury. These T cells are characterized best as Th1 (expressing CD4 and Tbet) and Treg (expressing CD4 and Foxp3) cells. Although we observed trends toward increased expression of IL-17 in the CLF-1-treated animals, we did not observe any differences in the number of neutrophils in the lung. What could account for the accumulation of the predominantly Th1 and Treg phenotype of lymphocytes observed in the lungs of CLF-1-treated mice? Gene expression results indicated that CLF-1/CLC (without bleomycin) induced a fourfold increase in IL-12B and a fivefold increase in IFN-γ in mouse lungs. Among the top 200 up-regulated genes were 10 chemokine genes with increased expression (CCL1, CCL2, CCL3, CCL7, CCL12, CCL19, CXCL9, CXCL10, CXCL11, and XCL1), all of which have been implicated in the chemotaxis of T lymphocytes.54–62 CCL19 expression is particularly intriguing, because it functions in homing of T cells to secondary lymphoid organs,63 which may account, in part, for our observation that T cells organized into discrete aggregates in the lungs of CLF-1-treated mice. Whether CLF-1/CLC stimulates expression and secretion of CCL19 directly in type II alveolar epithelial cells is unknown, but CCL19 expression has been shown previously in type II alveolar epithelial cells.64
We found that CLF-1/CLC-treated, bleomycin-injured animals exhibited significant increases in both inflammation and mortality.65 To obtain an adequate number of animals for quantitative analysis of collagen deposition, we used a lower dose of bleomycin. At this lower dose, we observed significantly increased inflammation in the CLF-1/CLC-treated mice but less fibrosis. How is this apparently antifibrotic effect of CLF-1 explained? Enhanced inflammation in the mouse lung is often associated with increased fibrosis,20,65–69 although in certain contexts increased inflammation may be associated with decreased fibrosis.48,70,71
We hypothesize that the increased representation of CD4+Tbet+ T cells in the CLF-1/CLC-treated mice enriches Th1 tone in the lung and leads to an antifibrotic microenvironment in the bleomycin-injured lung.72 Supporting this hypothesis is the data set from our microarray analysis showing increased expression of the antifibrotic cytokine interferon IFN-γ in bleomycin-injured, CLF-1/CLC-treated mice, compared with the vehicle control. In addition, there was a twofold increase in Stat1 expression, which is necessary for IFN-γ signaling.73 We did not detect any differences in gene expression of the Th2 transcription factor GATA3 or the Th2 cytokines IL-4 and IL-13.
What is the contribution of the Foxp3+ T cells in the CLF-1-treated animals? In IPF, there are reduced numbers of CD4+Foxp3+ T cells in the peripheral blood and in the bronchoalveolar lavage of IPF patients, compared with normal controls.74,75 Furthermore, suppression of T-cell proliferation and activation were impaired using Treg cells derived from IPF patients, compared with cells isolated from healthy volunteers.74 In experimental pulmonary fibrosis, Ccr7−/− mice were protected from bleomycin-induced pulmonary fibrosis.76 This protection was associated with the formation of lymphoid aggregates in the lung and increased numbers of CD4+Foxp3+CD25+ cells in the lung and increased expression of indoleamine 2,3-dioxygenase (Ido1), an enzyme that is up-regulated in dendritic cells by inflammatory stimuli, including IFN-γ.77 Ido1 has been found to induce Treg differentiation and proliferation.78 We found that Ido1 expression was increased fourfold in CLF-1 and bleomycin-exposed mice. Trujillo et al76 suggest that the presence of Treg cells is protective against fibrosis. Taken together, these data suggest that CD4+Foxp3+ cells are associated with protection from fibrosis. The mechanism by which Treg cells exert an antifibrotic phenotype is unknown. It is possible that Treg cells actively suppress the proliferation and/or profibrotic activities of other T cells (eg, Th2 cells). Further studies will be necessary to determine the mechanism by which Treg cells contribute to protection from fibrosis in humans and animals.
Other potential mechanisms that may underlie the antifibrotic potential of CLF-1 include significantly increased expression of CXCL9, CXCL10, and CXCL11, chemokines that are induced downstream by several stimuli, including IFN-γ.79 These chemokines signal through CXCR3.80 Cxcr3−/− mice experience exaggerated pulmonary fibrosis, which is relieved by IFN-γ.80 CXCL10 and CXCL11 have also been shown to attenuate bleomycin-induced pulmonary fibrosis.81–83 We note that signaling through CXCR3 may be deficient in IPF.6
Whether CLF-1 expression in the lung is specific to IPF is not known. Furthermore, the stimulus for secretion of CLF-1 in the lung is also unknown. Localization of CLF-1 expression in hyperplastic type II alveolar epithelial cells and the capacity of CLF-1/CLC to stimulate inflammation in the lung suggest that CLF-1 secretion may represent a generalized response to any lung injury. In tonsillar fibroblasts, CLF-1 expression is increased by proinflammatory mediators IFN-γ, TNF-α, and IL-6.49 Further studies will be necessary to elucidate the stimuli that drive expression of CLF-1 in the lung.
In summary, we report data supporting a potentially important antifibrotic role for CLF-1 expression in IPF. We speculate that CLF-1 expression in the lung could be a potentially reparative response to fibrotic lung injury that is specific to type II alveolar epithelial cells. We suggest that the effect of CLF-1 in the lung is likely to be complex, involving Th1 and Treg lymphocytes and production of cytokines (eg, IFN-γ) and multiple chemokines. Determining whether CLF-1 has therapeutic potential as an antifibrotic agent will require further investigation.
Footnotes
Supported by the NIH (K08 HL083085 and P30 ES009089-11 to D.J.K. and R01 HL095397 to N.K.), the American Lung Association of the City of New York (D.J.K.), and the Pulmonary Fibrosis Foundation.
Disclosures: N.K. has active grant support from Gilead Sciences and has received grant support in the past from Centocor and Biogen-Idec. N.K. is also a consultant to Sanofi-Aventis and Stromedix and has patent applications on peripheral blood biomarkers and microRNAs in IPF. S.M.G. is employed by Merck Pharmaceuticals.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2012.01.010.
Supplementary data
References
- 1.Raghu G., Weycker D., Edelsberg J., Bradford W.Z., Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 2.Fernández Pérez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, Yi ES, Ryu JH: Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest 137:129–137 [DOI] [PMC free article] [PubMed]
- 3.Luzina I.G., Todd N.W., Iacono A.T., Atamas S.P. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol. 2008;83:237–244. doi: 10.1189/jlb.0707504. [DOI] [PubMed] [Google Scholar]
- 4.Selman M., King T.E., Pardo A., American Thoracic Society; European Respiratory Society; American College of Chest Physicians Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 5.King T.E., Jr, Albera C., Bradford W.Z., Costabel U., Hormel P., Lancaster L., Noble P.W., Sahn S.A., Szwarcberg J., Thomeer M., Valeyre D., du Bois R.M., INSPIRE Study Group Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. 2009;374:222–228. doi: 10.1016/S0140-6736(09)60551-1. [DOI] [PubMed] [Google Scholar]
- 6.Pignatti P., Brunetti G., Moretto D., Yacoub M.R., Fiori M., Balbi B., Balestrino A., Cervio G., Nava S., Moscato G. Role of the chemokine receptors CXCR3 and CCR4 in human pulmonary fibrosis. Am J Respir Crit Care Med. 2006;173:310–317. doi: 10.1164/rccm.200502-244OC. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu Y., Kuwabara H., Ono A., Higuchi S., Hisada T., Dobashi K., Utsugi M., Mita Y., Mori M. Intracellular Th1/Th2 balance of pulmonary CD4(+) T cells in patients with active interstitial pneumonia evaluated by serum KL-6. Immunopharmacol Immunotoxicol. 2006;28:295–304. doi: 10.1080/08923970600809389. [DOI] [PubMed] [Google Scholar]
- 8.Narayanan A.S., Whithey J., Souza A., Raghu G. Effect of gamma-interferon on collagen synthesis by normal and fibrotic human lung fibroblasts. Chest. 1992;101:1326–1331. doi: 10.1378/chest.101.5.1326. [DOI] [PubMed] [Google Scholar]
- 9.Clark J.G., Dedon T.F., Wayner E.A., Carter W.G. Effects of interferon-gamma on expression of cell surface receptors for collagen and deposition of newly synthesized collagen by cultured human lung fibroblasts. J Clin Invest. 1989;83:1505–1511. doi: 10.1172/JCI114045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada T., Sugie I., Aisaka K. Effects of gamma-interferon on collagen and histamine content in bleomycin-induced lung fibrosis in rats. Lymphokine Cytokine Res. 1993;12:87–91. [PubMed] [Google Scholar]
- 11.Giri S.N., Hyde D.M., Marafino B.J., Jr Ameliorating effect of murine interferon gamma on bleomycin-induced lung collagen fibrosis in mice. Biochem Med Metab Biol. 1986;36:194–197. doi: 10.1016/0885-4505(86)90124-6. [DOI] [PubMed] [Google Scholar]
- 12.Hyde D.M., Henderson T.S., Giri S.N., Tyler N.K., Stovall M.Y. Effect of murine gamma interferon on the cellular responses to bleomycin in mice. Exp Lung Res. 1988;14:687–704. doi: 10.3109/01902148809087837. [DOI] [PubMed] [Google Scholar]
- 13.Saito A., Okazaki H., Sugawara I., Yamamoto K., Takizawa H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol. 2003;132:168–176. doi: 10.1159/000073718. [DOI] [PubMed] [Google Scholar]
- 14.Fichtner-Feigl S., Strober W., Kawakami K., Puri R.K., Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 15.Kimura T., Ishii Y., Yoh K., Morishima Y., Iizuka T., Kiwamoto T., Matsuno Y., Homma S., Nomura A., Sakamoto T., Takahashi S., Sekizawa K. Overexpression of the transcription factor GATA-3 enhances the development of pulmonary fibrosis. Am J Pathol. 2006;169:96–104. doi: 10.2353/ajpath.2006.051160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moodley Y.P., Misso N.L., Scaffidi A.K., Fogel-Petrovic M., McAnulty R.J., Laurent G.J., Thompson P.J., Knight D.A. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol. 2003;29:490–498. doi: 10.1165/rcmb.2002-0262OC. [DOI] [PubMed] [Google Scholar]
- 17.Moodley Y.P., Scaffidi A.K., Misso N.L., Keerthisingam C., McAnulty R.J., Laurent G.J., Mutsaers S.E., Thompson P.J., Knight D.A. Fibroblasts isolated from normal lungs and those with idiopathic pulmonary fibrosis differ in interleukin-6/gp130-mediated cell signaling and proliferation. Am J Pathol. 2003;163:345–354. doi: 10.1016/S0002-9440(10)63658-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scaffidi A.K., Mutsaers S.E., Moodley Y.P., McAnulty R.J., Laurent G.J., Thompson P.J., Knight D.A. Oncostatin M stimulates proliferation, induces collagen production and inhibits apoptosis of human lung fibroblasts. Br J Pharmacol. 2002;136:793–801. doi: 10.1038/sj.bjp.0704769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mozaffarian A., Brewer A.W., Trueblood E.S., Luzina I.G., Todd N.W., Atamas S.P., Arnett H.A. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J Immunol. 2008;181:7243–7253. doi: 10.4049/jimmunol.181.10.7243. [DOI] [PubMed] [Google Scholar]
- 20.Saito F., Tasaka S., Inoue K., Miyamoto K., Nakano Y., Ogawa Y., Yamada W., Shiraishi Y., Hasegawa N., Fujishima S., Takano H., Ishizaka A. Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am J Respir Cell Mol Biol. 2008;38:566–571. doi: 10.1165/rcmb.2007-0299OC. [DOI] [PubMed] [Google Scholar]
- 21.Nogee L.M., Dunbar A.E., 3rd, Wert S.E., Askin F., Hamvas A., Whitsett J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 22.Thomas A.Q., Lane K., Phillips J., 3rd, Prince M., Markin C., Speer M., Schwartz D.A., Gaddipati R., Marney A., Johnson J., Roberts R., Haines J., Stahlman M., Loyd J.E. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 23.Steele M.P., Speer M.C., Loyd J.E., Brown K.K., Herron A., Slifer S.H., Burch L.H., Wahidi M.M., Phillips J.A., 3rd, Sporn T.A., McAdams H.P., Schwarz M.I., Schwartz D.A. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005;172:1146–1152. doi: 10.1164/rccm.200408-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diaz de Leon A, Cronkhite JT, Katzenstein AL, Godwin JD, Raghu G, Glazer CS, Rosenblatt RL, Girod CE, Garrity ER, Xing C, Garcia CK: Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One 5:e10680 [DOI] [PMC free article] [PubMed]
- 25.Coward WR, Watts K, Feghali-Bostwick CA, Jenkins G, Pang L: Repression of IP-10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol Cell Biol 30:2874–2886 [DOI] [PMC free article] [PubMed]
- 26.Sanders Y.Y., Pardo A., Selman M., Nuovo G.J., Tollefsbol T.O., Siegal G.P., Hagood J.S. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:610–618. doi: 10.1165/rcmb.2007-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baumgartner K.B., Samet J.M., Coultas D.B., Stidley C.A., Hunt W.C., Colby T.V., Waldron J.A., Centers Collaborating Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Am J Epidemiol. 2000;152:307–315. doi: 10.1093/aje/152.4.307. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz D.A., Van Fossen D.S., Davis C.S., Helmers R.A., Dayton C.S., Burmeister L.F., Hunninghake G.W. Determinants of progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1994;149:444–449. doi: 10.1164/ajrccm.149.2.8306043. [DOI] [PubMed] [Google Scholar]
- 29.Kass D.J., Kaminski N. Evolving genomic approaches to idiopathic pulmonary fibrosis: moving beyond genes. Clin Transl Sci. 2011;4:372–379. doi: 10.1111/j.1752-8062.2011.00287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pardo A., Gibson K., Cisneros J., Richards T.J., Yang Y., Becerril C., Yousem S., Herrera I., Ruiz V., Selman M., Kaminski N. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005;2:e251. doi: 10.1371/journal.pmed.0020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuo F., Kaminski N., Eugui E., Allard J., Yakhini Z., Ben-Dor A., Lollini L., Morris D., Kim Y., DeLustro B., Sheppard D., Pardo A., Selman M., Heller R.A. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA. 2002;99:6292–6297. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh M, Handley D, Richards T, Selman M, Watkins SC, Pardo A, Ben-Yehudah A, Bouros D, Eickelberg O, Ray P, Benos PV, Kaminski N: Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 182:220–229 [DOI] [PMC free article] [PubMed]
- 33.Bridges R.S., Kass D., Loh K., Glackin C., Borczuk A.C., Greenberg S. Gene expression profiling of pulmonary fibrosis identifies Twist1 as an antiapoptotic molecular “rectifier” of growth factor signaling. Am J Pathol. 2009;175:2351–2361. doi: 10.2353/ajpath.2009.080954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elson G.C., Lelièvre E., Guillet C., Chevalier S., Plun-Favreau H., Froger J., Suard I., de Coignac A.B., Delneste Y., Bonnefoy J.Y., Gauchat J.F., Gascan H. CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat Neurosci. 2000;3:867–872. doi: 10.1038/78765. [DOI] [PubMed] [Google Scholar]
- 35.Factor P., Mutlu G.M., Chen L., Mohameed J., Akhmedov A.T., Meng F.J., Jilling T., Lewis E.R., Johnson M.D., Xu A., Kass D., Martino J.M., Bellmeyer A., Albazi J.S., Emala C., Lee H.T., Dobbs L.G., Matalon S. Adenosine regulation of alveolar fluid clearance. Proc Natl Acad Sci USA. 2007;104:4083–4088. doi: 10.1073/pnas.0601117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kass D., Bridges R.S., Borczuk A., Greenberg S. Methionine aminopeptidase-2 as a selective target of myofibroblasts in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2007;37:193–201. doi: 10.1165/rcmb.2006-0352OC. [DOI] [PubMed] [Google Scholar]
- 37.Zahurak M., Parmigiani G., Yu W., Scharpf R.B., Berman D., Schaeffer E., Shabbeer S., Cope L. Pre-processing Agilent microarray data. BMC Bioinformatics. 2007;8:142. doi: 10.1186/1471-2105-8-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu W., Dave N., Tseng G.C., Richards T., Xing E.P., Kaminski N. Comparison of normalization methods for CodeLink Bioarray data. BMC Bioinformatics. 2005;6:309. doi: 10.1186/1471-2105-6-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Segal E., Friedman N., Kaminski N., Regev A., Koller D. From signatures to models: understanding cancer using microarrays. Nat Genet. 2005;37(Suppl):S38–S45. doi: 10.1038/ng1561. [DOI] [PubMed] [Google Scholar]
- 40.Davé N.B., Kaminski N. Analysis of microarray experiments for pulmonary fibrosis. Methods Mol Med. 2005;117:333–358. doi: 10.1385/1-59259-940-0:333. [DOI] [PubMed] [Google Scholar]
- 41.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 42.Ihaka R., Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- 43.Spiegelhalter D.J., Thomas A., Best N.G. Computation on Bayesian graphical models: Bayesian Statistics 5: Proceedings of the Fifth Valencia International Meeting, June 5–9, 1994. In: Bernardo J.M., Berger J.O., Dawid A.P., Smith A.F.M., editors. Oxford University Press; Oxford: 1995. pp. pp 407–pp 425. [Google Scholar]
- 44.Lelièvre E., Plun-Favreau H., Chevalier S., Froger J., Guillet C., Elson G.C., Gauchat J.F., Gascan H. Signaling pathways recruited by the cardiotrophin-like cytokine/cytokine-like factor-1 composite cytokine: specific requirement of the membrane-bound form of ciliary neurotrophic factor receptor alpha component. J Biol Chem. 2001;276:22476–22484. doi: 10.1074/jbc.M101681200. [DOI] [PubMed] [Google Scholar]
- 45.Kass D.J. Cytokine-like factor 1 (CLF1): Life after development. Cytokine. 2011;55:325–329. doi: 10.1016/j.cyto.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forger N.G., Prevette D., deLapeyrière O., de Bovis B., Wang S., Bartlett P., Oppenheim R.W. Cardiotrophin-like cytokine/cytokine-like factor 1 is an essential trophic factor for lumbar and facial motoneurons in vivo. J Neurosci. 2003;23:8854–8858. doi: 10.1523/JNEUROSCI.23-26-08854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt-Ott K.M., Yang J., Chen X., Wang H., Paragas N., Mori K., Li J.Y., Lu B., Costantini F., Schiffer M., Bottinger E., Barasch J. Novel regulators of kidney development from the tips of the ureteric bud. J Am Soc Nephrol. 2005;16:1993–2002. doi: 10.1681/ASN.2004121127. [DOI] [PubMed] [Google Scholar]
- 48.Sakamoto H., Zhao L.H., Jain F., Kradin R. IL-12p40(-/-) mice treated with intratracheal bleomycin exhibit decreased pulmonary inflammation and increased fibrosis. Exp Mol Pathol. 2002;72:1–9. doi: 10.1006/exmp.2001.2409. [DOI] [PubMed] [Google Scholar]
- 49.Elson G.C., Graber P., Losberger C., Herren S., Gretener D., Menoud L.N., Wells T.N., Kosco-Vilbois M.H., Gauchat J.F. Cytokine-like factor-1, a novel soluble protein, shares homology with members of the cytokine type I receptor family. J Immunol. 1998;161:1371–1379. [PubMed] [Google Scholar]
- 50.Herholz J., Meloni A., Marongiu M., Chiappe F., Deiana M., Herrero C.R., Zampino G., Hamamy H., Zalloum Y., Waaler P.E., Crisponi G., Crisponi L., Rutsch F. Differential secretion of the mutated protein is a major component affecting phenotypic severity in CRLF1-associated disorders. Eur J Hum Genet. 2011;19:525–533. doi: 10.1038/ejhg.2010.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dagoneau N., Bellais S., Blanchet P., Sarda P., Al-Gazali L.I., Di Rocco M., Huber C., Djouadi F., Le Goff C., Munnich A., Cormier-Daire V. Mutations in cytokine receptor-like factor 1 (CRLF1) account for both Crisponi and cold-induced sweating syndromes. Am J Hum Genet. 2007;80:966–970. doi: 10.1086/513608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alexander W.S., Rakar S., Robb L., Farley A., Willson T.A., Zhang J.G., Hartley L., Kikuchi Y., Kojima T., Nomura H., Hasegawa M., Maeda M., Fabri L., Jachno K., Nash A., Metcalf D., Nicola N.A., Hilton D.J. Suckling defect in mice lacking the soluble haemopoietin receptor NR6. Curr Biol. 1999;9:605–608. doi: 10.1016/s0960-9822(99)80266-8. [DOI] [PubMed] [Google Scholar]
- 53.DeChiara T.M., Vejsada R., Poueymirou W.T., Acheson A., Suri C., Conover J.C., Friedman B., McClain J., Pan L., Stahl N., Ip N.Y., Yancopoulos G.D. Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell. 1995;83:313–322. doi: 10.1016/0092-8674(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida R., Nagira M., Kitaura M., Imagawa N., Imai T., Yoshie O. Secondary lymphoid-tissue chemokine is a functional ligand for the CC chemokine receptor CCR7. J Biol Chem. 1998;273:7118–7122. doi: 10.1074/jbc.273.12.7118. [DOI] [PubMed] [Google Scholar]
- 55.Jinquan T., Quan S., Feili G., Larsen C.G., Thestrup-Pedersen K. Eotaxin activates T cells to chemotaxis and adhesion only if induced to express CCR3 by IL-2 together with IL-4. J Immunol. 1999;162:4285–4292. [PubMed] [Google Scholar]
- 56.Loetscher P., Seitz M., Clark-Lewis I., Baggiolini M., Moser B. Monocyte chemotactic proteins MCP-1, MCP-2, and MCP-3 are major attractants for human CD4+ and CD8+ T lymphocytes. FASEB J. 1994;8:1055–1060. doi: 10.1096/fasebj.8.13.7926371. [DOI] [PubMed] [Google Scholar]
- 57.Stanford M.M., Issekutz T.B. The relative activity of CXCR3 and CCR5 ligands in T lymphocyte migration: concordant and disparate activities in vitro and in vivo. J Leukoc Biol. 2003;74:791–799. doi: 10.1189/jlb.1102547. [DOI] [PubMed] [Google Scholar]
- 58.Kennedy J., Kelner G.S., Kleyensteuber S., Schall T.J., Weiss M.C., Yssel H., Schneider P.V., Cocks B.G., Bacon K.B., Zlotnik A. Molecular cloning and functional characterization of human lymphotactin. J Immunol. 1995;155:203–209. [PubMed] [Google Scholar]
- 59.Jia G.Q., Gonzalo J.A., Lloyd C., Kremer L., Lu L., Martinez-A C., Wershil B.K., Gutierrez-Ramos J.C. Distinct expression and function of the novel mouse chemokine monocyte chemotactic protein-5 in lung allergic inflammation. J Exp Med. 1996;184:1939–1951. doi: 10.1084/jem.184.5.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pribul P.K., Harker J., Wang B., Wang H., Tregoning J.S., Schwarze J., Openshaw P.J. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol. 2008;82:4441–4448. doi: 10.1128/JVI.02541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herold S., von Wulffen W., Steinmueller M., Pleschka S., Kuziel W.A., Mack M., Srivastava M., Seeger W., Maus U.A., Lohmeyer J. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J Immunol. 2006;177:1817–1824. doi: 10.4049/jimmunol.177.3.1817. [DOI] [PubMed] [Google Scholar]
- 62.Colantonio L., Iellem A., Sinigaglia F., D'Ambrosio D. Skin-homing CLA+ T cells and regulatory CD25+ T cells represent major subsets of human peripheral blood memory T cells migrating in response to CCL1/I-309. Eur J Immunol. 2002;32:3506–3514. doi: 10.1002/1521-4141(200212)32:12<3506::AID-IMMU3506>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 63.Ngo V.N., Tang H.L., Cyster J.G. Epstein-Barr virus-induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J Exp Med. 1998;188:181–191. doi: 10.1084/jem.188.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marchal-Sommé J., Uzunhan Y., Marchand-Adam S., Kambouchner M., Valeyre D., Crestani B., Soler P. Dendritic cells accumulate in human fibrotic interstitial lung disease. Am J Respir Crit Care Med. 2007;176:1007–1014. doi: 10.1164/rccm.200609-1347OC. [DOI] [PubMed] [Google Scholar]
- 65.Lawson W.E., Polosukhin V.V., Stathopoulos G.T., Zoia O., Han W., Lane K.B., Li B., Donnelly E.F., Holburn G.E., Lewis K.G., Collins R.D., Hull W.M., Glasser S.W., Whitsett J.A., Blackwell T.S. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice after intratracheal bleomycin. Am J Pathol. 2005;167:1267–1277. doi: 10.1016/S0002-9440(10)61214-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gasse P., Mary C., Guenon I., Noulin N., Charron S., Schnyder-Candrian S., Schnyder B., Akira S., Quesniaux V.F., Lagente V., Ryffel B., Couillin I. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117:3786–3799. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA: Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207:535–552 [DOI] [PMC free article] [PubMed]
- 68.Tan R.J., Fattman C.L., Niehouse L.M., Tobolewski J.M., Hanford L.E., Li Q., Monzon F.A., Parks W.C., Oury T.D. Matrix metalloproteinases promote inflammation and fibrosis in asbestos-induced lung injury in mice. Am J Respir Cell Mol Biol. 2006;35:289–297. doi: 10.1165/rcmb.2005-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hao Z., Hampel B., Yagita H., Rajewsky K. T cell-specific ablation of Fas leads to Fas ligand-mediated lymphocyte depletion and inflammatory pulmonary fibrosis. J Exp Med. 2004;199:1355–1365. doi: 10.1084/jem.20032196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pochetuhen K., Luzina I.G., Lockatell V., Choi J., Todd N.W., Atamas S.P. Complex regulation of pulmonary inflammation and fibrosis by CCL18. Am J Pathol. 2007;171:428–437. doi: 10.2353/ajpath.2007.061167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corsini E., Luster M.I., Mahler J., Craig W.A., Blazka M.E., Rosenthal G.J. A protective role for T lymphocytes in asbestos-induced pulmonary inflammation and collagen deposition. Am J Respir Cell Mol Biol. 1994;11:531–539. doi: 10.1165/ajrcmb.11.5.7946383. [DOI] [PubMed] [Google Scholar]
- 72.Xu J., Mora A.L., LaVoy J., Brigham K.L., Rojas M. Increased bleomycin-induced lung injury in mice deficient in the transcription factor T-bet. Am J Physiol Lung Cell Mol Physiol. 2006;291:L658–L667. doi: 10.1152/ajplung.00006.2006. [DOI] [PubMed] [Google Scholar]
- 73.Silva C.M., Lu H., Weber M.J., Thorner M.O. Differential tyrosine phosphorylation of JAK1, JAK2, and STAT1 by growth hormone and interferon-gamma in IM-9 cells. J Biol Chem. 1994;269:27532–27539. [PubMed] [Google Scholar]
- 74.Kotsianidis I., Nakou E., Bouchliou I., Tzouvelekis A., Spanoudakis E., Steiropoulos P., Sotiriou I., Aidinis V., Margaritis D., Tsatalas C., Bouros D. Global impairment of CD4+CD25+FOXP3+ regulatory T cells in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:1121–1130. doi: 10.1164/rccm.200812-1936OC. [DOI] [PubMed] [Google Scholar]
- 75.Shimizu Y, Dobashi K, Endou K, Ono A, Yanagitani N, Utsugi M, Sano T, Ishizuka T, Shimizu K, Tanaka S, Mori M: Decreased interstitial FOXP3(+) lymphocytes in usual interstitial pneumonia with discrepancy of CXCL12/CXCR4 axis. Int J Immunopathol Pharmacol 23:449–461 [DOI] [PubMed]
- 76.Trujillo G, Hartigan AJ, Hogaboam CM: T regulatory cells and attenuated bleomycin-induced fibrosis in lungs of CCR7-/- mice. Fibrogenesis Tissue Repair 3:18 [DOI] [PMC free article] [PubMed]
- 77.Romani L., Fallarino F., De Luca A., Montagnoli C., D'Angelo C., Zelante T., Vacca C., Bistoni F., Fioretti M.C., Grohmann U., Segal B.H., Puccetti P. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 78.Johnson B.A., 3rd, Baban B., Mellor A.L. Targeting the immunoregulatory indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy. 2009;1:645–661. doi: 10.2217/IMT.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loos T., Dekeyzer L., Struyf S., Schutyser E., Gijsbers K., Gouwy M., Fraeyman A., Put W., Ronsse I., Grillet B., Opdenakker G., Van Damme J., Proost P. TLR ligands and cytokines induce CXCR3 ligands in endothelial cells: enhanced CXCL9 in autoimmune arthritis. Lab Invest. 2006;86:902–916. doi: 10.1038/labinvest.3700453. [DOI] [PubMed] [Google Scholar]
- 80.Jiang D., Liang J., Hodge J., Lu B., Zhu Z., Yu S., Fan J., Gao Y., Yin Z., Homer R., Gerard C., Noble P.W. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J Clin Invest. 2004;114:291–299. doi: 10.1172/JCI16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tager A.M., Kradin R.L., LaCamera P., Bercury S.D., Campanella G.S., Leary C.P., Polosukhin V., Zhao L.H., Sakamoto H., Blackwell T.S., Luster A.D. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. 2004;31:395–404. doi: 10.1165/rcmb.2004-0175OC. [DOI] [PubMed] [Google Scholar]
- 82.Jiang D., Liang J., Campanella G.S., Guo R., Yu S., Xie T., Liu N., Jung Y., Homer R., Meltzer E.B., Li Y., Tager A.M., Goetinck P.F., Luster A.D., Noble P.W. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J Clin Invest. 2010;120:2049–2057. doi: 10.1172/JCI38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burdick M.D., Murray L.A., Keane M.P., Xue Y.Y., Zisman D.A., Belperio J.A., Strieter R.M. CXCL11 attenuates bleomycin-induced pulmonary fibrosis via inhibition of vascular remodeling. Am J Respir Crit Care Med. 2005;171:261–268. doi: 10.1164/rccm.200409-1164OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.