

Abstract
Influenza A virus M2 (A/M2) forms a homotetrameric proton selective channel in the viral membrane. It has been the drug targets of antiviral drugs such as amantadine and rimantadine. However, most of the current virulent influenza A viruses carry drug resistant mutations alongside the drug binding site, such as S31N, V27A, and L26F etc., each of which might be dominant in a given flu season. Among these mutations, the V27A mutation was prevalent among transmissible viruses under drug selection pressure. Until now, V27A has not been successfully targeted by small molecule inhibitors, despite years of extensive medicinal chemistry research efforts and high throughput screening. Guided by molecular dynamics (MD) simulation of drug binding and the influence of drug binding on the dynamics of A/M2 from earlier experimental studies, we designed a series of potent spirane amine inhibitors targeting not only WT, but also both A/M2-27A and L26F mutants with IC50s similar to that seen for amantadine's inhibition of the WT channel. The potencies of these inhibitors were further demonstrated in experimental binding and plaque reduction assays. These results demonstrate the power of MD simulations to probe the mechanism of drug binding as well as the ability to guide design of inhibitors of targets that had previously appeared to be undruggable.
Introduction
Influenza virus infections present a major health problem on an annual basis, particularly in years of large-scale pandemics. The approved classes of small molecule drugs for treatment of influenza viral infections include neuraminidase inhibitors (zanamivir and oseltamivir) and M2 channel blockers (amantadine and rimantadine).1 Resistance to both classes is a problem: widespread resistance to the only orally bioavailable neuraminidase inhibitor, oseltamivir (Tamiflu), was encountered in the 2008-20092 and 2009-2010 flu season,3 and although amantadine was successfully used for over three decades, resistance is now so pervasive that the Center of Disease Control and Prevention (CDC) has advised against its continued use.4,5 Thus, there is an urgent need to develop novel orally bioavailable antivirals capable of targeting resistant strains of influenza A viruses. While progress has been made in the area of neuraminidase inhibitors,6-8 the design of inhibitors that address highly resistant forms of the M2 channel has proven to be challenging.1,9
Influenza A virus M2 proton channel (A/M2) forms a homotetrameric channel in the viral membrane that selectively conducts protons.10-12 Once viruses enter the infected cell by receptor-mediated endocytosis, the low pH in the endosome activates A/M2 and facilitates proton influx and disruption of the interaction of the viral RNA with its matrix.13 Another function of A/M2 is to equilibrate the pH across the Golgi in order to prevent the premature conformational change of the hemagglutinin, which acts as a pH-dependent fusogen.14-16
Although a variety of mutations can lead to amantadine-resistance in vitro,17-19 only three mutants, S31N, V27A and L26F, are generally observed in transmissible viruses that infect pigs, birds and humans.20-22 These substitutions map alongside the physiologically relevant drug binding site in the pore of the channel.23 A very extensive survey of pore-lining mutants of the M2 channel24 suggested a rationale for the surprising fitness of these mutants relative to the many less transmissible drug-resistant variants that are generated in patients during the course of amantadine or rimantadine treatment.19,25 L26F, S31N, and V27A are relatively unique in terms of retaining near-native proton flux and pH activation curves,24 which appear to be parameters finely tuned to respond to the properties of a given virus' hemagglutinin protein while minimizing toxicity for the parent cell until viral production is complete. Although S31N is the substitution found in current resistant strains, in other years V27A has predominated.26 Recent studies showed that the current predominance of S31N is not the result of drug selection pressure, because S31N was prevalent before the introduction of amantadine, and has become widespread in regions where amantadine was never used.20,22 Instead V27A was identified to be the major mutation emerging from drug selection pressure. While the L26F and S31N mutation cause a 10 to 20-decrease in the IC50s for amantadine inhibition, the corresponding V27A mutation renders the channel entirely resistant to both amantadine and rimantadine.27 We therefore focused on this particularly challenging mutant.
Over the last four decades, systematic studies of amantadine analogues and library screening have elucidated structure-activity relationships, and helped to identify potent channel-blockers.1,9 However, there have been no confirmed reports of small organic molecules that potently target highly amantadine-resistant variants of A/M2. Here we use a combination of molecular dynamics (MD) and classical medicinal chemistry approaches to design very potent inhibitors of V27A and L26F. MD was used to explore the mechanism of binding of amantadine to WT and of the designed inhibitor to V27A, thereby informing the mechanism and potency of the designed compounds. Potent inhibitors in general require an ammonium group, which associates with discrete, water-lined sites that might be hotspots for stabilizing a diffusing hydronium ion. These sites appear to be retained in the L26F and V27A mutants, and formed the basis for the design of inhibitors that target these variants while maintaining affinity for WT.
Materials and Methods
Materials
All starting material chemicals were purchased from commercial vendors and used without purification. Reactions were carried out using HPLC grade solvents under N2 atmosphere. Compounds were purified by silica gel flash column chromatography and characterized by ESI-MS, 1H-NMR and 13C-NMR. Details about the inhibitor synthesis procedure and characterization can be found in the supporting information.
Inhibitor Synthesis
Detailed synthesis procedure and compound characterization can be found in the supporting information.
Two electrode voltage clamp (TEVC) assay and plaque reduction assay
The inhibitors were tested via a two-electrode patch clamp (TEVC) assay using Xenopus laevis frog oocytes microinjectected with RNA expressing the A/M2 protein as in a previous report.27 The potency of the inhibitors was expressed as the percentage inhibition of A/M2 current observed after 2 min of incubation with 100 μM compounds, and IC50 values were collected for selected potent compounds.
Peptide synthesis
A/M2(22-46)-V27A peptide with A27, V28 and G34 selectively 15N and 13C labeled were manually synthesized using Fmoc chemistry at elevated temperature (75°C for both coupling and deprotection) in a semi-automated Quest synthesizer using Rink Amide Chemmatrix resin (Matrix Innovation Inc, Canada). 5 eq amino acid, 5 eq HCTU, 10 eq DIEA in NMP were used for 5 mins coupling. 5% piperazine and 0.1 M HOBt in DMF were used as the deprotection solution in order to minimize aspartamide formation. The peptide was cleaved from the resin using 95% TFA, 2.5% Tris, 2.5% H2O and precipitated from ether after removal of TFA. Ether was decanted after centrifugation and the peptide was washed with cold ether again. Final peptide was dissolved in 50% B' (59.9% isopropanol, 30% acetonitrile, 10% H2O, and 0.1% TFA)/50% A (99.9% H2O, 0.1% TFA) and purified by preparative C4 reverse phase HPLC with a linear gradient of 70% B' to 85% B'. The peptide was eluted at 78% B'. The purity and identity of the peptide was confirmed by analytical HPLC and MALDI-MS. Calculated MS: 2782.38, Observed MS: 2782.90.
Molecular Dynamics simulations
All simulations in this work were performed on the transmembrane region of the A/M2 bundle, spanning residues 25 to 46 and identified in the following as M2TM. The high-resolution structure of M2TM from Acharya et al28 was used as initial configuration. M2TM was embedded in a hydrated membrane of palmitoyl-oleoyl-phosphatidyl-choline (POPC) molecules, and periodic boundary conditions were applied with a 80 Å x 80 Å x 77 Å periodic box, corresponding to a 38 Å thick layer of water between two periodic images of the bilayer. The hydrated bilayer was neutralized by adding 27 K+ and 28 Cl- ions, respectively (KCl concentration of about 150 mM). The protein and the lipids were modeled using the CHARMM27 force field,29 and water molecules using the TIP3P force field.30 The electrostatic potential was solved by the particle mesh Ewald (PME) method31 with an accuracy threshold of 10-6, a real space spherical cutoff of 12 Å, and a fast Fourier transform (FFT) grid spacing of 0.8 Å. Lennard-Jones interactions were cut off at 12 Å, with a switching function starting from 10 Å. The equations of motion were solved with the velocity Verlet integrator using a timestep of 1.5 fs. The lengths of all bonds involving hydrogen atoms were kept constrained with the SHAKE method.32 The system was run at 310 K and 1 atm using Langevin temperature33 and Langevin piston pressure34,35 coupling schemes. Decay times for the thermostat and barostat were chosen to be 1 ps and 0.1 ps, respectively. MD simulations were performed with NAMD.36
Solid-state NMR (ssNMR)
1,2-dimyristoyl-sn-glycero-3-phosphoch-line (DMPC) was used to reconstitute A27, V28 and G34 labeled A/M2(22-46)-V27A by detergent dialysis as described before.37 The dry lipid powder was suspended in 1 mL of pH 7.5 phosphate buffer (10 mM NaH2PO4/Na2HPO4, 1 mM EDTA, and 0.01 mM NaN3), votexed, and free-thawed 8 times to create uniform lipid vesicles. The peptide powder was codissolved with ∼20 mg octyl-β-D-glucopyranoside (OG) in 1 mL of the same buffer. The solution was then mixed with 1 mL lipid vesicle solution, votexed for 2 hours and dialyzed with a 3.5-kDa molecular weight cutoff against 1 L buffer at 4°C for 3 days. The buffer was changed every 8-12 hours to remove the detergent. The protein-lipid precipitate appeared after one day. The proteoliposome solution was centrifuged at 150,000g and 6 °C for 4 hours to yield a membrane pellet with ∼40 wt% water. The final peptide : lipid molar ratio was 1 : 8. Drugs were directly titrated into the membrane pellet in the NMR rotor to a ratio of four per tetramer.
Solid-state NMR experiments were carried out on a 400 MHz (9.4 Tesla) and a 600 MHz (14.1 Tesla) Bruker AVANCE spectrometer using 4 mm MAS probes. Typical radio-frequency fields were 40-50 kHz for 13C and 15N and 70 kHz for 1H. 13C and 15N chemical shifts were referenced to the α-Gly CO signal at 176.49 ppm on the TMS scale and the 15N signal of N-acetyl-valine at 122 ppm on the liquid ammonia scale, respectively. 2D 15N-13C correlation spectra were measured using a REDOR sequence with a 0.8 ms mixing time for 13C-15N coherence transfer.38 2D 13C-13C correlation spectra were measured using a 1H-driven 13C spin diffusion sequence39 with a 40 ms 13C mixing time. All experiments were performed at 273 K and under 7 kHz spinning.
Results and Discussion
MD simulations of drug-binding
A/M2 channel blockers bind to a site within the N-terminal half of the pore, displacing water from a pore that leads to the pH-sensing H37 residues. Drugs bind most tightly when first incubated near neutral pH, where the channel has been most extensively characterized in bilayers and micelles by ssNMR,37,40-42 solution NMR40,43and X-ray crystallography.23,28 The transmembrane, TM, domain has been studied with and without an additional C-terminal cytoplasmic helix, and in the presence and absence of drugs. Although some details vary between structures, particularly in the cytoplasmic helices, the TM domain of all structures show striking similarities: the pore begins with a steric occlusion near the exterior-facing V27 residue, the V27-valve, then widens to form an approximately 12 Å N-terminal pore lined by small residues, A30, S31, and G34. The N-terminal pore leads to the gate-keeping H37 and W41 residues.11 Experimental studies, including very recent solution and ssNMR measurements place amantadine in the N-terminal aqueous pore with its hydrophobic adamantyl group docked against the V27 valve and the ammonium projecting downward towards H37.40
To obtain additional insight into the mode of binding, we performed classical MD simulations on A/M2 in complex with amantadine, in a fully hydrated aqueous bilayer. The newly available 1.65 Å crystal structure (PDB: 3LBW) was used as a starting point (Figure 1a),28 and the drug was initially placed as in the closely related ssNMR structure (PDB: 2KQT) of the amantadine-A/M2 complex37 in phospholipids vesicles. Focusing on the binding site, we analyzed the conformational distribution of the drug and water molecules. To examine the spatial relationship of water, drug, and other atomic groups in the time-averaged structure of the complex, we examined the density of these atoms averaged over snapshots taken between 20 and 80 nsec of the simulations. To further facilitate analysis, two-dimensional density maps were generated by rotational averaging about the pseudo-fourfold axis of the channel (Figure 1c,e,f).
Figure 1.
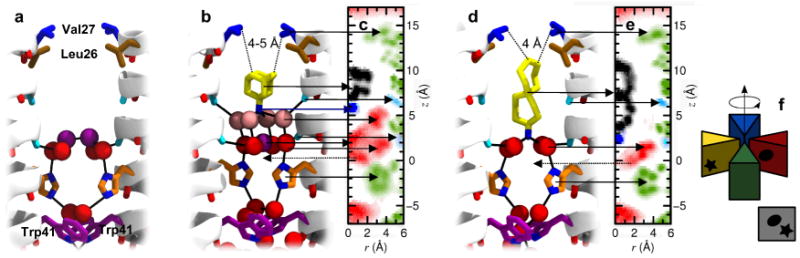
Layering of water molecules in the M2 channel pore. (a) Structure of the pore of A/M2 in the 1.65 Å X-ray structure (PDB: 3LBW) at intermediate pH28: the side chains Leu26, Val27, His37 and Trp41 are shown in brown, blue, orange and purplish, respectively. Two layers of crystallographically resolved water molecules are shown as red and purple spheres, while backbone carbonyls involved in hydrogen bonds with such water molecules (Ala30 and Gly34) are in light blue. (b) Representative MD snapshot of amantadine (yellow) within the pore of M2, with water molecules in red, purple and pink. (c) Density map in cylindrical coordinates of the heavy atoms of M2 (green), water (red), and amantadine (black, blue): oxygen atoms of residues 30 and 34 are in light blue. The density is averaged over the four monomers (see (f) for a graphical description of the average over the azimuthal angle shown in the density maps) and over the entire MD trajectory: two spots of the water density are alternatively occupied, and a dashed arrow shows the one not represented in (b). (d, e) MD snapshot and density map of M2 in complex with compound 1. Note that while amantadine inserts its charged ammonium group into the outer layer of water molecules (b, c), larger molecules such as compound 1 can only be accommodated when such water molecules are displaced (d, e).
Amantadine remains stable during the simulations, primarily rotating about its symmetry axis, with its apolar adamantyl group snugly bound against a constricted, hydrophobic V27 valve, and its ammonium group projecting downwards towards the water cluster. The water molecules show nsec fluctuations in which they hop between preferred (but not invariant) positions, observable as regions of high density in the two-dimensional radial projections. On average, amantadine's ammonium group is hydrated by four water molecules (Figure 1b,c) in a square pyramidal arrangement. This positively charged ammonium hydrate is stabilized by forming hydrogen bonds with a carbonyl box, formed by the four carbonyl groups of A30. Each carbonyl group tilts slightly away from the α-helical axis to allow interaction with water while retaining a helical hydrogen bond to an i+4 backbone amide, as is commonly observed in water-exposed sites of helices.44,45 Interestingly, a similar situation is repeated one helical turn down the channel, this time with a water molecule at the apex of the pyramid; the four water molecules at the base vertices of the square pyramid are stabilized by a carbonyl crown formed by G34, and strengthened by interactions with the imidazole nitrogens from the H37-box. This “lower” tetrad of water molecules is clearly seen in the electron density of the crystal structure of the protein in the drug-free form, while only diffuse density from solvent is seen near A30 in the absence of drugs. MD simulations at room temperature of both the G34A mutant of the crystal structure33 and the WT protein confirm that the two tetrads are both mobile, but water molecules associated to G34 and H37 are more stable than those associated to A30. Thus, the upper site might be more easily displaced than the lower sub-site near G34/H37 by channel-blocking drugs.
We therefore examined whether longer and more extended drugs might place their ammonium substituents deeper in the channel, occupying the apical site of the lower water cluster associated with G34 carbonyl box and H37-box. To test this idea we explored the consequences of systematically lengthening a previously described series of spiro-bicyclic amine inhibitors,27 ultimately resulting in an elongated inhibitor with two spiro-fused cycloheptane rings 1 (Figure 1d,e) as described in more detail below. The computed geometry for this molecule bound to WT is shown in Figure 1d,e. The site is constrained in one direction by the V27 valve, forcing the polar end of the more elongated inhibitor into the lower aqueous site (Figure 1d,e). Thus, these calculations suggested that different hydrophobic amine inhibitors might access one of two ammonium-binding sites, depending on the steric properties of their aliphatic substituents. This apparent flexibility provided a crucial insight to enable design of dual-specificity inhibitors for WT as well as V27A.
The V27A and L26F mutants have larger, more solvent-exposed N-terminal pores
To investigate how V27A and L26F mutations affect the size and shape of the binding site, we built theoretical models of these mutants in the drug-free form using the recently solved high resolution structure of A/M228 (PDB: 3LBW) as an initial configuration. Initial models were obtained by replacing the sidechains of V27 and L26 with Ala and Phe, respectively, and refined by performing classical MD simulations in a bilayer of 1,2-dimyristoyl-sn-glycero-3-phosphocho- line (DMPC) lipids hydrated by explicit solvent molecules. After a relaxation phase of several ns with the protein gradually released, the structures of the mutants were observed to be stable over a timespan of at least several tens of ns (backbone RMSD of the order of 1 Å). As expected, the mutation of the valines into alanines at position 27 does not perturb significantly the overall structure of the bundle. Therefore the effect of the mutation can be easily rationalized by observing that replacing the bulky Val sidechain with a small Ala methyl moiety results into an expansion of the pore radius near the top of the amantadine binding site (Figure 2c,d). A solution NMR structure of this mutant,46 which was published subsequent to the completion of these calculations, is in agreement with this conclusion and helps validate the modeling protocol. Particularly noteworthy and somewhat less anticipated, is the fact that a similar net effect is observed upon mutation of L26, which is not pore-lining, into phenylalanine (Figure 2e,f). In this case the introduction of the phenyl groups results in less efficient packing near the N-terminus of the helix. As a result, the entry region of the pore is more disordered and features, on average, a larger radius.
Figure 2.
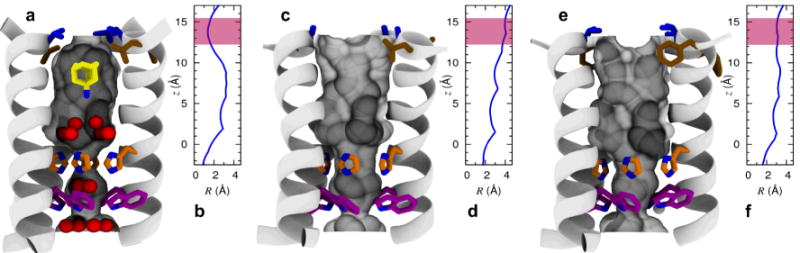
Shape modulation of the drug-binding pocket in the pore of A/M2 channel. A cartoon representation of the A/M2-TM helix bundle is shown for WT (a,b), V27A (c,d), and L26F (e,f). The molecular surface of the channel pore is highlighted and pore-lining residues Leu/Phe26 (brown), Val/Ala27 (blue), His37 (orange), and Trp41 (violet) are shown as sticks. Pore water molecules and amantadine are shown for wt as red spheres and sticks, respectively. The positions of water oxygens and amantadine are obtained from the crystal structure28 (PDB: 3LBW), and the ssNMR structure of the A/M2-amantadine complex29 (PDB: 2KQT), respectively. For each structure, the radius of the pore (computed using Hole47) is plotted as a function of the displacement along the channel axis in the region between His37 and Val-Ala27; the range of values corresponding to the entry region of the channel pore is highlighted in pink.
Most importantly, while these mutations resulted in large changes in the upper region of the pore, the structure of the channel and the water appeared largely unaffected near A30, G34, and H37. Thus, the challenge was to design molecules that were larger than amantadine and able to fill the increased volume of the upper pore created by V27A and L26F mutant, while still being accommodated within the WT structure. The ability of the pore to accommodate alkyl-ammonium hydrates near either A30 or G34's carbonyl groups provided flexibility in the design of molecules that could penetrate different depths depending on the volume of the upper pore of the mutants.
Design and synthesis of inhibitors targeting A/M2-V27A
Previously, we discovered a potent inhibitor of WT A/M2 (2; IC50 = 0.9 ± 0.1 μM) (Table 1) while investigating structure-activity relationships of a random screen hit (BL-174348). Although compound 2 was ineffective against V27A,27,49 ssNMR characterization of this compound in complex with WT A/M2 in bilayers showed that this spiro-piperidine 2 had a greater impact on the dynamics and magnetic environment of the pore than amantadine, and that it interacted over a more extended site within the channel. Thus, it appeared to be an attractive scaffold upon which to build functional groups to fill in the more spacious vestibule created by the V27A mutation. Indeed, a one carbon extension of the spiro-piperidine 2 gave spirane amine 3, which was the first molecule to show weak but saturable inhibition of V27A (IC50 = 84.9 ± 13.6 μM).27 Encouraged by this result, the length of 3 was further extended by inserting a second carbon spacer, via a simple methylene in 4 and its methyl-substituted analogue, 5. Their ability to inhibit proton currents was measured in Xenopus laevis oocytes expressing either WT A/M2 or A/M2-V27A. Encouragingly, 4 and 5 had potencies similar to amantadine against WT; more importantly they had similar low IC50 values against V27A, which amantadine is completely unable to inhibit.
Table 1. Effect of extending the length of the 6,6-spirane core on potency against WT A/M2 and A/M2-V27A.
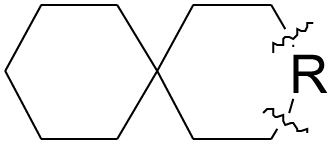 | |||
|---|---|---|---|
| Compound | R | IC50 WT A/M2 (μM) | IC50A/M2-V27A (μM) |
| 2 | 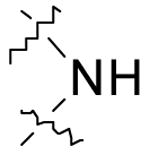 |
0.9 ± 0.1 | > 200 |
| 3 | 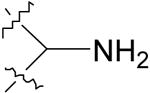 |
12.6 ± 1.1 | 84.9 ± 13.6 |
| 4 | 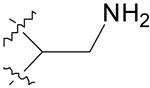 |
8.9 ± 0.6 | 31.1 ± 2.6 |
| 5 | 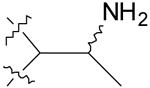 |
16.0 ± 1.8 | 25.2 ± 1.8 |
| Amantadine | 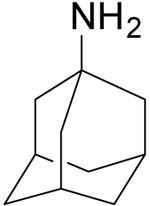 |
15.7 ± 1.2 | > 500 |
IC50 values were measured versus A/M2 from the Udorn strain of the virus, or the V27A mutant expressed in Xenopus laevis oocytes. All compounds are tested in hydrochloride form. The proton currents and extent of inhibition in the presence of various concentrations of drug were measured as described in methods.
Similar potency increases could be achieved by homologating the 6-membered rings of 3. The potency increased as the fully aliphatic ring was expanded from six to seven carbon atoms (7) (Table 2), and the same trend was observed when the amine-containing ring was expanded from six to seven carbon atoms (8). Combining these substitutions in the bis-cycloheptyl-spirane amine, 1, resulted in a compound with a two-fold higher potency than amantadine against WT A/M2, and great potency against the amantadine-insensitive mutant V27A with IC50 of 11.3 ± 0.7 μM. As a negative control, the 6-membered aliphatic ring of compound 3 was converted to the five-membered ring in 6, which only showed minimal inhibition against V27A mutant.
Table 2. Effect of ring size on potency of spirane amine inhibitors against WT A/M2 and A/M2-V27A.
| Compound | Structure | IC50 WT A/M2 (μM) | IC50A/M2-V27A (μM) |
|---|---|---|---|
| 6 | 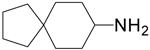 |
3.3 ± 0.2 | 319.1 ± 57.4 |
| 3 | 12.6 ± 1.1 | 84.9 ± 13.6 | |
| 7 | 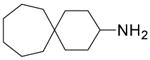 |
13.0 ± 1.1 | 28.1 ± 5.1 |
| 8 | 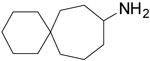 |
13.2 ± 1.0 | 68.1 ± 10.3 |
| 1 | 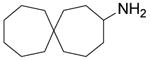 |
8.7 ± 0.6 | 11.3 ± 0.7 |
| Amantadine | 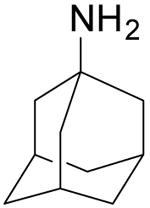 |
15.7 ± 1.2 | > 500 |
IC50 values were measured versus A/M2 from the Udorn strain of the virus, or the V27A mutant expressed in Xenopus laevis oocytes. All compounds were tested in hydrochloride salt form. The proton currents and extent of inhibition in the presence of various concentrations of drug were measured as described in methods.
Design of potent spiro-adamantane inhibitor of A/M2-V27A and L26F
MD simulations were used to further probe the mode of binding of amantadine and the potent bis-cycloheptyl-spirane amine 1 to both WT and V27A. Amantadine failed to bind a unique site when placed within V27A, and instead bound with its ammonium occupying either the upper or the lower aqueous sites. Moreover, its apolar adamantane cage was significantly less well dehydrated in V27A versus WT, explaining the loss in potency for the mutant. This behavior contrasts with that seen for the bis-cycloheptyl-spirane amine 1. As expected, the drug shifted its position upward in V27A, to allow its alkyl group to fill the larger cavity near the channel entrance (Figure 3e). Its ammonium group occupies the lower aqueous site in WT, where it forms solvent-mediated hydrogen bonds with the carbonyl of G34, versus A30 in V27A (Figure 3d,e). Although its apolar portion is more effectively dehydrated than amanatadine in V27A, the diffuse density of the upper ring of 1, when bound to V27A (Figure 3d), indicates that it averages between multiple orientations and does not fully fill the cavity. Thus, we suspected that its affinity might be increased further by modulating the steric bulk of its apolar substituent. We first considering converting the “upper” cyclohexane ring of 3 to a bicyclo[3.3.1]nonane (Intermediate I, Figure 4a), which can be further constrained by a methylene linkage to give spiroadamantane 9 (Figure 4a). This inhibitor was simulated against WT, V27A and L26F (Figure 3f,g). It was found that 9 had a similar pose as 1 in V27A with the hydrophobic adamantane filling in the extra space near A27. In the case of WT and L26F, compound 9 was pushed lower toward H37 and forms water mediated hydrogen bonding with H37. It is noted that high density of 9 was observed in all three variants (including L26F, not shown), suggesting tight binding with each mutant.
Figure 3.
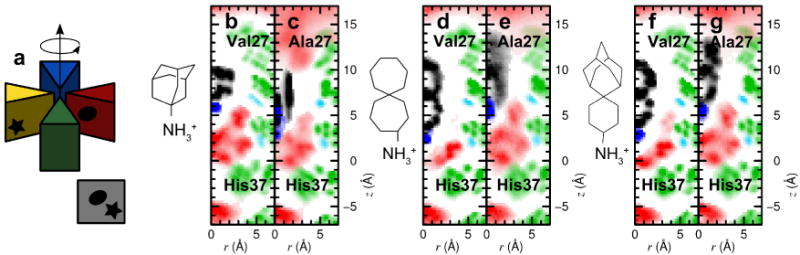
Density profiles for amantadine, 1 and 9 for WT and V27A. (a) Graphical description of the average over the azimuthal angle shown the density maps. (b-g) Density of protein (green), water (red) and drug (blue, black) heavy atoms, computed from MD simulations, and averaged over the four monomers (see illustration in (a). Carbonyl groups from A30 and G34 are shown in light blue. Panel (b) and (c) show densities for the complex between amantadine and WT, A/M2-V27A respectively, (d) and (e) between 1 and WT, A/M2-V27A, respectively, (f) and (g) between 9 and WT, A/M2-V27A, respectively.
Figure 4.
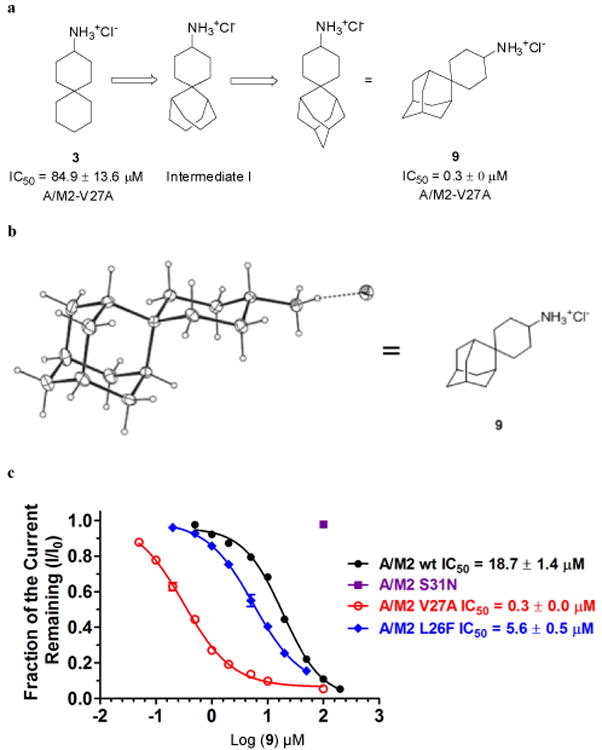
Spiroadamantane inhibitor 9 design and its dose response curve on WT, V27A, L26F and S31N inhibition. (a) Structure-based design of spiroadamantane 9. (b) X-ray crystal structure of spiroadamantane 9. (c) Dose response curve of 9 against WT, V27A and S31N A/M2 inhibition.
Compound 9 was then synthesized using a Robinson annulation reaction starting from 2-adamantanecarboaldehyde (Supporting Scheme 2), and its structure was confirmed by X-ray crystallography (Figure 4b Cambridge Crystallographic Data Centre deposition number: 824269). Spiroadamantane 9 proved to be the most potent V27A inhibitor-showing more than 280-fold lower IC50 for V27A than 3 (Figure 4c). Furthermore, this inhibitor was also highly active against another amantadine-resistant mutant L26F (Figure 4c), consistent with homology modeling and MD results showing that L26F and V27A have similarly expanded central cavities.
Activities in plaque reduction assay
The inhibitory effect of four of the most potent inhibitors on A/M2-V27A mutant was confirmed by plaque reduction assay of influenza A virus. Both the size and number of plaque formation were significantly reduced by all four inhibitors at 10 μM concentration (Figure 5). Consistent with the electrophysiology result, 9 was most potent and inhibited plaque formation dramatically at concentrations as low as 1 μM. As a negative control, amantadine showed no inhibition of A/M2-V27A virus replication even at 50 μM concentration. Thus, the inhibitory potency of the compounds against amantadine-resistant A/M2-V27A mutant channels seen in electrophysiology assays results in potent inhibition of the replication of influenza A virus.
Figure 5.
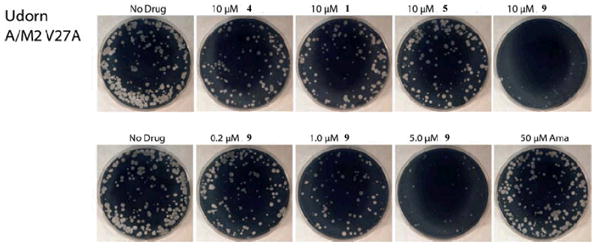
Plaque reduction assay of spirane amines on A/M2-V27A mutant virus. Effects of compounds on influenza A virus (A/Udorn/72) V27A mutant were evaluated by plaque formation on MDCK cells in the presence or absence of the compounds (10 μM or dose dependent) as described previously.27 9 is able to significantly reduce A/M2-V27A replication in as low as 5 μM conc. As control, amantadine has no effect on virus replication at up to 50 μM.
Direct binding of spirane amine to the TM domain of V27A
Solid-state NMR (ssNMR) spectroscopy provides a powerful probe of the structure of proteins in phospholipid bilayer environments. We synthesized A/M2-V27A TM peptide (22-46, M2TM) containing uniformly 15N, 13C-labeled A27, V28 and G34 (Supporting information). The labeled peptide was reconstituted into DMPC bilayers and 13C and 15N chemical shifts were measured using 2D magic-angle-spinning correlation experiments to examine drug-induced perturbation. Figure 6a and b show that the V28 15N resonance was sharpened and shifted downfield by 2 ppm upon incubation with excess 9. By contrast, incubation of A/M2TM-V27A with the weaker-binding 1 resulted in two partially overlapping peaks: the less intense peak has a chemical shift close to that seen in the absence of the inhibitor, indicative of either less complete binding or a minor conformation that is similar to the one found in the uncomplexed form. The more intense resonance in the presence of 1 has a strongly perturbed chemical shift, although the perturbation was less pronounced than that occasioned by the binding of 9. Furthermore, the 15N and 13C chemical shifts of G34 have previously been shown to be broad, heterogeneous and sensitive to drug-binding in WT A/M2 (Figure 6a).50 As was the case for binding spirane inhibitors to WT M2TM,49 incubation of V27A with 9 or 1 sharpened the peak, altered the intensity ratios, and shifted the chemical shift positions for the resonances associated with G34 in V27A (Figure 6b, c). Finally, compound 9 caused greater peak sharpening to the A27 peak than 1. These findings indicate a direct interaction of the drug with V27A, and suggest that the 10-fold higher potency of 9 compared to 1 is a result of a tighter and more extended interaction between the drug and the channel.
Figure 6.
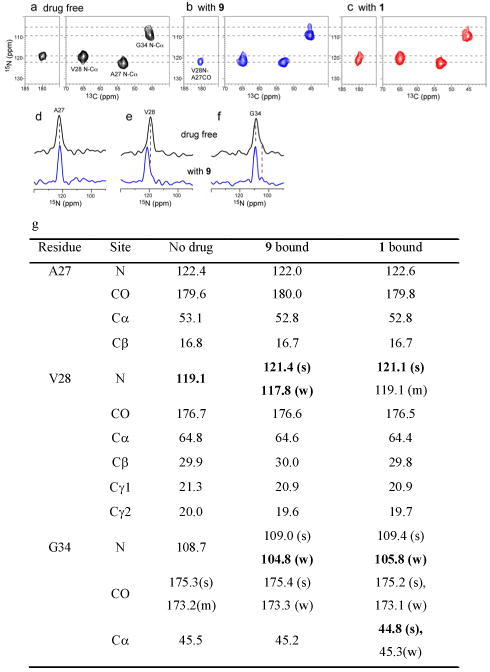
2D 15N-13C correlation spectra of A/M2-V27A in DMPC bilayers without and with spirane amine 9 and 1 bound. (a) Drug-free peptide. (b) With 9 bound. (c) With 1 bound. Four drugs per tetramer were used in (b-c). (d-f) 1D 15N cross sections without drug (top) and with spirane amine 9 (bottom). (d) A27. (e) V28. (f) G34. (g) 13C and 15N chemical shifts (ppm) of A/M2-V27A without drug and with drugs 9 and 1. Where two peaks are observed per site, intensities are labeled as strong (s), weak (w) or medium (m).
Conclusion
Despite extensive efforts, there have been no well-documented examples of inhibitors that target amantadine-resistant A/M2 mutants. This difficulty reflects the rather small size of the binding site in the WT protein, and the fact that resistant mutants tend to increase the polarity of the pore-lining residues and/or the hydration of the mouth of the channel. Thus, it was challenging to devise inhibitors that simultaneously targeted both mutant and WT forms, which was further exacerbated by the important role of relatively mobile pore water molecules, for both proton transduction as well as drug-binding. Thus, our early attempts to use automated docking to discover new inhibitors failed, and we turned to MD simulations to gain insight into the mechanism of proton transduction and its relation to drug-binding. Previous MD simulations from our group focused on methyl-ammonium, which was chosen because unsubstituted ammonium enhances proton conduction, possibly by mimicking hydronium ion in the conduction mechanism.51 Simulations of methyl-ammonium traversing the channel showed a saw-toothed potential of mean force, whose local minima are now seen to coincide with preferred locations of the charged ammonium in the drugs simulated here. The minima correspond to square planar arrays of carbonyl groups that stabilize the mobile hydrated ammonium groups in a manner analogous to the stabilization of hydrated cations in cation-specific channels.11,52 We hypothesized that more hydrophobic amine drugs obtain additional interactions by deepening the energy wells such that they act as inhibitors rather than enhancers of proton conduction as is the case for unsubstituted ammonia.53. Thus, we hypothesized that amine drugs act as reaction intermediate analogues, tapping into the ability of the protein to stabilize cations at specific locations of the channel. This insight paved the way to an understanding of the potential mode of binding of our initial hits for V27A, and to enhance their affinity by maximizing the fit with this mutant while simultaneously retaining affinity for WT through a related but distinct predicted binding mode.
In summary these data show the central role of MD simulations probing the mechanism of conduction and inhibition of a previously “undruggable” target. Relatively long simulations have allowed one to address mobile water as well as conformational mobility and resulted in a deeper understanding of the mechanism of inhibition that translated into tight-binding inhibitors of V27A.
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM56423 and AI74571 to W.F.D. and GM088204 to M. H. J. W. thank Dr. Patrick J. Carroll (University of Pennsylvania) for assistance in obtaining X-ray crystallographic data.
Footnotes
Supporting Information. Supplementary information and chemical compounds information is available at the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lagoja IM, De Clercq E. Med Res Rev. 2008;28:1–38. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moscona A. N Engl J Med. 2009;360:953–956. doi: 10.1056/NEJMp0900648. [DOI] [PubMed] [Google Scholar]
- 3.Baz M, Abed Y, Papenburg J, Bouhy X, Hamelin MÃv, Boivin G. N Engl J Med. 2009;361:2296–2297. doi: 10.1056/NEJMc0910060. [DOI] [PubMed] [Google Scholar]
- 4.Bright RA, Shay D, Bresee J, Klimov A, Cox N, Ortiz J. MMWR Morb Mortal Wkly Rep. 2006;55:44–46. [Google Scholar]
- 5.Fiore AE, Shay DK, Broder K, Iskander JK, Uyeki TM, Mootrey G, Bresee JS, Cox NJ. MMWR Recomm Rep. 2008;57:1–60. [PubMed] [Google Scholar]
- 6.Mitrasinovic PM. Curr Drug Targets. 2010;11:319–326. doi: 10.2174/138945010790711932. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Zhang J, Xu W. Curr Med Chem. 2007;14:2872–2891. doi: 10.2174/092986707782360024. [DOI] [PubMed] [Google Scholar]
- 8.von Itzstein M. Curr Opin Chem Biol. 2008;12:102–108. doi: 10.1016/j.cbpa.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 9.De Clercq E. Nat Rev Drug Discov. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinto LH, Holsinger LJ, Lamb RA. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Qiu JX, Soto C, DeGrado WF. Curr Opin Struct Biol. 2011;21:68–80. doi: 10.1016/j.sbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chizhmakov IV, Geraghty FM, Ogden DC, Hayhurst A, Antoniou M, Hay AJ. J Physiol-London. 1996;494:329–336. doi: 10.1113/jphysiol.1996.sp021495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin K, Helenius A. Cell. 1991;67:117–130. doi: 10.1016/0092-8674(91)90576-k. [DOI] [PubMed] [Google Scholar]
- 14.Sugrue RJ, Bahadur G, Zambon MC, Hallsmith M, Douglas AR, Hay AJ. EMBO J. 1990;9:3469–3476. doi: 10.1002/j.1460-2075.1990.tb07555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakaguchi T, Leser GP, Lamb RA. J Cell Biol. 1996;133:733–747. doi: 10.1083/jcb.133.4.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grambas S, Hay AJ. Virology. 1992;190:11–18. doi: 10.1016/0042-6822(92)91187-y. [DOI] [PubMed] [Google Scholar]
- 17.Abed Y, Goyette N, Boivin G. Antimicrob Agents Chemother. 2005;49:556–559. doi: 10.1128/AAC.49.2.556-559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown AN, McSharry JJ, Weng Q, Driebe EM, Engelthaler DM, Sheff K, Keim PS, Nguyen J, Drusano GL. Antimicrob Agents Chemother. 2010;54:3442–3450. doi: 10.1128/AAC.01385-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li D, Saito R, Suzuki Y, Sato I, Zaraket H, Dapat C, Caperig-Dapat IM, Suzuki H. J Clin Microbiol. 2009;47:466–468. doi: 10.1128/JCM.00892-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furuse Y, Suzuki A, Oshitani H. Antimicrob Agents Chemother. 2009;53:4457–4463. doi: 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bright RA, Medina MJ, Xu XY, Perez-Oronoz G, Wallis TR, Davis XHM, Povinelli L, Cox NJ, Klimov AI. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 22.Furuse Y, Suzuki A, Kamigaki T, Oshitani H. Virol J. 2009;6 doi: 10.1186/1743-422X-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Nature. 2008;452:380–380. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balannik V, Carnevale V, Fiorin G, Levine BG, Lamb RA, Klein ML, DeGrado WF, Pinto LH. Biochemistry. 2009;49:696–708. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiraishi K, Mitamura K, Sakai-Tagawa Y, Goto H, Sugaya N, Kawaoka Y. J Infect Dis. 2003;188:57–61. doi: 10.1086/375799. [DOI] [PubMed] [Google Scholar]
- 26.Saito R, Sakai T, Sato I, Sano Y, Oshitani H, Sato M, Suzuki H. J Clin Microbiol. 2003;41:2164–2165. doi: 10.1128/JCM.41.5.2164-2165.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balannik V, Wang J, Ohigashi Y, Jing XH, Magavern E, Lamb RA, DeGrado WF, Pinto LH. Biochemistry. 2009;48:11872–11882. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, Degrado WF, Klein ML. Proc Natl Acad Sci U S A. 2010;107:15075–80. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foloppe N, MacKerell JAD. J Comp Chem. 2000;21:86–104. [Google Scholar]
- 30.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 31.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 32.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 33.Adelman SA, Doll JD. J Chem Phys. 1976;64:2375–2388. [Google Scholar]
- 34.Feller SE, Zhang YH, Pastor RW, Brooks BR. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 35.Martyna GJ, Tobias DJ, Klein ML. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 36.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong M, Griffin RG. J Am Chem Soc. 1998;120:7113–7114. [Google Scholar]
- 39.Takegoshi K, Nakamura S, Terao T. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 40.Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. J Am Chem Soc. 2011;133:4274–4284. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu F, Luo W, Hong M. Science. 2010;330:505–508. doi: 10.1126/science.1191714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schnell JR, Chou JJ. Nature. 2008;451:591–U12. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manas ES, Getahun Z, Wright WW, DeGrado WF, Vanderkooi JM. J Am Chem Soc. 2000;122:9883–9890. [Google Scholar]
- 45.Walsh STR, Cheng RP, Wright WW, Alonso DOV, Daggett V, Vanderkooi JM, DeGrado WF. Protein Sci. 2003;12:520–531. doi: 10.1110/ps.0223003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pielak RM, Chou JJ. Biochem Biophys Res Commun. 2010;401:58–63. doi: 10.1016/j.bbrc.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MSP. J Mol Graph. 1996;14:354–360. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 48.Kurtz S, Luo G, Hahnenberger KM, Brooks C, Gecha O, Ingalls K, Numata K, Krystal M. Antimicrob Agents Chemother. 1995;39:2204–2209. doi: 10.1128/aac.39.10.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Cady SD, Balannik V, Pinto LH, DeGrado WF, Hong M. J Am Chem Soc. 2009;131:8066–8076. doi: 10.1021/ja900063s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu F, Luo W, Cady SD, Hong M. Biochim Biophys Acta. 2011;1808:415–423. doi: 10.1016/j.bbamem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carnevale V, Fiorin G, Levine BG, DeGrado WF, Klein ML. J Phys Chem C. 2010;114:20856–20863. doi: 10.1021/jp107431g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gouaux E, MacKinnon R. Science. 2005;310:1461–1465. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- 53.Mould JA, Drury JE, Frings SM, Kaupp UB, Pekosz A, Lamb RA, Pinto LH. J Biol Chem. 2000;275:31038–31050. doi: 10.1074/jbc.M003663200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.