

Abstract
Platelets are the primary cell mediator of thrombosis. A deficiency of platelets can result in life threatening bleeding defects. ‘Over active’ platelets contribute to life threatening outcomes in diseases such as heart attack, stroke, and cancer. The use of platelet inhibitors for thrombosis prevention must seek a delicate balance between inhibiting platelet activation and an associated increased bleeding risk. There are currently few platelet inhibitors available for clinical use making the search for novel anti-platelet drugs an ongoing priority. Several newly identified pathways of platelet activation may hold hope in this area. In addition, important roles for platelets beyond hemostasis have gained recent attention. A recognition that platelets contribute to the pathogenesis of inflammatory diseases such as arthritis and hepatitis has expanded our appreciation for the importance of understanding platelet function and platelet derived inflammatory mediators. It has also heightened interest in a continued search for new platelet inhibitors and presents new opportunities for platelet inhibitors to be used in a wide array of disease treatment strategies.
Keywords: platelet, inflammation, thrombosis, aggregation, activation, therapy
Introduction
Platelets are small (~1–2 μm), anucleate, megakaryocyte derived circulating cells present in high numbers in the blood. Typical platelet counts are 100,000–200,000/μL, making platelets the second most numerous cell in the blood. The most well understood function of platelets is its role as the primary cell mediator of thrombosis and a lack of platelets (thrombocytopenia), or defects in platelet function can result in major life threatening bleeding disorders. Untoward or unregulated platelet activation can also have severe outcomes by leading to thrombosis and loss of blood supply to major tissue beds. Cardiovascular disease and stroke are leading causes of morbidity and mortality in the Western world, and although many processes play a role in the development of vascular disease, thrombosis is the primary immediate event that precipitates stroke and acute coronary syndromes. There are currently few platelet specific anti-thrombotic agents available making direct platelet inhibitors, such as aspirin and clopidogrel (Plavix), mainstays in the medicine cabinet of cardiovascular patients.
Thrombosis is the result of a complex set of interactions between platelets, coagulation factors, and the vessel wall. Thrombosis proceeds through what is often described as a stereotypic set of steps: platelets are first activated, adhere to the vessel wall, and then aggregate with other platelets to form a stable thrombus [1,2]. There are also many intermediate steps important in efficient thrombus formation, such as loose contacts between platelets before firm adhesion. This creates micro-environments with high local concentrations of pro-thrombotic mediators [3]. Platelets can be activated by vessel wall exposure of extracellular matrix components, by activation of the coagulation cascade generating platelet agonists (e.g. thrombin), or by factors released from activated endothelial cells and platelets (e.g. ADP, thromboxane A2, vWF) [4–8]. Activation of receptors on resting platelets triggers a variety of intra-platelet signaling pathways, leading to subsequent steps of platelet activation, including conformational changes in receptors (e.g. GPIIb/IIIa), granule exocytosis and the secretion of vasoactive mediators.
Platelets have an obvious vital function in hemostasis, but they also have an under appreciated immune function. Many platelet granule constituents and secreted molecules have primary roles as immune mediators (Table 1) [9–12]. These platelet derived immune mediators recruit and activate leukocytes both at the site of platelet deposition and systemically. Platelets are not only activated by a denuded endothelium, but platelets are also activated by an intact inflamed endothelium and deposit at the site of vascular inflammation without forming a complete obstructive thrombus. We have demonstrated using two separate vascular inflammation based mouse models (a transplant model and a cerebral malaria model) that thrombi are present lining the lumen of inflamed blood vessels, without occluding the vessels (Figure 1). Interactions between an inflamed endothelium and platelets may be mediated by increased endothelial adhesion molecule expression (eg. P-selectin, ICAM, VCAM) serving to localize platelets and induce platelet secretion. These events perpetuate the cycle of inflammatory interactions between platelets, endothelial cells, and leukocytes.
Table 1.
| Location | Category | Molecule | Function |
|---|---|---|---|
| Alpha Granule | Adhesion | P-selectin (CD62) | Rolling, WBC attachment |
| vWf | Platelet and WBC adhesion | ||
| Chemokine | MIP-1 | Granulocyte recruitment and activation | |
| PF4 | WBC recruitment and activation | ||
| RANTES | WBC recruitment and activation | ||
| MCP | WBC recruitment and activation | ||
| PPBP/β-TG/NAP-2 | Granulocyte recruitment and activation | ||
| SDF-1 | Recruitment of WBC and stem cells | ||
| Cytokine | IL-1αand β | Pro-inflammatory effects on WBC, endothelial cells | |
| TGF-β | T-cell development, vascular remodeling | ||
| TNF-α | Pro-inflammatory effects on WBC, endothelial cells | ||
| Growth Factor | PDGF | Angiogenesis, immune activation | |
| VEGF | Angiogenesis, endothelial inflammation and cytokine induction | ||
| Other | GAS6 | Promotes WBC trafficking | |
| Complement | Complement cascade | ||
| Dense Granule | Glutamate | Cyclooxygenase activation | |
| ADP/ATP | Effects on T-cells and endothelial cells | ||
| Serotonin | Effects on T-cells and VSMC | ||
| Epinephrine | Augment WBC responses | ||
| Membrane | Integrin | GPIbα | Binds Mac-1 (CD11b/CD18 |
| GPIIb/GPIIIa | Binds fibrinogen | ||
| ICAM-2 | Binds WBC LFA-1 | ||
| JAM-1 and JAM-3 | Bind integrins such as LFA-1 and Mac-1 | ||
| Scavenger | CD36 | Receptor for Ox-LDL and malaria iRBC surface antigen | |
| ApoER2 (LRP8) | beta2-GPI binding | ||
| Cytoplasm | Induced Enzyme | Cyclooxygenase | Prostaglandins and Thromboxane |
Figure 1.
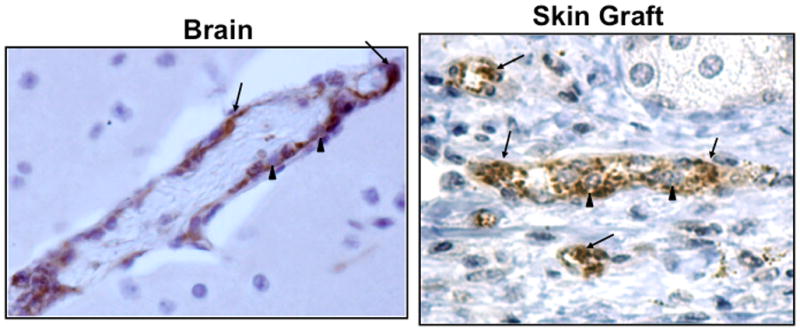
Platelet rich thrombi stained with anti-vWF antibody in brain of Plasmodium infected mouse (left side) and skin grafts (right side). Arrows point out thrombi, arrow heads leukocytes within the thrombi.
Activated platelets can also initiate interactions with quiescent endothelial cells and leukocytes through both contact dependent and independent mechanisms. Platelet inflammatory mediators include surface expressed adhesion molecules (P-selectin, integrins), secreted small molecules (ATP/ADP, serotonin, glutamate), as well as chemokines, and cytokines (Table 1). Platelet derived chemokines and cytokines, including Platelet Factor 4 (PF4/CXCL4), pro-platelet basic protein (ppbp and its breakdown product CXCL7/NAP-2), RANTES/CCL5, IL-1α/β and TGF-β can recruit and activate leukocytes distant from their site of deposition [13]. Other secreted mediators such as serotonin, ADP, and prostaglandins tend to exert pro-inflammatory effects in the local environment. Exteriorized or activated adhesion molecules on the surface of adherent platelets also assist in arresting leukocytes providing a contact dependent mechanism for platelets to exert pro-inflammatory effects.
A complete appreciation for both the thrombotic and pro-inflammatory functions of platelets, and the concept that platelets initiate, participate in, and sustain inflammation beyond the clot, has been slow to develop. This may be due in part to a misunderstood idea that because platelets are anucleate they cannot adapt to ‘environmental changes’. Because of their easily defined role in hemostasis, a long-standing focus of platelet researchers has been on platelet activation signaling pathways and integrin regulation. Recently the focus of platelet research has broadened to include new signaling mediators and previously unrecognized roles for platelets in vascular inflammatory diseases. We will highlight a few recent discoveries with the potential to lead to the development of new anti-thrombotics and impact the current use of platelet inhibitors. We will also highlight the current state of clinical anti-thrombotic therapy.
Our Changing View of Platelet Functions
Platelets and the synaptic termini of neurons are surprisingly similar in their composition. On the surface these are very different cells with very different functions. However, platelets and neurons share many similarities in receptor composition and molecular constituents. Each cell expresses proteins found in few other cell types such as synucleins, ADP/ATP receptors, TPO receptors, serotonin receptors, and glutamate receptors. Most platelet dense granule constituents are also neurotransmitters (glutamate, ADP, ATP, serontonin, dopamine, Ca2+ etc). This similarity may arise from both platelets and synaptic termini having unique functional demands due to their great distance from the ‘nuclear center’; the megakaryocyte in the bone marrow and the neuron cell body. Another major functional adaptation in common is that there are only two known places where pre-mRNA splicing occurs outside the nucleus; in synaptic termini and in platelets [14]. Platelets have stable pre-mRNA and upon platelet stimulation this pre-mRNA is spliced into mature mRNA that can be translated into protein. The importance of this discovery goes beyond just the demonstration of RNA splicing outside the nucleus, it provided a clear demonstration that despite the lack of a nucleus platelets have the ability to directly respond to environmental changes by altering their protein composition. Before this discovery it was known that platelets had RNA, but its significance was not appreciated, and it was largely viewed to be a by-stander effect of platelet budding from the megakaryocyte. With this paper the group headed by Andrew Weyrich helped to greatly advance a broader understanding that despite their anucleate status, platelets have a clear ability to up or down regulate protein expression. The relevance of this finding to health and disease remains to be demonstrated, but nonetheless, it represented a major advance in focusing platelet research in new directions. Recent publications, such as one demonstrating active platelet microRNA, has continued to build on this concept of peripheral platelet translational regulation [15].
Our lab has found that platelets, like neurons, express functional ionotropic glutamate receptors [16,17]. Glutamate is a major neurotransmitter that binds to NMDA, AMPA and Kainate (KA) receptors. Glutamate induces a Ca2+ influx through NMDA receptors and a Na+ influx when glutamate binds AMPAR and KAR. Others have described that platelet dense granules store and release glutamate, but a role for glutamate in platelet activation had not been described [18,19]. We found that glutamate is released in large concentrations within a developing thrombus and that AMPAR and KAR signaling amplifies platelet activation. Mice that lack AMPAR and KAR subunits or mice that are treated with receptor inhibitors have prolonged time to thrombus formation in vivo. Importantly, these mice do not have any bleeding diathesis, but form a delayed thrombus. This makes glutamate receptors an attractive target for the development of new anti-thrombotics. An important quality for a platelet inhibitor is an ability to blunt, but not totally block, platelet activation so as to not pose a large bleeding risk. We have found that AMPAR and KAR antagonists each prolong bleeding times and delay thrombus formation in mice and also reduce human platelet aggregation [17,16]. Many glutamate receptor antagonists have gone through clinical trials for treatment of stroke, but none to date has proven particularly effective. It remains to be seen through clinical trial if any of these existing glutamate receptor antagonists with established safety and toxicity data can find a new use as platelet inhibitors.
The use of mouse models has demonstrated that in addition to forming a coronary artery occlusion at the time of myocardial infarction (MI), platelets may have a prominent role in promoting the pathogenesis of atherosclerosis. Ley and colleagues demonstrated in an ApoE−/− mouse model that platelets deposit RANTES at the site of atherosclerotic lesions, one of the first clear mechanistic descriptions of how platelets may accelerate atherosclerosis [20,21]. This work and that by others in the mouse model have shown that platelets may be important early in atherosclerotic lesion development and its progression towards an unstable plaque. One of the major issues with mouse models of atherosclerosis is mice do not form unstable plaques, making an appreciation for the role of platelets in MI risk in humans difficult to extrapolate. However, there have been several human clinical studies indicating that platelets may drive lesion progression and platelets may be activated at sites of a growing atherosclerotic lesion [22,9] helping to validate this mouse model-based research.
Atherosclerosis is now seen as an immune mediated disease, with high cholesterol and oxidized lipid products well established risk factors at the heart of its pathogenesis. Recent work by Podrez et al has found that not only may platelets be activated at the site of atherosclerotic lesion development, but oxidized lipids may directly activate platelets via platelet CD36 [23]. This study puts platelets in the center of atherothrombosis by indicating that oxidized lipid mediated platelet activation may contribute immune mediators that promote lesion development locally and at sites distant from the actual lesion. Subsequent work by authors from this study has shown that the CD36 dependent platelet activation pathway functions through Mitogen Activated Protein Kinase (MAPK) signaling [24]. This knowledge helps to identify two new potential targets for anti-platelet drug development in the prevention and treatment of atherosclerosis; CD36 and MAPK members. However, both CD36 and MAP Kinases are expressed by many cells, making the development of platelet specific antagonists targeting these molecules potentially difficult.
Platelet Factor 4 (CXCL4) was the first CXC class chemokine identified and is the most abundant protein found in platelet α-granules. PF4 is prominently known for its role in heparin induced thrombocytopenia (HIT), but through the use of knockout mice its pro-inflammatory role is becoming better understood. PF4−/− mice on an ApoE−/− background have reduced atherosclerotic lesion development compared to control ApoE−/− mice [25]. This study indicates an important role for this platelet specific chemokine in the pathogenesis of atherosclerosis. Other studies have taken this knowledge into a potentially clinically relevant therapeutic development direction. PF4 forms a complex with RANTES and together create a highly pro-inflammatory environment. Through the use of peptides that disrupt PF4-RANTES associations investigators were also able to reduce lesion development in ApoE−/− mice [26].
With a more broad appreciation for platelet functions has come a more broad appreciation of the disease processes in which platelets may have a pathogenic role. Platelets may have a central role in the development of cerebral malaria. A large percentage of children infected with Plasmodium falciparum develop stroke-like symptoms resulting in long-term neurologic complications or death. We have found that platelet depletion of mice after infection with P. berghei ANKA leads to increased survival compared to control infected mice, and that this is in part mediated by PF4 [27]. Compared to WT mice, PF4−/− mice have improved survival, decreased inflammation, and less cerebral trafficking of T-cells and monocytes [27,28]. These data, and that of other labs, suggest that platelet antagonists may have potential utility in the treatment and prevention of cerebral malaria. Caution is needed however, as others have demonstrated that platelets may be able to directly kill intraerythrocytic parasites in uncomplicated infections [29]. Perhaps platelets have differential roles in response to different Plasmodium species or physiologic outcomes of infection.
Rheumatoid arthritis (RA) is a debilitating inflammatory joint disease. The pathogenesis of RA is not well understood and likely multi-factorial. In a recent paper platelets were shown to help drive the pathogenesis of RA in a mouse model [30]. In particular platelet derived microparticles were recovered from RA joints and contributed IL-1 to incite the inflammatory response. Blocking the signaling of IL-1 reduced lesion development. Similarly, transplant rejection begins at the vascular-immune interface and platelets may have a functional role in driving graft rejection. Platelet derived soluble CD154 in a cardiac allograft model has been shown sufficient to initiate cardiac allograft rejection independent of any cellular source for this molecule [31]. We have also found that platelets sustain vascular inflammation and recruit leukocytes to the transplant vasculature [32]. Can platelet inhibitors be useful in the treatment of RA and as adjunctive therapy for the prolongation of graft survival? More study is needed to address these potential clinical utilities for platelet inhibitors.
Current Clinical State of Anti-Platelet Therapies
While platelet investigators have made great strides in identifying new platelet activation pathways and relevant disease processes, clinical anti-platelet therapy has continued to focus on acute coronary syndromes. The search for agents that interfere with platelet aggregation and thrombus formation has been driven by percutaneous coronary innovations, with a particular focus on preventing thrombosis following stent implantation. This has led to the search for new classes and regimes of anti-platelet drugs that are currently in use and others that are being investigated. We will briefly discuss the current state of common anti-platelet therapies as well as emerging and investigational agents.
Aspirin
Aspirin is the current cornerstone of anti-platelet therapies. It acts by permanently inactivating prostaglandin H synthase 1 and synthase 2, or cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) as they are otherwise known. This inhibition leads to decreased synthesis of thromboxane A2 (TxA2) and subsequently TxA2-induced platelet aggregation [33]. Numerous studies have demonstrated the effectiveness of aspirin in the treatment and prevention of acute coronary syndromes, including myocardial infarction, stroke and peripheral artery disease [34].
An important issue in aspirin therapy is the concept of “aspirin resistance”, a term coined to describe the lack of a measureable change in platelet function ex vivo, inhibited TxA2 synthesis in vivo, or preventing thrombotic events in individuals [33]. Such patients are at higher risk of ischemic events. It has been suggested that these select patients may require more than the recommended 81 mg of aspirin daily to achieve an ideal protective effect, although it’s also been observed that other factors may be at play such as drug interactions and TxA2 production from other sources [34,33].
Thienopyridines
Along with aspirin, thienopyridines are the other common class of anti-platelet agents in clinical use today. After activation by the cytochrome P450 system, these drugs bond with the P2Y12 receptor on the platelet surface. ADP has an important role in the amplification of platelet aggregation and thienopyridines block the binding of ADP to the P2Y12 receptor. This class of drugs includes ticlopidine, clopidogrel, and prasugrel [34].
Ticlopidine is the prototype thienopyridine and its use was shown to be effective in treating stroke and myocardial infarction, particularly in combination with aspirin. However, it was also found to be linked to thrombotic thrombocytopenia purpura, neutropenia, and bone marrow aplasia [35,34]. Thus, it has since given way to the newer thienopyridine clopidogrel. The Clopidogrel Aspirin Stent Interventional Cooperative Study (CLASSICS) demonstrated clopidogrel to be as effective as ticlopidine with a lower incidence of side effects, and it has since become the first-line thienopyridine for dual anti-platelet therapy with aspirin [36].
As with aspirin, there is a subset of patients who harbor “resistance” or “non-responsiveness” to clopidogrel. The prevalence of clopidogrel resistance has been estimated by various sources to be between 8 and 30%, but depends on dose and time from ingestion. Studies suggests that this is due to insufficient levels of an active metabolite, possibly due to variations in P450 activity, as well as a genetic predisposition to decreased intestinal absorption [37,38]. There is some evidence that a 600 mg loading dose of clopidogrel, rather than the standard 300 mg loading dose, may be an effective strategy to counter clopidogrel resistance. Smaller studies have shown the effect of a maintenance dose of 150 mg, rather than the standard 75mg daily, as also promising for those resistant to clopidogrel [39,40]. The CURRENT/OASIS 7 study randomized over 25,000 patients with acute coronary syndrome to a 600 mg loading dose followed by a 150 mg maintenance dose for 7 days. In the two-thirds of the group that underwent percutaneous coronary intervention (PCI), in-stent thrombosis was reduced by 30% and myocardial infarction was reduced by 22% compared to the standard dose group. However, there was no benefit to the high dose in the group who did not undergo PCI [41]. Studies such as these demonstrate the need to learn more regarding the need to tailor therapy even with these very established anti-platelet drugs.
Prasugrel is a recently FDA approved third-generation thienopyridine for use in acute coronary syndromes treated with PCI. Phase I and II studies have shown that prasugrel has rapid effect onset, and generation of active metabolites superior to clopidogrel with less non-responsiveness and greater ADP inhibition. Prasugrel was approved after the TRITON-TIMI 38 trial revealed that it was superior to clopidogrel when combined with aspirin in reducing the primary endpoints of cardiovascular death, non-fatal MI, and nonfatal stroke [42,43,34]. However, it is associated with a significantly increased bleeding risk.
Glycoprotein IIb/IIIa Inhibitors
GPIIb/IIIa is a platelet receptor that has a central role in aggregation by cross-linking platelets via fibrinogen and is the target of a potent class of anti-thrombotic agents. These inhibitors are administered by intravenous route only, and are limited to acute settings, typically patients with acute coronary syndromes or who are undergoing PCI. The three drugs in this class are abciximab, a monoclonal antibody against the IIb/IIIa receptor, eptifibatide and tirofiban, peptide and non-peptide receptor inhibitors respectively [44]. All three of these agents have been tested extensively in clinical trials, and meta-analyses have shown benefit versus placebo. The TARGET trial is one of the few studies to directly compare these agents, having compared abciximab and tirofiban directly in patients undergoing PCI. Although abciximab was found superior at 30 days in various composite end points, at six months there was no significant difference. A sub-group analysis found that abciximab’s benefit was seen primarily in patients with acute coronary syndrome undergoing PCI at both 30 days and six months, but not at one year. This has been attributed to the dosing regimen of abciximab (a large bolus followed by continuous infusion) used in the trial, as well as its high degree of activity in the first hour after initiation, a critical period following PCI [44,45]. There is no evidence currently that abciximab is superior to tirofiban over the long term, although it does have more data from large clinical trials supporting its use. Eptifibitide’s advantage lies in its cost and its early onset of anti-platelet action similar to abciximab. Bleeding is a major complication with this class of drugs. Thrombocytopenia is the other major problem associated with these drugs, which appears to be more associated with abciximab than tirofiban or eptifibitide [46].
Emerging Therapies
There are currently compounds in development and clinical trials as platelet antagonists. Elinogrel is an investigational P2Y12 receptor inhibitor, which unlike other thienopyridines, is reversible in its blockade of ADP-mediated platelet aggregation with near complete reversal within 24 hours of administration [47]. Likewise, Ticagrelor is a cyclopentyltriazolopyrimidine derivative currently awaiting FDA approval that also reversibly acts on the P2Y12 receptor [48]. Protease-activated-receptor-1 (PAR-1) is another target of new drug development. In addition to its functions in the coagulation cascade, thrombin activates platelets by cleavage of PAR receptors. There are now two PAR-1 inhibitors under investigation, SCH530348 and E5555. SCH530348 is currently in two large, multi-center Phase III trials while E5555 in currently under Phase II study [49,50]. It remains to be seen if the promises of either compound gain approval for general clinical use.
The omega-3 fatty acids, eicosapentaenoic acid and docosahexaenoic acid, are found in oily fish and are compounds known to have anti-platelet effects. Although previous research into their potential anti-platelet effects has been inconclusive, a possible synergistic effect with standard dual anti-platelet therapy, namely aspirin and clopidogrel, may exist. Thus far a few small studies have shown that the addition of omega-3 fatty acids to clopidogrel and aspirin or aspirin alone were able to potentiate platelet response to these agents as measured by ex vivo platelet assays [51–53]. A retrospective study looked at patients treated with high-dose fish oil, aspirin and clopidogrel and found no statistically significant association with bleeding events compared to matched controls [54]. The approach of combining these omega-3 fatty acids with aspirin and clopidogrel appears to be safe and may represent a simple new means to increase responses to standard anti-platelet therapies without a large increase in bleeding risk.
Summary
Our knowledge of platelet activation pathways and platelet functions has greatly expanded in the past decade. The emerging role of platelets in driving vascular inflammation has likewise seen a great increase in significant publications. These findings will continue to fuel the development of platelet antagonists and the application of these drugs to many new disease processes.
Acknowledgments
Craig N. Morrell is supported by National Institutes of Health grants HL094547, HL093179, and R01HL093179-02S109. Dr. Block is supported by National Institutes of Health grant 1R21HL102582-01 and a research grant from GlaxoSmithKline. Dr. Ombrello is supported by 2T32HL007937-11 through the National Heart, Lung, and Blood Institute.
This publication was also made possible by Grant Number KL2 RR 024136 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. Information on NCRR is available at http://www.ncrr.nih.gov/. Information on Re-engineering the Clinical Research Enterprise can be obtained from http://nihroadmap.nih.gov/clinicalresearch/overview-translational.asp.
References
- 1.Andrews RK, Berndt MC. Platelet physiology and thrombosis. Thromb Res. 2004;114 (5–6):447–453. doi: 10.1016/j.thromres.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 2.Buller HR, Ten Cate T. Coagulation and platelet activation pathways. A review of the key components and the way in which these can be manipulated. Eur Heart J. 1995;16(Suppl L):8–10. doi: 10.1093/eurheartj/16.suppl_l.8. [DOI] [PubMed] [Google Scholar]
- 3.Brass LF, Zhu L, Stalker TJ. Minding the gaps to promote thrombus growth and stability. J Clin Invest. 2005;115 (12):3385–3392. doi: 10.1172/JCI26869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Catella-Lawson F. Vascular biology of thrombosis: Platelet-vessel wall interactions and aspirin effects. Neurology. 2001;57 (5 Suppl 2):S5–7. doi: 10.1212/wnl.57.suppl_2.s5. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald DJ. Vascular biology of thrombosis: The role of platelet-vessel wall adhesion. Neurology. 2001;57 (5 Suppl 2):S1–4. doi: 10.1212/wnl.57.suppl_2.s1. [DOI] [PubMed] [Google Scholar]
- 6.Fuse I. Disorders of platelet function. Crit Rev Oncol Hematol. 1996;22 (1):1–25. doi: 10.1016/1040-8428(94)00167-7. [DOI] [PubMed] [Google Scholar]
- 7.Coughlin SR. Molecular mechanisms of thrombin signaling. Semin Hematol. 1994;31 (4):270–277. [PubMed] [Google Scholar]
- 8.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3 (8):1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 9.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115 (12):3378–3384. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massberg S, Konrad I, Schurzinger K, Lorenz M, Schneider S, Zohlnhoefer D, Hoppe K, Schiemann M, Kennerknecht E, Sauer S, Schulz C, Kerstan S, Rudelius M, Seidl S, Sorge F, Langer H, Peluso M, Goyal P, Vestweber D, Emambokus NR, Busch DH, Frampton J, Gawaz M. Platelets secrete stromal cell-derived factor 1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J Exp Med. 2006;203 (5):1221–1233. doi: 10.1084/jem.20051772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gawaz M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res. 2004;61 (3):498–511. doi: 10.1016/j.cardiores.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 12.Langer H, May AE, Daub K, Heinzmann U, Lang P, Schumm M, Vestweber D, Massberg S, Schonberger T, Pfisterer I, Hatzopoulos AK, Gawaz M. Adherent platelets recruit and induce differentiation of murine embryonic endothelial progenitor cells to mature endothelial cells in vitro. Circ Res. 2006;98 (2):e2–10. doi: 10.1161/01.RES.0000201285.87524.9e. [DOI] [PubMed] [Google Scholar]
- 13.von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, Weber C. Rantes deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. 2001;103 (13):1772–1777. doi: 10.1161/01.cir.103.13.1772. [DOI] [PubMed] [Google Scholar]
- 14.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, Fratto CM, Tolley E, Kraiss LW, McIntyre TM, Zimmerman GA, Weyrich AS. Escaping the nuclear confines: Signal-dependent pre-mrna splicing in anucleate platelets. Cell. 2005;122 (3):379–391. doi: 10.1016/j.cell.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landry P, Plante I, Ouellet DL, Perron MP, Rousseau G, Provost P. Existence of a microrna pathway in anucleate platelets. Nat Struct Mol Biol. 2009;16 (9):961–966. doi: 10.1038/nsmb.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun H, Swaim A, Herrera JE, Becker D, Becker L, Srivastava K, Thompson LE, Shero MR, Perez-Tamayo A, Suktitpat B, Mathias R, Contractor A, Faraday N, Morrell CN. Platelet kainate receptor signaling promotes thrombosis by stimulating cyclooxygenase activation. Circ Res. 2009 doi: 10.1161/CIRCRESAHA.109.198861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrell CN, Sun H, Ikeda M, Beique JC, Swaim AM, Mason E, Martin TV, Thompson LE, Gozen O, Ampagoomian D, Sprengel R, Rothstein J, Faraday N, Huganir R, Lowenstein CJ. Glutamate mediates platelet activation through the ampa receptor. J Exp Med. 2008;205 (3):575–584. doi: 10.1084/jem.20071474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrarese C, Sala G, Riva R, Begni B, Zoia C, Tremolizzo L, Galimberti G, Millul A, Bastone A, Mennini T, Balzarini C, Frattola L, Beghi E. Decreased platelet glutamate uptake in patients with amyotrophic lateral sclerosis. Neurology. 2001;56 (2):270–272. doi: 10.1212/wnl.56.2.270. [DOI] [PubMed] [Google Scholar]
- 19.Berk M, Plein H, Ferreira D. Platelet glutamate receptor supersensitivity in major depressive disorder. Clin Neuropharmacol. 2001;24 (3):129–132. doi: 10.1097/00002826-200105000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Huo Y, Ley KF. Role of platelets in the development of atherosclerosis. Trends Cardiovasc Med. 2004;14 (1):18–22. doi: 10.1016/j.tcm.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein e. Nat Med. 2003;9 (1):61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 22.Michiels JJ, Gawaz M. Preface: Platelets in inflammation and atherothrombosis. Semin Thromb Hemost. 2007;33 (2):119–122. doi: 10.1055/s-2007-969024. [DOI] [PubMed] [Google Scholar]
- 23.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet cd36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13 (9):1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen K, Febbraio M, Li W, Silverstein RL. A specific cd36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res. 2008;102 (12):1512–1519. doi: 10.1161/CIRCRESAHA.108.172064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sachais BS, Turrentine T, Dawicki McKenna JM, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (pf4) from platelets reduces atherosclerosis in c57bl/6 and apoe−/− mice. Thromb Haemost. 2007;98 (5):1108–1113. [PubMed] [Google Scholar]
- 26.Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15 (1):97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- 27.Srivastava K, Cockburn IA, Swaim A, Thompson LE, Tripathi A, Fletcher CA, Shirk EM, Sun H, Kowalska MA, Fox-Talbot K, Sullivan D, Zavala F, Morrell CN. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe. 2008;4 (2):179–187. doi: 10.1016/j.chom.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava K, Field DJ, Aggrey A, Yamakuchi M, Morrell CN. Platelet factor 4 regulation of monocyte klf4 in experimental cerebral malaria. PLoS One. 2010;5(5):e10413. doi: 10.1371/journal.pone.0010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMorran BJ, Marshall VM, de Graaf C, Drysdale KE, Shabbar M, Smyth GK, Corbin JE, Alexander WS, Foote SJ. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science. 2009;323 (5915):797–800. doi: 10.1126/science.1166296. [DOI] [PubMed] [Google Scholar]
- 30.Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O’Donnell E, Farndale RW, Ware J, Lee DM. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 327(5965):580–583. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu H, Zhang X, Mannon RB, Kirk AD. Platelet-derived or soluble cd154 induces vascularized allograft rejection independent of cell-bound cd154. J Clin Invest. 2006;116 (3):769–774. doi: 10.1172/JCI27155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrell CN, Murata K, Swaim AM, Mason E, Martin TV, Thompson LE, Ballard M, Fox-Talbot K, Wasowska B, Baldwin WM., 3rd In vivo platelet-endothelial cell interactions in response to major histocompatibility complex alloantibody. Circ Res. 2008;102 (7):777–785. doi: 10.1161/CIRCRESAHA.107.170332. [DOI] [PubMed] [Google Scholar]
- 33.Patrono C, Garcia Rodriguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353 (22):2373–2383. doi: 10.1056/NEJMra052717. [DOI] [PubMed] [Google Scholar]
- 34.Gurbel PA, Tantry US. Combination antithrombotic therapies. Circulation. 2010;121 (4):569–583. doi: 10.1161/CIRCULATIONAHA.109.853085. [DOI] [PubMed] [Google Scholar]
- 35.Symeonidis A, Kouraklis-Symeonidis A, Seimeni U, Galani A, Giannakoulas N, Fragopanagou E, Tiniakou M, Matsouka P, Zoumbos N. Ticlopidine-induced aplastic anemia: Two new case reports, review, and meta-analysis of 55 additional cases. Am J Hematol. 2002;71 (1):24–32. doi: 10.1002/ajh.10150. [DOI] [PubMed] [Google Scholar]
- 36.Bertrand ME, Rupprecht HJ, Urban P, Gershlick AH. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting : The clopidogrel aspirin stent international cooperative study (classics) Circulation. 2000;102 (6):624–629. doi: 10.1161/01.cir.102.6.624. [DOI] [PubMed] [Google Scholar]
- 37.Staritz P, Kurz K, Stoll M, Giannitsis E, Katus HA, Ivandic BT. Platelet reactivity and clopidogrel resistance are associated with the h2 haplotype of the p2y12-adp receptor gene. Int J Cardiol. 2009;133 (3):341–345. doi: 10.1016/j.ijcard.2007.12.118. [DOI] [PubMed] [Google Scholar]
- 38.De Miguel A, Ibanez B, Badimon JJ. Clinical implications of clopidogrel resistance. Thromb Haemost. 2008;100 (2):196–203. [PubMed] [Google Scholar]
- 39.Bonello L, Camoin-Jau L, Armero S, Com O, Arques S, Burignat-Bonello C, Giacomoni MP, Bonello R, Collet F, Rossi P, Barragan P, Dignat-George F, Paganelli F. Tailored clopidogrel loading dose according to platelet reactivity monitoring to prevent acute and subacute stent thrombosis. Am J Cardiol. 2009;103 (1):5–10. doi: 10.1016/j.amjcard.2008.08.048. [DOI] [PubMed] [Google Scholar]
- 40.Tavassoli N, Voisin S, Carrie D, Lapeyre-Mestre M, Galinier M, Montastruc JL, Pathak A. High maintenance dosage of clopidogrel is associated with a reduced risk of stent thrombosis in clopidogrel-resistant patients. Am J Cardiovasc Drugs. 2010;10 (1):29–35. doi: 10.2165/11318260-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 41.Mehta SR, Bassand JP, Chrolavicius S, Diaz R, Fox KA, Granger CB, Jolly S, Rupprecht HJ, Widimsky P, Yusuf S. Design and rationale of current-oasis 7: A randomized, 2 × 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with st and non-st-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J. 2008;156 (6):1080–1088. doi: 10.1016/j.ahj.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 42.Wiviott SD, Braunwald E, McCabe CH, Horvath I, Keltai M, Herrman JP, Van de Werf F, Downey WE, Scirica BM, Murphy SA, Antman EM. Intensive oral antiplatelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary syndromes treated with percutaneous coronary intervention and stenting in the triton-timi 38 trial: A subanalysis of a randomised trial. Lancet. 2008;371 (9621):1353–1363. doi: 10.1016/S0140-6736(08)60422-5. [DOI] [PubMed] [Google Scholar]
- 43.Michelson AD, Frelinger AL, 3rd, Braunwald E, Downey WE, Angiolillo DJ, Xenopoulos NP, Jakubowski JA, Li Y, Murphy SA, Qin J, McCabe CH, Antman EM, Wiviott SD. Pharmacodynamic assessment of platelet inhibition by prasugrel vs. Clopidogrel in the triton-timi 38 trial. Eur Heart J. 2009;30 (14):1753–1763. doi: 10.1093/eurheartj/ehp159. [DOI] [PubMed] [Google Scholar]
- 44.Moliterno DJ. Advances in antiplatelet therapy for acs and pci. J Interv Cardiol. 2008;21(Suppl 1):S18–24. doi: 10.1111/j.1540-8183.2008.00409.x. [DOI] [PubMed] [Google Scholar]
- 45.Roffi M, Moliterno DJ, Meier B, Powers ER, Grines CL, DiBattiste PM, Herrmann HC, Bertrand M, Harris KE, Demopoulos LA, Topol EJ. Impact of different platelet glycoprotein iib/iiia receptor inhibitors among diabetic patients undergoing percutaneous coronary intervention: : Do tirofiban and reopro give similar efficacy outcomes trial (target) 1-year follow-up. Circulation. 2002;105 (23):2730–2736. doi: 10.1161/01.cir.0000018123.02672.c7. [DOI] [PubMed] [Google Scholar]
- 46.Tuhta AG, Yesildag O, Koprulu D. Tirofiban-associated acute thrombocytopenia. Acta Cardiol. 2006;61 (5):577–579. doi: 10.2143/AC.61.5.2017776. [DOI] [PubMed] [Google Scholar]
- 47.Berger JS, Roe MT, Gibson CM, Kilaru R, Green CL, Melton L, Blankenship JD, Metzger DC, Granger CB, Gretler DD, Grines CL, Huber K, Zeymer U, Buszman P, Harrington RA, Armstrong PW. Safety and feasibility of adjunctive antiplatelet therapy with intravenous elinogrel, a direct-acting and reversible p2y12 adp-receptor antagonist, before primary percutaneous intervention in patients with st-elevation myocardial infarction: The early rapid reversal of platelet thrombosis with intravenous elinogrel before pci to optimize reperfusion in acute myocardial infarction (erase mi) pilot trial. Am Heart J. 2009;158 (6):998–1004. doi: 10.1016/j.ahj.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 48.Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, Mahaffey KW, Scirica BM, Skene A, Steg PG, Storey RF, Harrington RA, Freij A, Thorsen M. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361 (11):1045–1057. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- 49.Goto S, Yamaguchi T, Ikeda Y, Kato K, Yamaguchi H, Jensen P. Safety and exploratory efficacy of the novel thrombin receptor (par-1) antagonist sch530348 for non-st-segment elevation acute coronary syndrome. J Atheroscler Thromb. 2010;17 (2):156–164. doi: 10.5551/jat.3038. [DOI] [PubMed] [Google Scholar]
- 50.Serebruany VL, Kogushi M, Dastros-Pitei D, Flather M, Bhatt DL. The in-vitro effects of e5555, a protease-activated receptor (par)-1 antagonist, on platelet biomarkers in healthy volunteers and patients with coronary artery disease. Thromb Haemost. 2009;102 (1):111–119. doi: 10.1160/TH08-12-0805. [DOI] [PubMed] [Google Scholar]
- 51.Gajos G, Rostoff P, Undas A, Piwowarska W. Effects of polyunsaturated omega-3 fatty acids on responsiveness to dual antiplatelet therapy in patients undergoing percutaneous coronary intervention: The omega-pci (omega-3 fatty acids after pci to modify responsiveness to dual antiplatelet therapy) study. J Am Coll Cardiol. 2010;55 (16):1671–1678. doi: 10.1016/j.jacc.2009.11.080. [DOI] [PubMed] [Google Scholar]
- 52.Lev EI, Leshem-Lev D, Mager A, Vaknin-Assa H, Harel N, Zimra Y, Bental T, Greenberg G, Dvir D, Solodky A, Assali A, Battler A, Kornowski R. Circulating endothelial progenitor cell levels and function in patients who experienced late coronary stent thrombosis. Eur Heart J. 2010:ehq184. doi: 10.1093/eurheartj/ehq184. [pii] [DOI] [PubMed] [Google Scholar]
- 53.Lev EI, Solodky A, Harel N, Mager A, Brosh D, Assali A, Roller M, Battler A, Kleiman NS, Kornowski R. Treatment of aspirin-resistant patients with omega-3 fatty acids versus aspirin dose escalation. J Am Coll Cardiol. 2010;55 (2):114–121. doi: 10.1016/j.jacc.2009.08.039. [DOI] [PubMed] [Google Scholar]
- 54.Watson PD, Joy PS, Nkonde C, Hessen SE, Karalis DG. Comparison of bleeding complications with omega-3 fatty acids + aspirin + clopidogrel--versus--aspirin + clopidogrel in patients with cardiovascular disease. Am J Cardiol. 2009;104 (8):1052–1054. doi: 10.1016/j.amjcard.2009.05.055. [DOI] [PubMed] [Google Scholar]