

Abstract
Objective
Genome-wide association (GWAS) methods have identified genes contributing to Parkinson disease (PD); we sought to identify additional genes associated with PD susceptibility.
Methods
A two stage design was used. First, individual level genotypic data from five recent PD GWAS (Discovery Sample: 4,238 PD cases and 4,239 controls) were combined. Following imputation, a logistic regression model was employed in each dataset to test for association with PD susceptibility and results from each dataset were meta-analyzed. Second, 768 SNPs were genotyped in an independent Replication Sample (3,738 cases and 2,111 controls).
Results
Genome-wide significance was reached for SNPs in SNCA (rs356165, G: odds ratio (OR)=1.37; p=9.3 × 10−21), MAPT (rs242559, C: OR=0.78; p=1.5 × 10−10), GAK/DGKQ (rs11248051, T:OR=1.35; p=8.2 × 10−9/ rs11248060, T: OR=1.35; p=2.0×10−9), and the HLA region (rs3129882, A: OR=0.83; p=1.2 × 10−8), which were previously reported. The Replication Sample confirmed the associations with SNCA, MAPT, and the HLA region and also with GBA (E326K OR=1.71; p=5 × 10−8 Combined Sample) (N370 OR=3.08; p=7 × 10−5 Replication sample). A novel PD susceptibility locus, RIT2, on chromosome 18 (rs12456492; p=5 × 10−5 Discovery Sample; p=1.52 × 10−7 Replication sample; p=2 × 10−10 Combined Sample) was replicated. Conditional analyses within each of the replicated regions identified distinct SNP associations within GBA and SNCA, suggesting that there may be multiple risk alleles within these genes.
Interpretation
We identified a novel PD susceptibility locus, RIT2, replicated several previously identified loci, and identified more than one risk allele within SNCA and GBA.
Introduction
Parkinson disease (PD) is the second most common adult-onset neurodegenerative disorder worldwide.1 Five genes have been identified with mutations that result in Mendelian forms of PD; however, mutations have been found in fewer than 5% of individuals with PD, suggesting additional genes contribute to disease risk.2 Many candidate gene studies and several GWAS have been performed to identify risk factors for PD, with growing evidence for the role of SNCA, MAPT, GBA, GAK/DGKQ, and the HLA region in disease susceptibility.3–12 Two recent studies found evidence for association with additional loci including ACMSD, STK39, MCCC1/LAMP3, SYT11, CCDC62/HIP1R, STX1B, FGF20, STBD1, GPNMB and PARK16.11,13 However, there is evidence that there are additional loci yet to be identified.
SUBJECTS AND METHODS
Discovery Sample
To identify additional genes associated with PD, we combined publicly available genotype level GWAS data obtained from dbGaP4,6 along with two new datasets that are not yet publicly available and were obtained directly from the investigator who performed the GWAS.5,7,8. All datasets employed standard UK Brain Bank criteria14 for the diagnosis of PD, with a modification to allow the inclusion of cases that had a family history of PD. This modification was made because it is believed that familial PD cases may have a stronger genetic contribution than sporadic PD, making them potentially more informative for genetic studies. PD cases with a reported age of onset below 18 years of age were removed from the dataset (n=17). When data were available, any PD cases known to harbor a causative mutation, either two parkin mutations or a single LRRK2 mutation, were excluded from further analysis (n=57).
An Illumina genotyping array was used by all studies. Individual level genotypic data was available and reviewed across studies to identify sample duplicates (see Supplemental Methods). Prior to performing imputation, each study was subjected to rigorous quality review and data cleaning (see Supplemental Methods for more details) and principal component analysis was used to control for population stratification. Imputation was then performed for all autosomes using MACH 1.0.15 The 2.5 million HapMap2 SNPs were analyzed using ProbABEL (http://mga.bionet.nsc.ru/~yurii/ABEL/) and a logistic regression model, that included sex and age, when appropriate (see Supplemental Methods). Meta-analysis was performed with METAL (http://www.sph.umich.edu/csg/abecasis/Metal/) using an inverse-variance weighting scheme. This allowed an overall effect size to be estimated. Genomic control was employed so that results were down-weighted if the study’s lambda exceeded 1.00. The Discovery Sample was large enough to have 80% power to detect relative risks as small as 1.14–1.18 with a relatively common risk allele (MAF 0.2–0.35).
SNP Selection for Replication Genotyping
A custom Illumina genotyping array was designed with 768 SNPs that included: Known causative mutations: SNPs that genotyped two common LRRK2 mutations in European populations (G2019S and R1441H); Known risk factors: GBA (N370S, L444P, E326K, T369M); Previous GWAS associations: PARK16, LRRK26,12, SNCA5–8,12, MAPT5–8, GAK5,8, the HLA region8; Sex confirmation: 3 SNPs on the Y chromosome and 6 SNPs on the X chromosome in addition to the sex-specific probes included in the GoldenGate custom oligonucleotide pool; Top priority association results from the meta-analysis: SNPs were selected based on increasing p-value. A SNP was removed from consideration if it was in linkage disequilibrium (LD) (r2>0.80) with a SNP having a smaller p-value or had an Illumina design score less than 0.40 (if p<1 × 10−5) or 0.60 (if p≥1 × 10−5). This approach identified 619 SNPs (all p < 3.2 × 10−4). In addition, 28 additional SNPs were selected in the highest priority regions (p<1 × 10−5), in case one of the SNPs in these regions failed quality assessment after being genotyped on the replication array (e.g., call rate <0.98, divergence from HWE in controls p<0.0001). Ancestry informative markers (AIMs): SNPs were selected based on fixation indices (FST) between the Ashkenazi and British population clusters as defined using annotated results from Eigenstrat (see Supplemental Figure 1). Markers were then ranked based on how well they differentiated between the two subpopulations, and 100 were selected in a manner similar to the 619 replication SNPs. A SNP was excluded from further consideration if it was in LD (r2>0.05) with any marker with a larger FST, or if it had an Illumina design score less than 0.80. Samples were genotyped by the Genetic Resource Core Facility SNP Center at Johns Hopkins University using Illumina GoldenGate chemistry16 and a custom panel of 768 SNPs (GS0012520-OPA) (see Supplemental Methods).
Replication Sample
The independent Replication Sample included 3,738 PD cases and 2,111 controls. Samples were obtained either from an established repository (Coriell Repositories or National Cell Repository for Alzheimer Disease) that assured the samples had appropriate consent for sample and data sharing or directly from the investigator who had collected the sample, and whose study was approved by the appropriate Human Subject Committee at their institution. All samples included in the Replication Sample were reported as white, non-Hispanic. All cases underwent a neurological evaluation that employed PD diagnostic criteria based broadly on the United Kingdom PD Society Brain Bank Criteria,17 although modified to allow a positive family history of PD. Three cases reported an age of onset ≤18 years and were excluded from further study. When information was available, cases were excluded if they were known to harbor a causative mutation (either 2 parkin mutations or a single LRRK2 mutation). Controls were selected, when possible, from the same study that also provided cases. Based on self-report, the control subjects did not have a personal history of PD.
The first level of data review focused on genotyping quality (SNP completeness). The second level focused on which samples and which SNPs would be included in analyses. The multidimensional scaling (MDS) algorithm implemented in PLINK was performed using the 100 AIMs and all other independent SNPs (SNPs with r2>0.30 were not included) to confirm that all samples were indeed white and non-Hispanic. Samples with a LRRK2 mutation were removed from further analysis, as were any that were cryptically identical to an individual in the Discovery sample. More details are available in the Supplemental Methods.
We utilized the same logistic regression model used in the initial meta-analysis to analyze the Replication Sample. The initial analysis included the 619 SNPs designed to replicate our top priority association results. Unlike the Discovery Sample in which each study included both cases and controls, the Replication Sample included some studies providing both cases and controls, while others provided only cases or only controls. Therefore, we could not analyze each sample separately as we had in the Discovery Sample analyses. Rather, the entire Replication Sample was analyzed together. The mean age at exam of the controls was later than the mean age at onset of the cases; therefore, we did not include age in the logistic regression model. There were statistically significant sex differences between the cases and controls. Therefore, the final analytic model included both sex as well as one principal component to adjust for the population stratification due to the disproportionate Ashkenazi Jewish ancestry of the cases. All analyses were performed using PLINK. Odds ratios and p-values were computed to assess the strength of the association. After excluding AIMs and considering linkage disequilibrium between SNPs as implemented in SimpleM,18 there were 530 effectively independent tests, requiring a corrected threshold of p < 9 × 10−5 for an association to be considered replicated in the Replication Sample
Joint Analyses
We performed a meta-analysis to combine the results of the independent Discovery and Replication Samples only for the SNPs successfully genotyped in the Replication Sample. We used the same analytic approach as in the Discovery Sample. An association was considered statistically significant if the p-value in the joint analyses exceeded genome-wide significance (p< 5 × 10−8).
To test the hypothesis that there might be more than one risk variant in a particular gene or gene region contributing to the association, we performed conditional analyses. For each statistically significant gene/region, we identified the SNP with the most extreme p-value in the combined Discovery and Replication samples. We then modified the logistic regression model to include not only sex and the principal component covariate, but also the genotype at the most significant SNP. We then reviewed the p-value for the other SNPs in the gene/region to determine if any other SNPs remained statistically significant (gene-wide empirical p<0.05 using permutation testing) after adjusting for the effect of the most significant SNP. In this way, we could identify genes/regions in which more than one SNP provided distinct evidence of association with PD susceptibility.
Ingenuity Pathway Analysis (IPA) software was used to search for biological relationships among the genes meeting genome-wide significance. A gene list (DGKQ, GAK, the HLA region, MAPT, SNCA, and RIT2) was entered into a “My Pathway” analysis in IPA. Restricting species to human and allowing for findings among chemicals, the Path Explorer tool under the Build tab was used to search among the Ingenuity knowledge base and external databases to identify the shortest pathways among the genes with either no or one intervening molecule. Links between genes represent protein-protein interactions or indicate one gene influences phosphorylation of the connected gene.
RESULTS
Discovery Sample
The final Discovery Sample used in the meta-analysis included 4,238 PD cases and 4,239 controls (Table 1). Meta-analysis was performed combining the results from each dataset to identify SNPs associated with PD susceptibility (see Figure 1; Table 2). Genome-wide significance (p<5 × 10−8) was reached for SNPs in SNCA (rs356165; OR=1.37; p=9.3 × 10−21), MAPT (rs242559; OR=0.77; p=1.5 × 10−10), GAK (rs11248051; OR=1.35; p=8.2 × 10−9)/DGKQ (rs11248060; OR=1.35; p=2.0x 10−9), and the HLA region (rs3129882; OR=1.21; p=1.2 × 10−8), which have been previously established in PD susceptibility. No other regions exceeded genome-wide thresholds of significance; however, 28 SNPs had association results with p<10−5 (see Supplemental Table 1 for complete results).
Table 1.
Summary properties of the studies included in the meta-analysis
| Variable | PROGENI/GenePD5 | NIA Phase I4 | NIA Phase II6 | HIHG7 | NGRC8 | All Studies |
|---|---|---|---|---|---|---|
| Platform | Illumina 370 | Illumina 250+300 | Illumina 550 | Illumina 610/1M/550 | Illumina Omni1 | |
| # of SNPsa | 324,989 | 514,260 | 521,070 | 484,712 | 788,882 | |
| Total N available | 1,739 | 523 | 1,206 | 1,262 | 3,986 | 8,716 |
| Cases used in analyses | 840 | 245 | 618 | 579 | 1,956 | 4,238 |
| Controls used in analyses | 862 | 256 | 520 | 619 | 1,982 | 4,239 |
| Lambda (genomic inflation) | 1.008 | 1.012 | 1.015 | 1.001 | 1.041 | |
| % of cases with family history of PD | 100.0% | 25.5% | 35.5% | 25.2% | 21.75 | |
| Age (direction) b | 4 × 10−29 (+) | 4 × 10−6 (−) | 0.003 (+) | 9 × 10−50 (−) | 8 × 10−128 (−) | 8 × 10−86 (−) |
| Male (direction) c | 6 × 10−16 (+) | 0.003 (+) | 5 × 10−12 (+) | 3 × 10−22 (+) | 3 × 10−72 (+) | 2 × 10−115 (+) |
Number of SNPs in filtered dataset prior to imputation. This is the number of unambiguous (i.e. no A/T or C/G pairings) SNPs passing all quality assessment.
A plus sign (+) for Age indicates an association between PD risk and older age. Those studies where the age at onset of the cases was significantly older than the age at exam of the controls are bolded; age was included as a covariate in these studies only.
A plus sign (+)_for Male indicates an association between older PD risk and male sex; sex was included as a covariate for all studies.
The individual level genotypes for PROGENI/GenePD, the NIA Phase I and II, and NGRC are all available through dbGaP.
Figure 1.
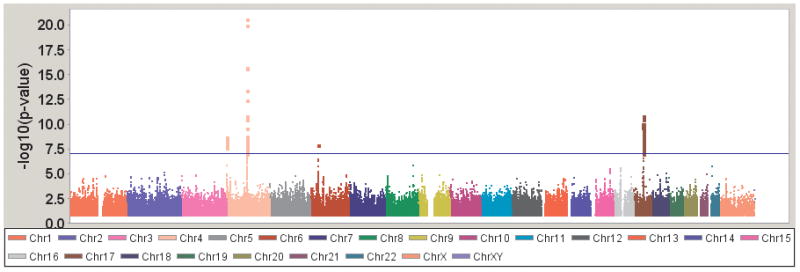
Genome-wide association results for PD susceptibility.
Table 2.
Five loci associated with PD at genome-wide significance in the Discovery Set
| Locus | Chr | SNP | bp | A1/A2 | A1 freq | Imp/geno1 | Odds Ratio | p-value | Direction of Effect in 5 studies2 |
|---|---|---|---|---|---|---|---|---|---|
| SNCA | 4q | rs356165 | 90856624 | G/A | 0.4099 | IIIII | 1.37 | 9 × 10−21 | +++++−−−−− |
| MAPT | 17q | rs242559 | 42198305 | C/A | 0.2165 | IIIIG | 0.78 | 1 × 10−10 | −−−−− |
| GAK | 4p | rs11248051 | 848332 | T/C | 0.1071 | GIIIG | 1.35 | 8 × 10−9 | +++++ |
| DGKQ | 4p | rs11248060 | 954359 | T/C | 0.1237 | GGGGG | 1.35 | 2 × 10−9 | +++++ |
| HLA region | 6p | rs3129882 | 32517508 | A/G | 0.4275 | GGGGG | 0.83 | 1 × 10−8 | −+ −−− |
Values for imputed (I) or genotyped (G) status; bold indicates genome-wide significance (p<5 × 10−8)
Direction of effects are listed in the following order: PROGENI/GenePD, NIA Phase I, NIA Phase II, HIHG, NGRC
Distinct clusters could be identified based on ancestry (see Supplemental Results). However, none of the genome-wide significant findings could be explained by ancestry. These were tested in three ways: 1) adjusting for principal components; 2) adjusting for cluster membership; and 3) stratifying by cluster membership.
Replication Sample
The Replication Sample is summarized in Table 3. Genotypes were successfully generated for 705 of 768 attempted SNPs (92%). The only notable SNP loss was the GBA L444P SNP, which failed to genotype presumably because of the homology with the neighboring pseudogene.
Table 3.
Replication Sample
| Study | # PD cases | # Controls | Total |
|---|---|---|---|
| Harvard NeuroDiscovery Center Biomarker Study26 | 441 | 247 | 658 |
| GenePD27–29 | 276 | 269 | 545 |
| PROGENI30 | 311 | 197 | 508 |
| Search31 | 357 | 150 | 507 |
| DATATOP32 | 359 | 0 | 359 |
| Partners | 358 | 0 | 358 |
| LOAD Study33 | 0 | 450 | 450 |
| Postcept34 | 318 | 2 | 320 |
| Core PD35 | 536 | 0 | 536 |
| JHU Udall | 125 | 0 | 125 |
| NetPD36 | 427 | 0 | 427 |
| Mayo Clinic Jacksonville | 74 | 87 | 161 |
| Other samples (from Coriell) | 186 | 709 | 895 |
| Total recruited | 3,738 | 2,111 | 5,849 |
| Cases | Controls | p-value | |
| Total number analyzed (n) | 3,223 | 2,035 | |
| Male:Female ratio | 2,069:1,154 | 897:1,138 | 1 × 10−46 |
| Age at Onset (case) | 56.3 +/− 12.2 | 1 × 10−78 A | |
| Age at evaluation | 65.6 +/− 10.2 | 64.1 +/− 15.2 | 0.0003 |
Comparison made to age at evaluation of controls.
Data was released for 5,794 study samples (>99% of attempted samples) and 123 blinded duplicate study samples. Detailed review of samples was performed to remove samples that were unexpected duplicates, poorly performing, or did not cluster as Caucasian, non-Hispanic (see Supplemental Results). All samples identified with a causative LRRK2 mutation were eliminated from further analysis (n=61 cases, 1 control).
Analysis of the Replication Sample confirmed the previously identified associations with SNCA, MAPT, the HLA region, and GBA (See Figure 2; complete results in Supplemental Table 2). Only the GAK/DGKQ region was not statistically significant (p=0.01). We replicated a novel locus on chromosome 18 within the RIT2 gene that is in LD with markers in nearby SYT4 (rs12456492; p=2 × 10−7; see Figure 3). Given the regional LD, determining if the underlying functional variation affects one gene product versus the other can be difficult to discern, as is the case with the GAK/DGKQ locus.
Figure 2. Manhattan plot of Results.
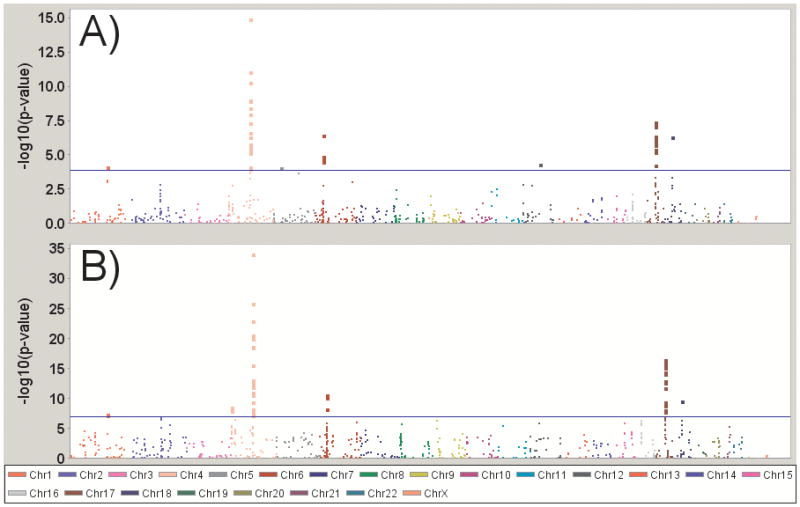
(A) Replication Sample results alone (B) Meta-analyzed with the Discovery Sample; the blue line indicates the study-wide significance level (p<9.4 × 10−5 for the replication stage alone, p<5 × 10−8 for the meta-analysis)
Figure 3. Forest plot of the novel RIT2 SNP (rs12456492).

Rsq values are a measure of imputation quality generated by MACH that range from 0 to 1, with 1 being highly accurate
Joint Analysis
With the power of the joint analysis of the Discovery and Replication Samples, GBA now reached genome-wide significance. Many additional SNPs in GAK/DGKQ, SNCA, the HLA region, and MAPT reached significance. Our newly identified locus, RIT2, also met genome-wide criteria in this joint analysis (OR=1.19; p=2 × 10−10) (Table 4).
Table 4.
Summary of the Statistically Significant SNPs from the Meta Analysis of the Discovery and Replication Samples and for Conditional Analyses of the Combined Sample
| Region | # of markers tested | Marker | Chr | Position | Alleles (Ref/Other) | Discovery
|
Replication
|
Combined Sample
|
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| p-value | Ref Freq | p-value | OR (95% CI) | # SNPs tagged | p-value | ||||||
| GBA | 7 | E326K | 1 | 153472791 | A/G | 2 × 10−5 | 0.017 | 0.0009 | 1.71 (1.55–1.89) | 2 | 5 × 10−8 |
| N370S† | 1 | 153472258 | C/T | NAa | 0.009 | 7 × 10−5 | 3.08 (2.32–4.09) | 2 | 7 × 10−5 | ||
| GAK | 13 | rs11248060 | 4 | 954359 | T/C | 1 × 10−9 | 0.131 | 0.045 | 1.26 (1.21–1.31) | 7 | 3 × 10−9 |
| SNCA | 33 | rs356220 | 4 | 90860363 | T/C | 9 × 10−21 | 0.414 | 1 × 10−15 | 1.38 (1.34–1.42) | 12 | 8 × 10−35 |
| rs356198† | 4 | 90901527 | A/G | 4 × 10−5 | 0.175 | 2 × 10−5 | 0.82 (0.79–0.84) | 3 | 5 × 10−9 | ||
| HLA region | 24 | rs2395163 | 6 | 32495787 | C/T | 3 × 10−7 | 0.197 | 1 × 10−5 | 0.81 (0.78–0.84) | 11 | 3 × 10−11 |
| MAPT | 40 | rs199515 | 17 | 42211804 | G/C | 2 × 10−11 | 0.187 | 4 × 10−7 | 0.76 (0.74–0.79) | 29 | 3 × 10−17 |
| RIT2 | 8 | rs12456492 | 18 | 38927378 | G/A | 4 × 10−5 | 0.340 | 5 × 10−7 | 1.19 (1.16–1.22) | 6 | 2 × −10 |
Indicates a conditional analysis that includes the most significant SNPs in the region as a covariate(additive model); odds ratios and p-values for these SNPs are from the conditional analysis. The same conditional analysis was then performed in the Discovery Sample and meta-analyzed.
N370S was not tagged in the discovery set and could not be included in a conditional analysis of that dataset.
To further explore the association results in each gene, we performed conditional analyses in the combined samples. We detected two distinct effects within the GBA locus (Table 4). The SNPs with the most extreme p-values in the Combined Sample were rs12726330 and the E326K variant, which both reached genome-wide significance (p=5 × 10−8). These two SNPs are in high LD with each other, so when one genotype is included in a logistic regression model, the other becomes non-significant. When E326K is included in the logistic model, another SNP remained statistically significant (N370S; p<7 × 10−5). All results included the principal component that accounts for Ashkenazi ancestry, which controls for the increased incidence of GBA mutations in the Ashkenazi population. Moreover, when individuals within the Ashkenazi cluster were excluded from the Discovery Sample and the Replication Sample, the association to rs12726330 and to E326K remained at genome-wide significance (Discovery Sample: 3,792 cases, 3,842 controls, p=2 × 10−5; Replication Sample: 3,025 cases, 1,931 controls, p=0.0005; Combined Sample: p<5 × 10−8).
We detected two distinct associations at the SNCA locus (Table 4); one association that is tagged by rs356220 and the other tagged by rs356198. The second association, tagged by rs356198, still exceeded genome-wide significance when conditioning on rs356220 (p=5 × 10−9). Our results are corroborated by other studies which identified independent associations within SNCA.9,10,12 See Supplementary Methods and Supplementary Table 6 for more information.
We assessed the biological relationships among the genome-wide significant genes identified in our study (Figure 4). Paths between genes represent protein-protein interactions or phosphorylation. This network suggests that GAK and RIT2 may be part of the same disease pathway as MAPT and SNCA, while DGKQ and the HLA region may influence risk of PD via another mechanism.
Figure 4.

Ingenuity Analysis
DISCUSSION
We performed a large meta-analysis including two studies not included in any reported meta-analysis. The Discovery and Replication samples were well characterized and established criteria were utilized for the diagnosis of PD. Both sporadic and familial PD cases were included. Cases with a known causative mutation were excluded (i.e. LRRK2 mutation; two parkin mutations). Using a rigorous two-stage design, we identified a novel locus, RIT2, associated with PD susceptibility. In addition, we also replicated loci previously associated with PD, including GAK, SNCA, the HLA region, and MAPT. Pathway analyses suggest that GAK and RIT2 may be part of the same disease pathway as MAPT and SNCA, while DGKQ and the HLA region may influence risk via another mechanism.
We detected genome-wide significant evidence of association to RIT2, a gene proposed in previous studies but which did not meet stringent statistical criteria as a risk factor for PD. The protein encoded by human RIT2 binds to the product of human calmodulin 1 (phosphorylase kinase, delta) CALM119. Of note, CALM1 binds to human SNCA and MAPT 20,21 Comparison of gene expression in brain tissue from neuropathologically confirmed PD cases and controls demonstrates reduced expression of RIT2 in the remaining portion of the substantia nigra.22 Results from our GWAS, pathway analysis and expression studies provide supporting biological evidence that RIT2 acts as a PD gene and suggest a starting point for functional analysis.
We also explored the role of GBA variants in PD susceptibility. E326K is sometimes considered a benign polymorphism, since in the homozygous or compound heterozygous state it is not sufficient to cause Gaucher disease. However, results of this study and a previous study23 indicate that E326K may be a susceptibility allele for PD. Most previous GWAS have not included all known GBA mutations in their analyses; for example, N370S is not included or tagged by GWAS arrays. However, we did ensure that this mutation was genotyped in our Replication Sample. Therefore, we were able to test in our Replication Sample for the association of GBA mutations and variants with PD susceptibility and then could utilize conditional analyses to determine that it was likely that there is more than one genetic factor in GBA influencing disease risk. Thus, our results suggest that additional analyses and potential functional studies are warranted to better delineate the role of GBA in PD susceptibility
We also detected evidence of at least two distinct genetic effects within SNCA, a well-known PD susceptibility gene. While the SNP with the most extreme p-value in the Discovery Sample (rs356165) failed to genotype in the Replication Sample, it is in complete LD (r2=1.0) with the most extreme p-value in the Replication sample (rs356220). Moreover, they belong to the same LD block as the top SNP in other studies (rs356219; r2=0.96).11 Two previous studies have reported high LD (D’=0.90) but low intermarker correlation (r2<0.10) between the primary SNCA finding, rs356220, and the deleterious Rep1-263 allele.24,25 Rep1 is a microsatellite marker with three predominant alleles (259, 261, and 263) that has consistently been associated with PD risk and often with age at onset. The independent signal reported here, rs356198, is in high LD with the inversely associated Rep1-259 allele (in the PROGENI dataset: D’=0.92, r2=0.48). It is possible that the two independent SNPs are tagging a functional effect of Rep1 or that Rep1 is not functional, but merely tagging the same underlying causal variant(s) as the 2 SNPs.
The SNP with the most extreme p-value in the HLA region (rs3129882) was the same SNP identified in the NGRC sample which initially reported this association. 8 This is to be expected, since that study is included in our meta-analysis. This SNP was successfully genotyped in the Replication Sample, but was not statistically significant (p=0.92). Rather a different SNP was statistically significant in the Replication Sample (rs2395163; p=1×10−5) and reached genome-wide significance in the Combined Sample (p=3×10−11). LD is high between the two SNPs (D’=0.92), but the correlation was low due to differing allele frequencies (MAF for rs3129882 =0.433; MAF for rs2395163 =0.197; r2=0.25). The allele frequency of rs2395163 is closer to that seen in the variant with the most extreme p-value in the another recent meta-analysis of PD11 (MAF for chr6:32588205=0.15) and is in moderate to high LD with that SNP in the 1000 Genomes data (D′=1.00; r2=0.71). There is evidence that rs2395163 and chr6:32588205 tag the same LD block and that the association with these SNPs is independent of the original rs3129882 finding (see Supplementary Table 5 and Supplemental Methods).
Recently, another group reported results from a meta-analysis of several existing GWAS.11,13 Two of the Discovery Samples are in common in both studies and there is some overlap among our Replication samples, although the extent is difficult to quantify. There are several regions in common between studies. For example, both our study and theirs confirmed the association of GAK, SNCA, the HLA region, and MAPT. The two recent meta-analyses reported ten new loci: ACMSD, STK39, MCCC1/LAMP3, SYT11, CCDC62/HIP1R, PARK16, NMD3, STBD1, GPNMB, FGF20, MMP16 and STX1B. Of note, SYT11 is within the same LD block on chromosome 1 as RIT1, whereas RIT2 is within the same LD block on chromosome 18 as SYT4. It remains to be seen which of these genes harbors the true susceptibility alleles and if they have an interaction within a common pathway leading to PD pathogenesis. Supplemental Table 3 summarizes the results in our study for the SNPs in the ten new loci. We have nominal significance (p<0.05) for all but one of these SNPs. Similar odds ratios in the same direction and for the same allele as presented in the original paper was observed when analyses were limited to the two datasets not included in the original manuscript (HIHG and NGRC), and all but three SNPs remained nominally significant.
Comparing our results to those of recent GWAS, one other previously reported locus could be replicated by our analyses. BST1 has been seen in multiple datasets.8,9,12 Although association to this locus did not meet our genome-wide criteria in either the Discovery or Combined Sample analyses, our results for SNPs in this gene did meet established criteria for replication of a previously reported association. The SNP rs4698412 had a p-value of 0.002 in our Discovery Sample, 5 × 10−5 in our Replication Sample, and 3 × 10−7 in the Combined Sample.
In summary, we completed a meta-analysis of existing available PD GWAS datasets and identified a novel susceptibility locus, RIT2, and confirmed the association of several known genes. Using our Replication and Discovery Samples, conditional analyses confirmed that in two genes, there are multiple risk alleles that have distinct effects on disease risk. These results have important implications as studies are being designed to sequence these regions to identify all potentially functional disease-associated variants.
Supplementary Material
Acknowledgments
This project was supported by R01CA141668, R01NS37167, R01NS065070, R56NS037167, MO1RR00750, R01NS36960, P50NS39764, R01NS064155, U10NS44482, NIH/NINDS 1RC2NS070276, NS057567, P50NS072187-02, IH NS36630, UL1 RR024156, NS050487, NS060113, U24AG026395, 1I01BX000531, NS24778, R01NS36711, Harvard NeuroDiscovery Center, The Michael J. Fox Foundation Edmond J Safra Michael J Fox Foundation Global Genetic Consortium Initiative, Bumpus Foundation, Robert P. and Judith N. Goldberg Foundation, National Parkinson Foundation; Parkinson’s Disease Foundation Joseph R. Mazzulli and Grace Bwala, Mayo Clinic Florida (MCF) Research Committee CR programs (MCF #90052018 and MCF #90052030) (ZKW), and the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch (MCF #90052031/PAU #90052) (ZKW, OAR), and Dystonia Medical Research Foundation (ZKW). Control samples and clinical data were provided from the National Cell Repository for Alzheimer’s Disease (U24 AG021886) and the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds). PD and control brain samples provided by the Sun Health Research Institute in Sun City, Arizona, which is supported by the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011 and 05-901 to the Arizona Parkinson’s Disease Consortium) and the Prescott Family Initiative of the Michael J. Fox Foundation for Parkinson’s Research; the Harvard Brain Tissue Resource Center, which is supported in part by PHS grant number R24 MH 068855, and the Human Brain and Spinal Fluid Resource Center VA West Los Angeles Healthcare Center, 11301 Wilshire Blvd. Los Angeles, CA 90073, which is sponsored by NINDS/NIMH, National Multiple Sclerosis Society, Department of Veterans.
We also thank Roxann Ashworth, Cheryl Halter, Jacqueline Jackson, Cherylyn Jauregui, Jennifer Lash, and Jane Romm. We thank all persons with and without PD for volunteering to participate in this research study.
PSG-PROGENI Investigators and Coordinators
Albany Medical College: S Factor, D Higgins, S Evans; Barrow Neurological Institute: H Shill, M Stacy, J Danielson, L Marlor, K Williamson; Baylor College of Medicine: J Jankovic, C Hunter; Beth Israel Deaconess Medical Center: D Simon, P Ryan, L Scollins; Beth Israel Medical Center: R Saunders-Pullman, K Boyar, C Costan-Toth, E Ohmann; Brigham & Women’s Hospital: L Sudarsky, C Joubert; Brown University (Memorial Hospital of RI): J Friedman, K Chou, H Fernandez, M Lannon; Cleveland Clinic Florida-Weston: N Galvez-Jimenez, A Podichetty, K Thompson; Clinical Neuroscience Center: P Lewitt, M DeAngelis; Colorado Neurological Institute: C O’Brien, L Seeberger, C Dingmann, D Judd; Columbia University Medical Center: K Marder, J Fraser, J Harris; Creighton University: J Bertoni, C Peterson; Evanston Northwestern Healthcare: M Rezak, G Medalle; Hotel-Dieu Hospital-Chum: S Chouinard, M Panisset, J Hall, H Poiffaut; Hunter Homes McGuire Veterans Medical Center: V Calabrese, P Roberge; Indiana University School of Medicine: J Wojcieszek, J Belden; Institute For Neurodegenerative Disorders: D Jennings, K Marek, S Mendick; Johns Hopkins University: S Reich, B Dunlop; London Health Sciences Centre: M Jog, C Horn; Mayo Clinic Jacksonville: R Uitti, M Turk; McFarland Neurosciences: T Ajax, J Mannetter; Medical College of Georgia: K Sethi, J Carpenter, B Dill, L Hatch, K Ligon, S Narayan; Medical College of Wisconsin: K Blindauer, K Abou-Samra, J Petit; Medical University of Ohio: L Elmer, E Aiken, K Davis, C Schell, S Wilson; Mount Sinai School of Medicine: M Velickovic, W Koller (deceased), S Phipps; North Shore-LIJ Health System: A Feigin, M Gordon, J Hamann, E Licari, M Marotta-Kollarus, B Shannon, R Winnick; Northwestern University: T Simuni, A Videnovic, A Kaczmarek, K Williams, M Wolff; Ochsner Clinic Foundation: J Rao, M Cook; Ohio State University: M Fernandez, S Kostyk, J Hubble, A Campbell, C Reider, A Seward; Oregon Health & Science University: R Camicioli, J Carter, J Nutt, P Andrews, S Morehouse, C Stone; Ottawa Hospital Civic Site: T Mendis, D Grimes, C Alcorn-Costa, P Gray, K Haas, J Vendette; Pacific Neuroscience Medical Group: J Sutton, B Hutchinson, J Young; Saskatoon Dist Health Board Royal Univ Hosp: A Rajput, A Rajput, L Klassen, T Shirley; Scott & White Hospital/Texas A&M University: B Manyam, P Simpson, J Whetteckey, B Wulbrecht; The Parkinson’s & Movement Disorder Institute: D Truong, M Pathak, K Frei, N Luong, T Tra, A Tran, J Vo; Toronto Western Hospital, University Health: A Lang, G Kleiner-Fisman, A Nieves, L Johnston, J So; UMDNJ-School of Osteopathic Medicine: G Podskalny, L Giffin; University of Alabama at Birmingham: P Atchison, C Allen; University of Alberta: W Martin, M Wieler; University of Calgary: O Suchowersky, S Furtado, M Klimek; University of California Irvine: N Hermanowicz, S Niswonger; University of California San Diego: C Shults (deceased), D Fontaine; University of California San Francisco: M Aminoff, C Christine, M Diminno, J Hevezi; University of Chicago: A Dalvi, U Kang, J Richman, S Uy, J Young; University of Cincinnati: A Dalvi, A Sahay, M Gartner, D Schwieterman; University of Colorado Health Sciences Center: D Hall, M Leehey, S Culver, T Derian; University of Connecticut: T Demarcaida, S Thurlow; University of Iowa: R Rodnitzky, J Dobson; University of Kansas Medical Center: K Lyons, R Pahwa, T Gales, S Thomas; University of Maryland School of Medicine: L Shulman, S Reich, W Weiner, K Dustin; University of Miami: K Lyons, C Singer, W Koller (deceased), W Weiner, L Zelaya; University of Minnesota: P Tuite, V Hagen, S Rolandelli, R Schacherer, J Kosowicz; University of New Mexico: P Gordon, J Werner; University of Puerto Rico School of Medicine: C Serrano, S Roque; University of Rochester: R Kurlan, D Berry, I Gardiner; University of South Florida: R Hauser, J Sanchez-Ramos, T Zesiewicz, H Delgado, K Price, P Rodriguez, S Wolfrath; University of Tennessee Health Science Center: R Pfeiffer, L Davis, B Pfeiffer; University of Texas Southwestern Medical Center: R Dewey, B Hayward, A Johnson, M Meacham, B Estes; Wake Forest University School of Medicine: F Walker, V Hunt, C O’Neill; Washington University: B Racette, L Good, M Rundle
GenePD Investigators and Coordinators
University Southern California School of Medicine: M Lew; University of Calgary: O Suchowersky; University of Lübeck, Germany: C Klein; UMDNJ-Robert Wood Johnson Medical School: L Golbe, MH Mark; Massachusetts General Hospital, Harvard Medical School: J Growdon, N Huggins; University of Virginia Health System: GF Wooten; University of Alabama at Birmingham: R Watts; University of Toronto: M Guttman; Washington University School of Medicine: B Racette, J Perlmutter; Barrow Neurological Institute: L Marlor; Sun Health Research Institute: H Shill; University of Miami: C Singer; Parkinson Institute, Istituti Clinici di Perfezionamento, Milano, Italy: S Goldwurm, G Pezzoli; Boston University School of Medicine: MH Saint-Hilaire, T Massood; Cleveland Clinic Foundation: K Baker, I Itin; University of Louisville School of Medicine: I Litvan; University of Sydney ANZAC Research Institute, Concord Hospital, Sydney, Australia: G Nicholson, A Corbett; Struthers Parkinson’s Center, Minneapolis: M Nance; Port City Neurology, Scarborough, ME: E Drasby; Parkinson’s Disease and Movement Disorder Center of Boca Raton: S Isaacson; Newcastle University, Newcastle upon Tyne, UK: D Burn, P Chinnery; General Regional Hospital Bolzano, Bolzano, Italy: P Pramstaller; University of Arkansas for Medical Sciences: J Al-hinti; Aarhus University Hospital, Aarhus, Denmark: A Moller, K Ostergaard; University of Arizona: S Sherman; Auckland City Hospital, Auckland, New Zealand: R Roxburgh, B Snow; University of Kentucky College of Medicine: J Slevin, F Cambi.
NGRC Investigators and Coordinators
New York State Department of Health Wadsworth Center: D Kay, J Montimurro, V Kusel; VA Puget Sound Health Care System and University of Washington: A Samii, E Martinez, D Yearout; Oregon Health and Sciences University: J Nutt; Evergreen Hospital Medical Center: P Agarwal, A Griffith; Virginia Mason Medical Center: JW Roberts; Samuel Stratton VA Medical Center and Albany Medical Center: DS Higgins. Albany Medical Center: Eric Molho, Emory University: Ami Rosen
HIHG Investigators and Coordinators
CA Jauregui, MA Nance, RL Watts, JP Hubble, WC Koller, K Lyons, R Pahwa, MB Stern, A Colcher, BC Hiner, J Jankovic, WG Ondo, FH Allen, Jr., CG Goetz, GW Small, D Masterman, F Mastaglia, BL Scott, C Singer, F Nahab, MA Pericak-Vance, and JL Haines who contributed to this study. Some of the samples used in this study were collected while the Udall PDRCE was at Duke University.
NetPD Investigators and Coordinators
University of Michigan: K Chou, K Wernette; Rush-Presbyterian-St. Luke’s Medical Center: K Shannon, J Jaglin; Oregon Health Sciences University: J Carter, P Andrews; Baylor College of Medicine: J Jankovic, J Shahed, C Hunter; University of Virginia: FG Wooten, MF Keller; The Parkinson’s Institute: C Tanner, T Stewart, C Lawrence, K Smith; University of Miami: C Singer, M Quesada; University of Pennsylvania: A Siderowf, S Reichwein; University of South Florida: R Hauser, S Wolfrath, G Piper; Washington University: B Racette, P Deppen, M Ammel; Johns Hopkins University School of Medicine: T Dawson, B Dunlop; University of Calgary: O Suchowersky, L Derwent; University of Southern California: MF Lew, M Villa; Georgia Health Sciences University: K Sethi, B Dill, S Narayan; Barrow Neurological Institute: R Burns, L Marlor; University of Alabama: RL Watts, R McMurray; Brigham & Women’s Hospital: L Sudarsky, G Hage; The Parkinson’s & Movement Disorder Institute: D Truong, AH Tran, M Tran; University of California San Francisco: MJ Aminoff, J Roth; Beth Israel Deaconess Medical Center: DK Simon, L Scollins, L Paul, P Rose; Northwestern University: T Simuni, K Williams; University of Texas Southwestern Medical Center: R Dewey, B Hayward, A Johnson, A Gode; University of Colorado Health Sciences Center: M Leehey, J Bainbridge; Duke University Medical Center: B Scott, J Field; LSU Health Science Center Shreveport: R Zweig, C Hilliard; SUNY Downstate Medical Center: I Bodis-Wollner, S Glazman; University of Vermont: R Hamill, C Homan, S Lennox, C Potter; Southern Illinois University: R Elble, C Young, B Lokaitis, D Kelly; Pacific Health Research & Education Institute: GW Ross, S Terashita; Thomas Jefferson University: J Schneider, S Sendek; Thomas Jefferson University/Llankenau Hospital: S Gollomp, R Gollomp, S Raza; Dartmouth Hitchcock Medical Center: DJ Coffey, PR Leblanc; University of Kentucky: F Cambi, R Wagner; Medical University of South Carolina: K Bergman, V Hinson, V Salak; LSU Medical Center: J Rao, M Cook.
CorePD Investigators and Coordinators
Columbia University: RN Alcalay, E Caccappolo, H Mejia-Santana, M-X Tang, L Rosado, MO Reilly, D Ruiz, B Ross, M Verbitsky, S Kisselev, ED Louis, H Andrews, C Waters, S Fahn, L Cote, S Frucht, B Ford, R Ottman, LN Clark; Rush University: C Comella; University of Pennsylvania: A Colcher, A Siderowf; Institute of Neurodegenerative Disorders: D Jennings; Park Nicollet Clinic: M Nance; Beth Israel: S Bressman; University of Miami: WK Scott; Parkinson’s Institute: C Tanner; Marshfield Clinic: S Mickel; Neuroscience Institute at Central DuPage Hospital: M Rezak; Northwestern University: K Novak; Butler Hospital: JH Friedman; University of Tennessee: R Pfeiffer; Johns Hopkins University: L Marsh; Medical College of Wisconsin: B Hiner.
LOAD Investigators and Coordinators
The following investigators and Alzheimer’s Disease Centers participated in the Study: Boston University: R Green, N Kowall, L Farrer; Columbia University: J Williamson, V Santana; Duke University: D Schmechel, P Gaskell, K Welsh-Bohmer, M Pericak-Vance; Indiana University: B Ghetti, MR Farlow, K Horner; Massachusetts General Hospital JH Growdon, D Blacker, RE Tanzi, BT Hyman; Mayo Clinic-Rochester: B Boeve, K Kuntz, L Norgaard, N Larson; Mayo Clinic-Jacksonville: D Kistler, F Parfitt, J Haddwow; Mount Sinai School of Medicine: J Silverman, M Schnaider Beeri, M Sano, J Wang, R Lally; Northwestern University: N Johnson, M Mesulum, S Weintraub, E Bigio; Oregon Health and Science University: J Kaye, P Kramer, J Payne-Murphy; Rush University: D Bennett, H Jacobs, J-S Chang, D Arends; University of Alabama at Birmingham: L Harrell; University of California, Los Angeles: G Bartzokis, J Cummings, PH Lu, U Toland; University of Kentucky: W Markesbery, C Smith, A Brickhouse; University of Pennsylvania: J Trojanowski, V Van Deerlin, EM Wood; University of Pittsburgh: S DeKosky, R Sweet, E Weamer; University of Southern California: IH Chui, A Varpetian; University of Texas Southwestern: R Diaz-Arrastia, R Rosenberg, B Davis; University of Washington: T Bird, M Rumbaugh, GD Schellenberg, M Raskind; Washington University at St Louis: A Goate, JMorris, J Norton, D Levitch, B Grant, M Coats.
DATATOP Investigators and Coordinators
Participating Investigators -- University of Kansas: W Koller; University of South Florida: CW Olanow; University of Iowa: R Rodnitzky; Massachusetts General Hospital: JS Fink, JH Growdon; Ohio State University: G Paulson; University of Rochester: R Kurlan; Roger Williams General Hospital: JH Friedman; Oregon Health Sciences University: S Gancher, J Nutt; University of Saskatchewan: AH Rajput; University of Virginia: JB Bennett, GF Wooten; Sinai Hospital, Detroit: P LeWitt; Rush-Presbyterian-St Luke’s Medical Center: C Goetz, C Tanner, K Shannon; University of Calgary: O Suchowersky; Columbia-Presbyterian Medical Center: MF Brin, SB Bressman; University of Miami: WJ Weiner and J Sanchez-Ramos; Baylor College of Medicine: J Jankovic; University of Michigan: JB Penney; Toronto Hospital: A Lang; St Luke’s Hospital, Denver: M Hoehn; California Parkinson’s Foundation: JTetrud; Ottawa Civic Hospital: JD Grimes; University of Nebraska: R Pfeiffer; University of California, SanDiego: C Shults (deceased), L Thal; Montreal General Hospital-McGill University: S Gauthier; University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School: LI Golbe; Washington University, St Louis: JS Perlmutter; Johns Hopkins University: H Moses III, SG Reich; Graduate Hospital and University of Pennsylvania: HI Hurtig, M Stern.
Site Coordinators -- R Barter and B Vetere-Overfield, Kansas City, Kans; L Gauger and T Malapira, Tampa, Fla; J Dobson, Iowa City, Iowa; S Atamian, M Tennis, JB Cohen, G Desclos, and E Hoffman, Boston; L Denio, S Huber, T Woike, K Zoog, R Mendell, and K Dudte, Columbus, Ohio; J Behr, IF Gardiner, Rochester, NY; M Lannon, Providence, RI; J Carter andS Northrup, Portland, Oreg; B Kanigan, Saskatoon, Sask; M Turk and E Landow, Charlottesville, Va; P Schlick andK Mistura, Detroit; VS Carroll and JA Thelen, Chicago; C Demong, Calgary, Alta.; L Winfield and C Moskowitz, New York; A Ingenito, C Sheldon, and L Cornelius, Miami; D Heibergand C Dunne, Houston; J Brady, Ann Arbor, Mich; C Kierans, L Belle-Scantlebury, and J Duff, Toronto; H Weber, Denver; D Savoini, P Lewis, and SJ Kutner, San Jose, Calif; PGray, Ottawa; C Glaeske and R Hofman, Omaha, Neb; MM Payand D Salmon, San Diego, Calif; F McFaul and D Amyot, Montreal; M Bergen, New Brunswick, NJ; L McGee-Minnich, St Louis; P O’Donnell, Baltimore; and S Ferrise and K Shallow, Philadelphia.
Coordination and Data Center -- University of Rochester Medical Center: RM Pelusio (program manager); A Rudolph (senior study coordinator); C Miller (nurse clinician); M Linsner, J Connorton, J Nusbaum, and C Casaceli (analyst-programmers); C Irvine, C Orme, and GJ Wixsom (information analysts); M Schirazi, J Sotack, and H Randolph (data-control clerks); R Nobel, D Baker, D LaDonna, ME Rothfuss, L Doerr (deceased), L Rumfola, and B Kavanaugh (secretarial staff); and J Wendel (CLINFO manager).
Biostatistics Center -- Department of Biostatistics, University of Rochester Medical Center: C Odoroff (deceased)and D Oakes (chief biostatisticians); M McDermott and S Eberly(biostatisticians); S Plumb (lead programmer); and A Watts, L Yorkey, A Choi, and K Gerwitz (analyst-programmers).
Pharmacy Center -- Strong Memorial Hospital: P Evans (chief pharmacist); and L Dellapena and V Singletary(pharmacy technicians).
Safety Monitoring Committee -- R Herndon (chair, January 1,1987, to June 30, 1988), Portland, Oreg; P Tariot (chair, July 1, 1988, to present), Rochester, NY; and E Bell, RC Griggs, and WJ Hall, Rochester, NY.
Scientific Advisory Committee -- CD Marsden (chair), London; TN Chase, Bethesda; G Cohen, J Fleiss, and R Mayeux, New York; L Jacobs and AJ Moss, Rochester, NY; and E Melamed, Tel Aviv, Israel.
Assay Standards Committee -- R Roth (chair), New Haven CT; M Galloway, Detroit; I Irwin, San Jose CA; P LeWitt, Detroit; and G Vatassery, Minneapolis.
Neuropsychological Testing Committee -- P Como (chair), Rochester, NY; J St Cyr, Toronto; Y Stern and J Williams, New York; and R Wilson, Chicago.
Monitoring Committee, National Institute of Neurological Disorders and Stroke -- EM Stadlan (chair), Bethesda; M Alter, Philadelphia; K Bergmann, New Hyde Park, NY; J Cedarbaum, White Plains, NY; J Ellenberg, Bethesda; and R Kibler, Atlanta.
CSF Assay Center -- Lafayette Clinic, Wayne State University: M.P. Galloway (director); M. Kaplan (deceased), R. Lodhi, M.J. Keegan, B. Matthews, E.A. Novak.
Deprenyl Metabolites Assay Center -- Institute for Medical Research, San Jose CA: I. Irwin (director).
Tocopherol Assay Center -- Our Lady of Mercy Medical Center, Bronx, NY: E. Norkus (director).
Specimen Repository -- Department of Neurology, University of Rochester: D Flood (director), T McNeill, N Harary, L Koek.
Laboratory Surveillance Testing -- SciCor Laboratories, Indianapolis: RL Creveling (director).
Massachusetts General Hospital Investigators and coordinators
JR Mazzulli, G Bwala
Harvard NeuroDiscovery Center Biomarker Study (HBS)
Investigators and study coordinators -- Harvard NeuroDiscovery Center: CR Scherzer, BT Hyman, AJ Ivinson, NE Maher, AK Sarokhan, KC Lockhart, A Santarlasci; Brigham and Women’s Hospital: LR Sudarsky, MT Hayes, E Hart; Massachusetts General Hospital: JH Growdon, MA Schwarzschild, AY Hung, AW Flaherty, D Blacker, A-M Wills, US Sohur, VK Unni, NI Mejia, A Viswanathan, SN Gomperts, MW Albers, KE Swords, RK Rudel, JT Hirschberger; OA Padilla; University of Ottawa: M Schlossmacher; Harvard School of Public Health: A Ascherio; Biogen Idec: BM Ravina;
Data coordination staff -- Harvard NeuroDiscovery Center: B Khadka, OA Padilla, B Zheng, JJ Locascio;
Biobank management staff -- Harvard NeuroDiscovery Center: SS Roderick, CG Kan, Z Liao.
SEARCH Steering Committee Members
CM Tanner, GW Ross, RD Abbott, JW Langston.
Enrolling Institutions, Investigators and Coordinators (listed by number of exams contributed) -- The Parkinson’s Institute, Sunnyvale, CA: CM Tanner, D Roucoux, S Yerabati, A Smith; University of South Florida, Tampa, FL: RA Hauser, D Delaney, SC Wolfrath, J Nemeth, T Mclain; Baylor College of Medicine, Houston, TX: J Jankovic, C Hunter, L Shinawi, KR Flores; Emory University School of Medicine, Atlanta, GA: SA Factor, C Ingram; Beth Israel Medical Center, New York, NY: S Bressman, A Deligtisch, K Boyar, A Wolff; Toronto Western Hospital, Toronto, Ontario, Canada: C Marras, M Enjati; University of Kansas Medical Center, Kansas City, KA: K Lyons, C Mack, A Langhammer, T Gales; Veterans Affairs Pacific Islands Health Care System, Honolulu, HI: GW Ross, FK Wakashige.
Parkinson’s Institute Coordination, Interviewer and Analytic Staff -- M Lu, K Comyns, M Korell, L Sterling, R VanVeghten, M Padua, J Wright, B Allred, L Bushman, M Bynum, C Cage, A Carroll, M Chang, R Cook, E Ferriter, M Franco, K Gorman, M Kemp, S Knutson, S Lavoie, E Mills, C Nguyen, P Niedle, J Pia, B Priestley, V Sims, JA Soetmelk, B Song, B Stang, M Staver, A Stover.
References
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–35. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 2.Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. Journal of Geriatric Psychiatry and Neurology. 2010;23:228–42. doi: 10.1177/0891988710383572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maraganore DM, et al. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77:685–93. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fung HC, et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–6. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 5.Pankratz N, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon-Sanchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards TL, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamza TH, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42:781–5. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saad M, et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum Mol Genet. 2011;20:615–27. doi: 10.1093/hmg/ddq497. [DOI] [PubMed] [Google Scholar]
- 10.Spencer CC, et al. Dissection of the genetics of Parkinson’s disease identifies an additional association 5’ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet. 2011;20:345–53. doi: 10.1093/hmg/ddq469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.International Parkinson Disease Genomics Consortium. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–9. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 13.International Parkinson Disease Genomics Consortium. A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 2011;7:e1002142. doi: 10.1371/journal.pgen.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–4. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan JB, Chee MS, Gunderson KL. Highly parallel genomic assays. Nat Rev Genet. 2006;7:632–44. doi: 10.1038/nrg1901. [DOI] [PubMed] [Google Scholar]
- 17.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–52. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao X, Starmer J, Martin ER. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol. 2008;32:361–9. doi: 10.1002/gepi.20310. [DOI] [PubMed] [Google Scholar]
- 19.Lee CH, Della NG, Chew CE, Zack DJ. Rin, a neuron-specific and calmodulin-binding small G-protein, and Rit define a novel subfamily of ras proteins. J Neurosci. 1996;16:6784–94. doi: 10.1523/JNEUROSCI.16-21-06784.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee D, Lee SY, Lee EN, Chang CS, Paik SR. alpha-Synuclein exhibits competitive interaction between calmodulin and synthetic membranes. J Neurochem. 2002;82:1007–17. doi: 10.1046/j.1471-4159.2002.01024.x. [DOI] [PubMed] [Google Scholar]
- 21.Padilla R, Maccioni RB, Avila J. Calmodulin binds to a tubulin binding site of the microtubule-associated protein tau. Mol Cell Biochem. 1990;97:35–41. doi: 10.1007/BF00231699. [DOI] [PubMed] [Google Scholar]
- 22.Bossers K, et al. Analysis of gene expression in Parkinson’s disease: possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain Pathol. 2009;19:91–107. doi: 10.1111/j.1750-3639.2008.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nichols WC, et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology. 2009;72:310–6. doi: 10.1212/01.wnl.0000327823.81237.d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pankratz N, et al. Alpha-synuclein and familial Parkinson’s disease. Mov Disord. 2009;24:1125–31. doi: 10.1002/mds.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mata IF, et al. SNCA variant associated with Parkinson disease and plasma alpha-synuclein level. Arch Neurol. 2010;67:1350–6. doi: 10.1001/archneurol.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.