

Abstract
Context:
Congenital adrenal hyperplasia (CAH) is one of the inborn errors of metabolic disorder inherited in an autosomal recessive manner caused by the defects in the steroid 21 hydroxylase CYP21A2 gene. We analyzed the genotype of 62 patients with classic CAH.
Aims:
To find out the underlying mutations of CYP21A2 gene.
Settings and Design:
Cohort of CAH patients.
Materials and Methods:
Sixty-two patients with CAH were recruited from the endocrine clinic at AIIMS. Electrochemiluminiscence method was used for estimating the levels of cortisol. Radioimmunoassay kit-based method was used for estimating the 17 OHP levels. Polymerase chain reaction amplification was done using specific primers to amply the CYP21A2 gene.
Statistical Analysis Used:
Statistical analysis was done by using Epi Info Version 3.5.1.2008.
Results:
Out of 62 patients, 50 were simple virilizers (SV) and 12 were salt wasters (SW). Fifty-six were females and six were males. Five 46, XX children were reared as males. Age at presentation varied from 8 months to 38 years. Molecular genetic analysis revealed that the highest number of patients harboured (In 2) IVS2-13 A/C > G (48%), followed by p.P30L (46%), p.Q318X (35%), (D 8 bp) deletion 8 bp (26%), p.I172N (26%), and p. R356W (20%) mutations.
Conclusion:
This is among the few studies to analyze the mutational spectrum of CYP21A2 gene in a large CAH cohort from India. Molecular diagnosis of CYP21A2 gene should be considered as part of the CAH evaluation to assess the risk of the patients/parents/siblings and to offer genetic counseling.
Keywords: Ambiguous genitalia, CYP21A2 gene, phenotype, salt wasting, simple virilizing
INTRODUCTION
Congenital adrenal hyperplasia (CAH) is an autosomal recessive inherited disorder caused by steroid 21-hydroxylase deficiency. The deficiency of this enzyme leads to impaired cortisol production and increased 17-OH-progesterone (17 OHP) biosynthesis. The increased androgen levels during embryonic development cause virilization of the external genitalia in females. Depending on the enzyme activity, patients with CAH are categorized into classic and nonclassic type.
The gene encoding the 21-hydroxylase enzyme (CYP21A2) and a nonfunctional pseudogene (CYP21A1P) are located within the human leukocyte antigen class III gene region on the short arm of chromosome 6 (6p21.3), closely adjacent in tandem arrangement with the C4A and C4B genes encoding for the fourth component of the serum complement.[1–3] These two genes consist of 10 exons and show a high homology with a nucleotide identity of 98% in their exon and 96% in their intron sequences.[4,5] Most of the inactivating mutations are generated by unequal crossing over or gene conversion between the functional (CYP21A2) gene and a nonfunctional (CYP21A1P) pseudogene.[6,7] As a result, complete gene deletions/large gene conversions/ 8-bp/single point mutations are manifested with severe phenotypic anomalies in patients with CAH.[2,8]
To date, many studies from different ethnic groups around the world have reported the ethnic specific frequency of CYP21A2 gene mutations and their impact on the clinical features of CAH patients. The classic CAH disease has a worldwide incidence of about 1:10,000 to 1:16,000 live births[9,10] and a carrier frequency of 1:60.[11] Only two studies are available on the frequency of CYP21A2 gene mutations from India.[12,13] Therefore, in this study we analyzed the genotype of patients with classic CAH from the Indian subcontinent.
MATERIALS AND METHODS
Patients
This study was approved by AIIMS Ethics Committee. Patients with CAH were recruited from the endocrine clinic at AIIMS. These patients were classified into the salt wasting, and simple virilizing as per their phenotype, clinical history, and hormonal profile. Detailed family history of the patient was taken. Physical examination was done including prader staging for genital appearance, sex of rearing, and hirsutism status.
Methods
Hormonal analysis
Electrochemiluminiscence method was used for estimating the levels of cortisol, adrenocorticotropic hormone, testosterone, and dehydroepiandrosterone sulfate. Radioimmunoassay kit-based method was used for estimating the 17 OHP levels (Diagnostic Systems Laboratories, Inc., Webster, TX, supplied by Immunotech Marcelle, France).
Cytogenetic analysis
Conventional cytogenetic analysis was carried out on peripheral blood using standard techniques. Karyotyping was carried out on G-banded metaphases obtained from 72-h cultures.
Molecular analysis
Informed consent to carry out molecular genetic studies was obtained from the patient/parents. DNA was isolated from blood samples by using the Qiagen DNA isolation kit as per the manufacturer's instructions. DNA was quantified and subjected to polymerase chain reaction (PCR) amplification using specific primers to amply the CYP21A2 gene.[14] This gene was amplified in two segments using specific primers as shown in [Figure 1]. Using fragment 1 as template, we performed a secondary PCR with the appropriate primers (Figure 1., upper left) to detect p.P30L, In2, and ? 8 bp mutations. Fragment 2 was digested with ApaL1 restriction enzyme [Table 1] to detect both p.V281L and p.R339H mutations at the same time. Fragment 2 was also used as template to perform a secondary PCR with the appropriate primers [Figure 1, lower right] to detect p.I172N, p.Q318X, p.R356W, and p.P453S mutations.
Figure 1.
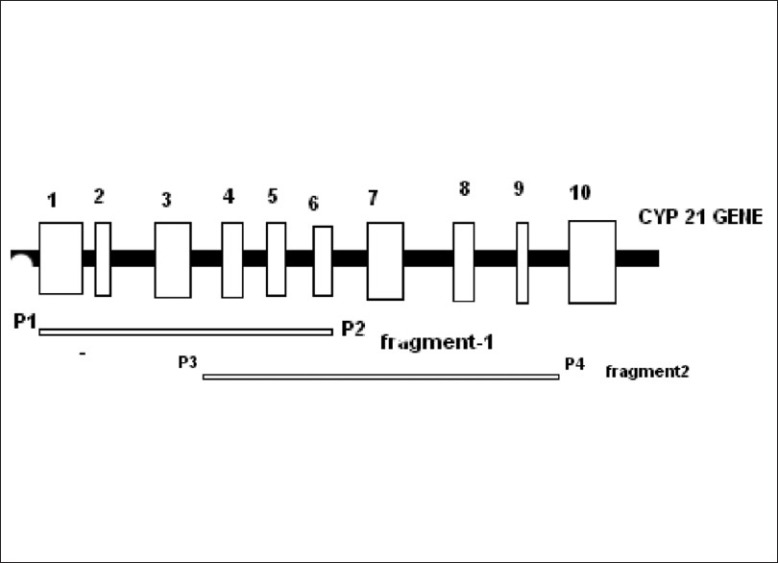
Location of the PCR primers used to detect the mutations in CYP21A2 gene
Table 1.
Primers and Restriction Enzymes used for the detection of CYP21A2 gene mutations
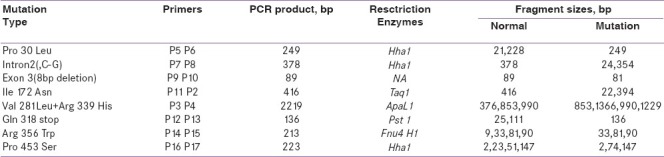
Secondary PCR products were digested with the appropriate restriction enzymes [Table 1] to identify the common mutations using restriction fragment length polymorphism method[15–21] followed by 1% agarose gel electrophoresis/10% polyacrylamide gel electrophoresis to separate the restriction fragments.
Genotype classification
These patients were divided into three groups as described previously.[22] Group 0/Null consisted of mutations [Figures 2 and 3] with complete loss of enzyme activity (deletions, conversions, Δ 8 bp deletion, p.Q318X, and p.R356W). Group A consisted of In2 mutation with low enzyme activity and group B consisted of p.I172N with relatively low normal enzyme activity.
Figure 2.
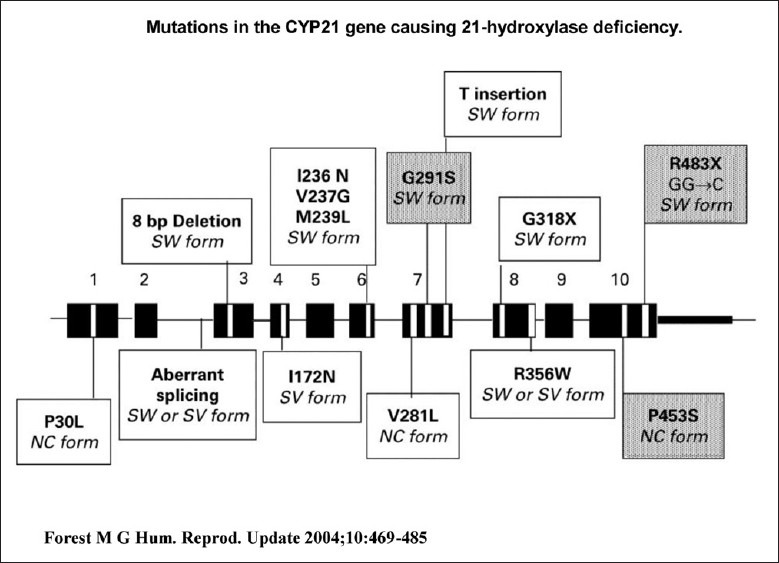
Approximate location of the CYP21A2 gene mutations
Figure 3.
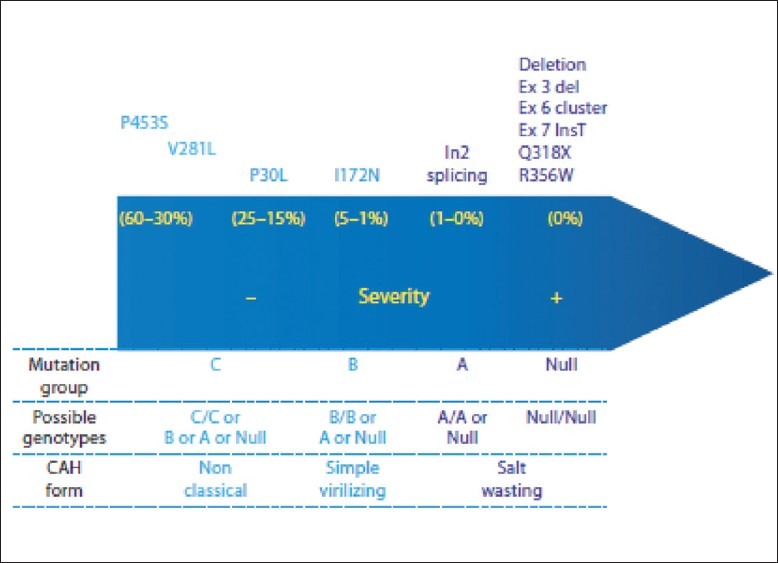
Resudual enzymatic activity according to in vitro CYP21A2 fundctional sudies. The mutation groups refer to the Krone et al. (2000) classification and correspond to the genotypes clustered in the underscrored line
RESULTS
Out of 62 patients, 56 were females and 6 were males. Five 46, XX children were reared as males. Seventeen were siblings. Five patients were from consanguineous marriage. Age at presentation varied from 8 months to 38 years. The mean age was 15.39 ± 10.44 years. The mean 17 OHP and cortisol levels were 17.1 ± 32.93 ng/ml and 8.59 ± 7.63 μg/dl, respectively. Except nine patients, all were on treatment at the time of sample collection for hormonal and molecular analysis. Among these 62 CAH patients, 56 (90%) have ambiguous genitalia. Fifty patients (81.0%) were simple virilizing type and 12 patients (19.0%) were salt wasting type.
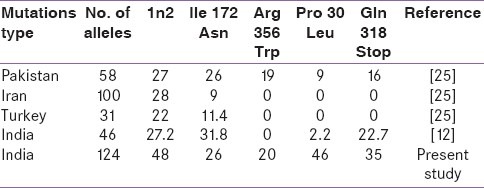
Molecular genetic analysis [Table 2] revealed that 46 patients (74.2%) were found to have abnormal genotype. Out of these 46 patients, 37 (74.0%) were simple virilizers (SV) and eight (66.7%) were salt wasters (SW). The highest number of patients harboured In2 (48%), followed by p.P30L (46%), p.Q318X (35%), Δ 8 bp (26%), p.I172N (26%), and p. R356W (20%) mutations. However, in SW group, both In2 (50%) and p.Q318X (50%) mutations were found to be high followed by p.P30L (25%), R356W (25.0%), Δ 8 bp (12.5%), and p.I172N (12.5%), whereas in SV group the highest frequency of mutations were to found to be p.P30L (51.3%) and In2 (50%) followed by p.Q318X (32%), Δ 8 bp (30%), p.I172N (30%), and p.R356W (20.0%). The allele frequencies of these mutations were compared with [Table 3] different populations.
Table 2.
Distribution of common mutations in classic CAH patients
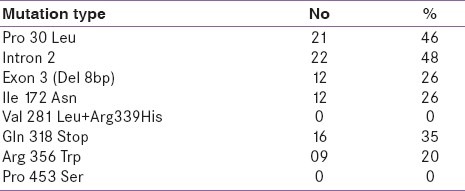
Table 3.
Allele frequency of the common mutations in CYP21A2 gene in Different Populations
DISCUSSION
Most of our patients sought medical consultation during puberty due to ambiguous genitalia and primary amenorrhea. SV type is more prevalent than SW type. This may be due to the salt wasting crisis leading to infant mortality before the diagnosis. Moreover, females were higher than males as evident from the literature that most of the male children miss the correct diagnosis during infancy until and unless they are tested for precocious puberty.
We observed an interesting pattern of mutational spectrum in CYP21A2 gene in our patients. Out of 124 alleles, we identified mutations in 92 alleles (74.2%) in our study population which is almost similar to the previous study.[12] Among the nine common mutations analyzed, In2 was the most prevalent mutation and the similar findings were reported in East Indians, Hong Kong Chinese, Singapore Chinese, Indians and Malays, Japanese, Iranian, French, Pakistani Taiwanese, and in American populations.[13,23–30]
This single-nucleotide point mutation In2 associated with the classic 21-OHD by the aberrant splicing of In2 leading to a null activity of CYP21A2. Other disease causing severe mutations like, p.Q318X, Δ 8 bp, and p.R356W were also identified in an increasing frequency, and the similar pattern was reported in East India, France, Mexico, and Spain.[12,13,31,32] In SV type, p.I172N mutation was found to be high and this trend was reported in many studies.[22,25,33,34] I172N is the only one mutation specifically associated with the SV form of the disease, and mutation of this hydrophobic residue to a polar residue results in an enzyme with approximately 1% or normal activity.[21,35] The frequency of p.P30L mutation was relatively high in our study, when compared with other studies.[12,13,36,37] This mutation was first described in a nonclassic CAH patient and considered as a part of larger gene conversion or a chimeric CYP21A1P/CYP21A2 gene. Subsequently, this mutation has been described in SV, NC, and also in SW CAH patients.[37,38]
This study is among the few studies to analyze the mutational spectrum of CYP21A2 gene in a large CAH cohort from India. Direct sequencing is required to characterize the complete gene. Molecular diagnosis of CYP21A2 gene should be considered as part of the CAH evaluation in order to assess the risk of the patients/parents/siblings and to offer genetic counseling.
ACKNOWLEDGEMENT
This study is being supported by the Department of Biotechnology (Ref.N0.BT/PR7205/Med/12/277/06), Government of India.
Footnotes
Source of Support: Department of Biotechnology (Ref.N0.BT/PR7205/Med/12/277/06), Government of India.
Conflict of Interest: None declared.
REFERENCES
- 1.Donohoue PA, Parker K, Migeon CJ. Congenital adrenal hyperplasia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. Vol. 7. New York: McGraw-Hill; 1995. pp. 2929–66. [Google Scholar]
- 2.White PC, Tusie-Luna MT, New MI, Speiser PW. Mutations in steroid 21-hydroxylase (CYP21) Hum Mutat. 1994;3:373–8. doi: 10.1002/humu.1380030408. [DOI] [PubMed] [Google Scholar]
- 3.White PC, Grossberger D, Onufer BJ, Chaplin DD, New MI, Dupont B, et al. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci U S A. 1985;82:1089–93. doi: 10.1073/pnas.82.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci U S A. 1986;83:5111–5. doi: 10.1073/pnas.83.14.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: A pseudogene and a genuine gene. Proc Natl Acad Sci U S A. 1986;83:2841–5. doi: 10.1073/pnas.83.9.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White PC, Vitek A, Dupont B, New MI. Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 1988;85:4436–40. doi: 10.1073/pnas.85.12.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higashi Y, Tanae A, Inoue H, Fujii-Kuriyama Y. Evidence for frequent gene conversion in the steroid 21-hydroxylase P- 450 (C21) gene: Implications for steroid 21-hydroxylase deficiency. Am J Hum Genet. 1988;42:17–25. [PMC free article] [PubMed] [Google Scholar]
- 8.Wedell A. Molecular genetics of congenital adrenal hyperplasia (21-hydroxylase deficiency): Implications for diagnosis, prognosis and treatment. Acta Paediatr. 1998;87:159–64. doi: 10.1080/08035259850157598. [DOI] [PubMed] [Google Scholar]
- 9.Therrell BL, Jr, Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, Prentice L, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101:583–90. doi: 10.1542/peds.101.4.583. [DOI] [PubMed] [Google Scholar]
- 10.Van der Kamp HJ, Wit JM. Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol. 2004;151(Suppl 3):U71–5. doi: 10.1530/eje.0.151u071. [DOI] [PubMed] [Google Scholar]
- 11.Balsamo A, Cacciari E, Piazzi S, Cassio A, Bozza D, Pirazzoli P, et al. Congenital adrenal hyperplasia: Neonatal mass screening compared with clinical diagnosis only in the Emilia-Romagna region of Italy, 1980-1995. Pediatrics. 1996;98:362–7. [PubMed] [Google Scholar]
- 12.Mathur R, Menon PS, Kabra M, Goyal RK, Verma IC. Molecular characterization of mutations in Indian children with congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency. J Pediatr Endocrinol Metab. 2001;14:27–35. doi: 10.1515/jpem.2001.14.1.27. [DOI] [PubMed] [Google Scholar]
- 13.Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Najmabadi H, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. 2007;90:414–21. doi: 10.1016/j.ymgme.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oriola J, Isabel P, Isabel M, Carles P, Francisca RF. Rapid screening method for detecting mutations in the 21-hydroxylase gene. Clin Chem. 1997;43:557–61. [PubMed] [Google Scholar]
- 15.Tusie-Luna MT, Speiser PW, Dumic M, New MI, White PC. A mutation (Pro-30 to Leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Mol Endocrinol. 1991;5:685–92. doi: 10.1210/mend-5-5-685. [DOI] [PubMed] [Google Scholar]
- 16.Higashi Y, Tanae A, Inoue H, Hiromasa T, Fujii-Kuriyama Y. Aberrant splicing and missense mutations cause steroid 21-hydroxylase [P-450(C21)] deficiency in humans: Possible gene conversion products. Proc Natl Acad Sci U S A. 1988;85:7486–90. doi: 10.1073/pnas.85.20.7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amor M, Parker KL, Globerman H, New MI, White PC. Mutation in the CYP21B gene (Ile-172-Asn) causes steroid 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 1988;5:1600–4. doi: 10.1073/pnas.85.5.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Speiser PW, New MI, White PC. Molecular genetic analysis of nonclassic steroid 21-hydroxylase deficiency associated with HLA-B14, DR1. N Engl J Med. 1988;319:19–23. doi: 10.1056/NEJM198807073190104. [DOI] [PubMed] [Google Scholar]
- 19.Helmberg A, Tusie-Luna MT, Tabarelli M, Kofler R, White PC. R339H and P453S: CYP21 mutations associated with nonclassic steroid 21-hydroxylase deficiency that are not apparent gene conversions. Mol Endocrinol. 1992;6:1318–22. doi: 10.1210/mend.6.8.1406709. [DOI] [PubMed] [Google Scholar]
- 20.Globerman H, Amor M, Parker KL, New MI, White PC. Nonsense mutation causing steroid 21-hydroxylase deficiency. J Clin Invest. 1988;82:139–44. doi: 10.1172/JCI113562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiou SH, Hu MC, Chung BC. A missense mutation at Ile172Asn or Arg356Trp causes steroid 21-hydroxylase deficiency. J Biol Chem. 1990;265:3549–52. [PubMed] [Google Scholar]
- 22.Wedell A, Thilén A, Ritzén EM, Stengler B, Luthman H. Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: Implications for genetic diagnosis and association with disease manifestation. J Clin Endocrinol Metab. 1994;78:1145–52. doi: 10.1210/jcem.78.5.8175971. [DOI] [PubMed] [Google Scholar]
- 23.Chan AO, But WM, Ng KL, Wong LM, Lam YY, Tiu SC, et al. Molecular analysis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency in Hong Kong Chinese patients. Steroids. 2011;76:1057–62. doi: 10.1016/j.steroids.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Loke KY, Lee YS, Lee WW, Poh LK. Molecular analysis of CYP-21 mutations for congenital adrenal hyperplasia in Singapore. Horm Res. 2001;55:179–84. doi: 10.1159/000049992. [DOI] [PubMed] [Google Scholar]
- 25.Khan AH, Aban M, Raza J, Ul Haq N, Jabbar A, Moatter T. Ethnic disparity in 21-hydroxylase gene mutations identified in Pakistani congenital adrenal hyperplasia patients. BMC Endocr Disord. 2011;11:5. doi: 10.1186/1472-6823-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barbat B, Bogyo A, Raux-Demay MC, Kuttenn F, Boué J, Simon-Bouy B, et al. Screening of CYP21 gene mutations in 129 French patients affected by steroid 21-hydroxylase deficiency. Hum Mutat. 1995;5:126–30. doi: 10.1002/humu.1380050205. [DOI] [PubMed] [Google Scholar]
- 27.Lee HH, Lee YJ, Wang YM, Chao HT, Niu DM, Chao MC, et al. Low frequency of the CYP21A2 deletion in ethnic Chinese (Taiwanese) patients with 21-hydroxylase deficiency. Mol Genet Metab. 2008;93:450–7. doi: 10.1016/j.ymgme.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 28.Asanuma A, Ohura T, Ogawa E, Sato S, Igarashi Y, Matsubara Y, et al. Molecular analysis of Japanese patients with steroid 21-hydroxylase deficiency. J Hum Genet. 1999;44:312–7. doi: 10.1007/s100380050167. [DOI] [PubMed] [Google Scholar]
- 29.Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. 1992;90:584–95. doi: 10.1172/JCI115897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finkielstain GP, Chen W, Mehta SP, Fujimura FK, Hanna RM, Van Ryzin C, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96:161–72. doi: 10.1210/jc.2010-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mornet E, Crété P, Kuttenn F, Raux-Demay MC, Boué J, White PC, et al. Distribution of deletions and seven point mutations on CYP21B genes in three clinical forms of steroid 21-hydroxylase deficiency. Am J Hum Genet. 1991;48:79–88. [PMC free article] [PubMed] [Google Scholar]
- 32.Ordoñez-Sánchez ML, Ramírez-Jiménez S, López-Gutierrez AU, Riba L, Gamboa-Cardiel S, Cerrillo-Hinojosa M, et al. Molecular genetic analysis of patients carrying steroid 21-hydroxylase deficiency in the Mexican population: Identification of possible new mutations and high prevalence of apparent germ-line mutations. Hum Genet. 1998;102:170–7. doi: 10.1007/s004390050672. [DOI] [PubMed] [Google Scholar]
- 33.Marino R, Ramirez P, Galeano J, Perez Garrido N, Rocco C, Ciaccio M, et al. Steroid 21-hydroxylase gene mutational spectrum in 454 Argentinean patients: Genotype-phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2011;75:427–35. doi: 10.1111/j.1365-2265.2011.04123.x. [DOI] [PubMed] [Google Scholar]
- 34.Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab. 2000;85:1059–65. doi: 10.1210/jcem.85.3.6441. [DOI] [PubMed] [Google Scholar]
- 35.Tusie-Luna MT, Traktman P, White PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265:20916–22. [PubMed] [Google Scholar]
- 36.L’Allemand D, Tardy V, Grüters A, Schnabel D, Krude H, Morel Y. How a patient homozygous for a 30-kb deletion of the C4-CYP 21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2000;85:4562–7. doi: 10.1210/jcem.85.12.7018. [DOI] [PubMed] [Google Scholar]
- 37.Dolzan V, Sólyom J, Fekete G, Kovács J, Rakosnikova V, Votava F, et al. Mutational spectrum of steroid 21-hydroxylase and the genotype-phenotype association in Middle European patients with congenital adrenal hyperplasia. Eur J Endocrinol. 2005;153:99–106. doi: 10.1530/eje.1.01944. [DOI] [PubMed] [Google Scholar]
- 38.Araujo RS, Billerbeck AE, Madureira G, Mendonca BB, Bachega TA. Substitutions in the CYP21A2 promoter explain the simple-virilizing form of 21-hydroxylase deficiency in patients harbouring a P30L mutation. Clin Endocrinol (Oxf) 2005;62:132–6. doi: 10.1111/j.1365-2265.2005.02184.x. [DOI] [PubMed] [Google Scholar]