

Abstract
Adrenocortical cancers in childhood are very rare tumors. The tumors have varied presentation — either virilizing forms or presentation with Cushing's syndrome, or both. In children, due to the rapid development of symptoms they come to attention early, however, if not diagnosed and treated early can have a downhill course. The last decade has seen the emergence of new diagnostic imaging modalities. There is also intense ongoing research in newer treatment modalities as these tumors can be unresectable or have a high recurrence rate.
Keywords: Childhood adrenocortical carcinoma, Cushing's syndrome, virilizing tumors
INTRODUCTION
Adrenocortical carcinoma (ACC) are rare tumors that have a bimodal distribution, the first peak is in children less than five years and the second around the fifth decade.[1] Although most adult ACC are non-functional, in the pediatric age group, nearly 95% are functional.[2] Virilization is the most common abnormality and Cushing's syndrome and hyperaldosteronism are less frequent.[3] We begin the review with a case vignette and subsequently discuss the clinical presentation, diagnosis, and management. Newer imaging modalities and treatment are discussed.
CASE REPORT
A two-year-old girl presented to a pediatrician for poor growth and obesity. The pediatrician suspected Cushing's syndrome and referred her to an endocrinologist, but the parents did not comply. The parents reported again after two years. During this period the girl's growth velocity was poor; she had become fatter, developed acne, and had excessive hair growth over body, hence was referred to this tertiary care center.
On examination her height was 78 cm (SDS — 2.6, growth velocity 3 cm / year), weight was 26 kgs (SDS + 6.03), and blood pressure was 124 / 92 mmHg (normal < 88 / 50 mmHg). She had Cushingoid facies, signs of virilization in the form of pustular acne, hirsutism (Ferriman Gallaway score 20), presence of axillary hairs, puberache (Tanner pubic stage 4), and clitoromegaly. There was a large mass in the right side of the abdomen. There were no neurocutaneous markers. There was no family history of malignancy. Her bone age was 6.4 years (TW 2 method).. Laboratory investigations revealed normal hematological and biochemical parameters. Morning (0800 hours) serum cortisol was 44 μg / dL, and was non-suppressible with dexamethasone (cortisol-35 μg / dL). Other hormonal levels were as follows: dihyrdro-epiandrosterone sulfate 777 μg / dL (22 – 184), 17-OH-progesterone 7.7 ng / mL (< 1.0), and testosterone 0.29 ng / mL (< 0.4). Ultrasound of abdomen revealed a heteroechoiec mass occupying the most of right side of the abdomen and calcification could be appreciated in the centre of the mass. Computed tomography (CT) of the abdomen showed a heteroechoiec right suprarenal mass measuring 13 × 10 × 11 cm, with cystic and calcific areas and an attenuation of 35 Hounsefield Units (HU). Fine needle aspiration cytology (FNAC) from the mass was suggestive of an adrenocortical tumor [Figure 1]. A Positron emission tomography (PET)–CT confirmed the tumor, which had heterogeneous Fluorodeoxyglucose (FDG) uptake with a standardized uptake value (SUV) of 7.2 compared to the liver SUV of 1.5. Both lungs revealed multiple, mildly FDG avid end arterial pulmonary nodules scattered in both the lungs [Figure 2]. With the abovementioned findings the lung lesions were taken as distant metastasis. In view of the stage IV disease, the prognosis was explained to the parents and the child was offered cisplatin-based palliative chemotherapy and ketoconazole, which the parents refused.
Figure 1.
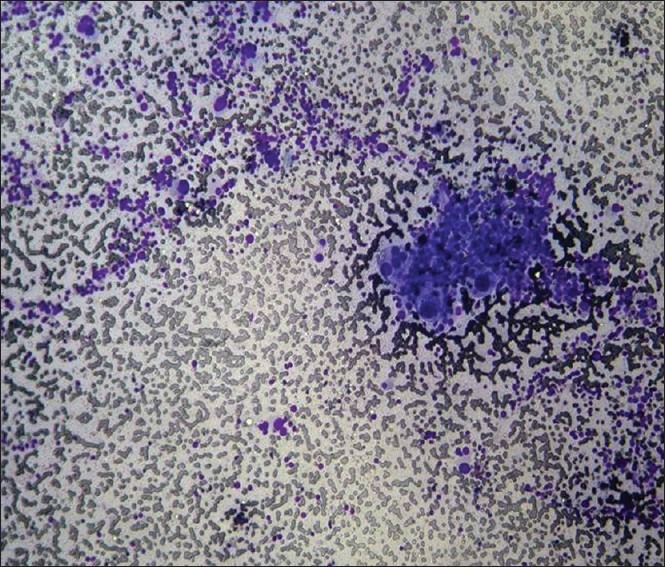
Photomicrograph of fine needle aspiration from the right adrenal mass, showing sheets of dispersed cells with foamy fragile cytoplasm, uniform, enlarged and hyperchromatic nuclei with inclusions and multi-lobed nucleoli
Figure 2.
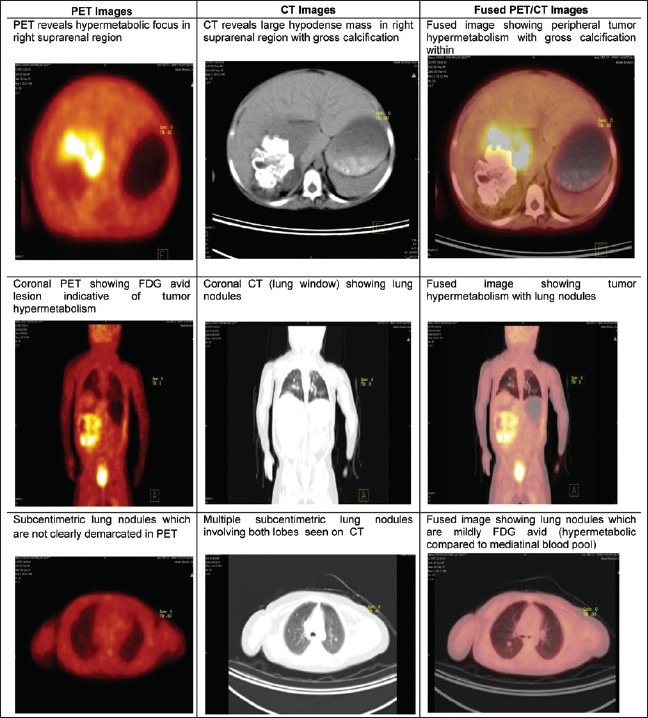
Fused Fluorodeoxyglucose / Positron emission tomography-CT image showing an Fluorodeoxyglucose-avid lesion in the right adrenal gland with distant metastasis in the lungs
DISCUSSION
The case described herewith is a classical presentation of childhood ACC. ACC is a very rare tumor in the pediatric population. Most of the tumors are sporadic; however, a fraction of them might be familial. Familial ACC can be associated with Li–Fraumeni syndrome, Weidemann–Beckwith syndrome, multiple endocrine neoplasia type 1 or Carney complex.[4] The etiopathogenesis of ACC is not completely understood. Abnormalities in the insulin-like growth factor II (IGF II),[5] p53 tumor suppressor gene,[5] steroidogenic factor 1 overexpression,[6] and mutations of the β catenin gene have been implicated.[7]
Most childhood ACC present with virilization (84.2%), with purely virilizing in 55%. This might be associated with overproduction of other adrenocortical hormones. Isolated Cushing's syndrome in the pediatric age group in seen in about 5%. About 10% are non-functional.[2] This in contrast to adults where most of the ACC are non-functional, and if functional, most of them tend to be Cushing's syndrome. Our patient was one of the rare case, where both androgen and glucocorticoid excess have started simultaneously, as she had poor growth inspite of virilization, which tend to cause growth acceleration. Cushing's syndrome is more common when the tumor size is more than 10 cm as seen in our patient.[8]
There is often a delay of about eight months between onset of symptoms and thus a delay in diagnosis.[4] ACC tends to grow and metastasize rapidly if untreated. It is very unfortunate in this girl as her first visit to the pediatrician was at the age of 2 years but the second visit only at 4 years of age. ACC tend to grow and metastasize rapidly if untreated and this is what happened in this young girl.
Workup in a patient suspected of ACC begins with hormonal estimation. The following tests are performed:[4]
Low-dose dexamethasone cortisol suppression test (LDDST). In pediatric patients we give 0.5 mg dexamethasone, six hourly, for 48 hours, and subsequently, with serum cortisol sampling at 0 and 48 hours in children weighing more than 40 kgs. If the child weighs less than 40 kgs the dose of dexamethasone is 30 μg / kg / day in divided doses. An overnight DST (ONDST) can be performed as a screening test using dexamethasone in a dose of 15 μg / kg / day. A normal child will suppress serum cortisol to less than 1.8 μg / dL (50 nmol / L) at 48 hours. To localize the cause of Cushing's syndrome one can use high dose DST (HDDST) in a dose of 120 μg / kg / day
Twenty-four-hour urinary free cortisol excretion. This test can be unreliable in the pediatric age group, as complete collection is an issue, moreover, it is required to be performed at least thrice. Although it has a good sensitivity its specificity is poor
Midnight serum cortisol of more than 4.4 μg / dL has a sensitivity of 99.9% in diagnosing Cushing's syndrome
Baseline serum dehydroepiandrosterone sulfate (DHEAS), 17-hydroxyprogesterone. These represent the adrenal androgen secretion. The normal values are based upon the age and pubertal status of the child
Baseline serum estradiol in boys
Baseline serum testosterone, androstenedione
Serum aldosterone and plasma renin activity
Twenty-four-hour urinary catecholamines or metanephrines excretion. This is not needed in all patients. The need to perform this should also be guided by the clinical profile of the patient. However, before FNAC of an adrenal mass is performed, pheochromocytoma should be ruled out
The next step in evaluation is imaging. Imaging is essential to localize the tumor, plan the surgical procedure, and evaluate the locoregional spread, distant metastases, and staging of the disease. A CT of the abdomen will be the initial imaging modality. It is difficult to differentiate between a benign adenoma and ACC radiologically, but there are features that might favor ACC:[9]
Size of the tumor — > 4 cm is more likely to harbor ACC; suspicion becomes even stronger if it is > 6 cm. Our patient had a tumor > 10 cm, which also favors Cushing's phenotype
Shape — usually well-demarcated, but presence of irregular margins are seen in ACC
Texture — heterogeneous intensity due to hemorrhage, necrosis, and fibrosis, and can have a stellate appearance, which is considered an important finding. Calcifications are also an important finding[10]
Attenuation — in unenhanced images it is more than 10 HU
Vascularity — more vascular
Contrast washout — < 50% after 10 minutes of contrast administration
Ultrasonography and magnetic resonance imaging (MRI) are helpful in detecting the spread into the inferior vena cava.
The role of PET in the evaluation of adrenal mass is evolving.[11] PET has the advantage of determining the functional characteristics of the lesion. Various radiopharmaceuticals have been used. 131I 6β-Iodomethylnorcholesterol binds to the low density lipoprotein in the lipid-rich adenomas and has a specificity of 100% and sensitivity of 70% in detecting these lesions. FDG PET can differentiate between benign and malignant lesions. Adenomas will show an uptake that is less compared to that of the liver, while ACC has a higher uptake. 11C metomidate, which binds to 11 α hydroxylase can be used in ACC. 111In-octreotide has been used in Cushing's syndrome.
FDG-PET is being used increasingly in the initial evaluation of ACC and in post-operative follow up. Maurea et al.,[12] showed that FDG PET in non-secreting adrenal lesions had sensitivity, specificity, and a negative predictive value of 100%, to detect the malignant lesions. FDG-PET not only recognizes the malignant potential of the lesion, but also aids in detecting the distant metastases as shown by Becherer et al.[13] This becomes important in the staging of the disease and the therapy planned. FDG-PET has also been used successfully to detect local recurrences.[14] The intensity of the FDG uptake has also been used to prognosticate ACC. A maximal SUV of > 10 is associated with a poorer outcome. Likewise a FDG uptake volume of > 150 mL is associated with bad prognosis.[15]
Fine needle aspiration cytology can be performed during initial evaluation. FNAC can only differentiate between metastatic lesion to adrenals and an adrenocortical tumor. It cannot differentiate between a benign and malignant lesion, for which distant metastases has to be demonstrated. However, a scoring system like the updated Weiss system on histopathological examination can predict a malignant lesion. This system uses five parameters — > 5 mitoses / 50 high power fields, ≤ 25 % clear tumor cells in the cytoplasm, abnormal mitoses, necrosis, and capsular invasion. Each parameter is given one point if present. If the score is ≥ 3 it is suggestive of malignancy.[16] A recent guideline advices against FNAC with the possibility of capsular breach, causing seeding of tumor cells in the surrounding tissue.[17]
Staging in childhood ACC varies from that in adults. Complete surgical resection is the only hope for long-term remission in these tumors. The staging involves the size, weight, and amount of resection of the tumor. Stage I is a tumor < 5 cm, weighing < 200 g, with complete resection; stage II — > 5 cm, > 200 g, with complete resection; stage III — local spread to lymph nodes, kidney, inferior vena cava or incomplete resection, and stage IV — distant metastases to either lung or liver or to both.[8] Seventy-five percent of the children present in stage I / II, 10% in stage III, and the remaining in stage IV.
Surgery remains the cornerstone of treatment in stages I – III of ACC. Open surgery remains the gold standard and laparoscopic surgery is discouraged in view of excess locoregional recurrences. The aim of surgery should be to achieve margin negative, that is, R0 resection of the tumor. Even after this the recurrence rate can be as high as 80%.[17]
Those patients with severe symptoms, unresectable or recurrent tumors require some form of medical therapy. These are of two types. First, chemotherapy for the control of tumor growth and second, to control the symptoms of hormonal excess.The adrenolytic drug, mitotane is the most effective drug. Apart from its cytotoxic effect it also inhibits adrenal steroidogenesis and acts to control excess hormone production. The patient might require steroid replacement during therapy with mitotane. Palliative cisplatin-based chemotherapy and / or radiotherapy can be an alternative to surgical debulking. Ketoconazole or metyrapone is used to control hypercotisolemic symptoms. If the patient is unable to take it orally or the symptoms need to be controlled acutely, intravenous etomidate is used with concurrent steroid replacement.[17]
Newer modalities of treatment are being investigated to treat ACC:[18]
-
Targeted therapies:
- Insulin-like growth factor 2 (IGF2) – this hormone is among the most consistently expressed in ACC. Its action in mediated by binding to the IGF 1 receptor. Figitumumab, an anti-IGF 1R monoclonal antibody has shown encouraging results
- Multikinase inhibitors like sorafenib and sunitinib are also undergoing trials
- Epidermal growth factor receptor and vascular endothelial growth factor inhibitors, although they sound exciting because of their increased expression in ACC, have not yielded encouraging results
- Mammalian target of rapamycin signaling (mTOR) inhibitors, everolimus and temsirolimus, are being investigated
- Wnt signaling inhibitors and inverse agonist of steroidogenic factor 1 are undergoing in vitro evaluation for ACC. These drugs may be useful, especially in childhood ACC, where their expression is increased
Percutaneous radiofrequency ablation has been used for tumors less than 5 cm, for short-term local control of ACC
Radionucleotide therapy with iodine-labeled metomidate can be a futuristic modality
Coming back to the patient, the delayed interval for diagnosis proved unfortunate for the patient. With the use of PET we were able to detect distant metastases to the lungs and accordingly stage the patient. The parents of the child refused the chemotherapy offered.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Ng L, Libertino JM. Adrenocortical carcinoma: Diagnosis, evaluation and treatment. J Urol. 2003;169:5–11. doi: 10.1016/S0022-5347(05)64023-2. [DOI] [PubMed] [Google Scholar]
- 2.Michalkiewcz E, Sandrini B, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG, et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the international pediatric adrenocortical tumor registry. J Clin Oncol. 2004;22:838–45. doi: 10.1200/JCO.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 3.Cagle PT, Hough AJ, Pysher TJ, Johnson EH, Kirkland RT, Holcombe JH, et al. Comparison of adrenal cortical tumors in children and adults. Cancer. 1986;57:2235–7. doi: 10.1002/1097-0142(19860601)57:11<2235::aid-cncr2820571127>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 4.Sutter JA, Grimberg A. Adrenocortical tumors and hyperplasias in childhood - Etiology, genetics, clinical presentation and therapy. Pediatr Endocrinol Rev. 2006;4:32–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Patalano A, Brancato V, Mantero F. Adrenocortical cancer treatment. Horm Res. 2009;71:99–104. doi: 10.1159/000178049. [DOI] [PubMed] [Google Scholar]
- 6.Almeida MQ, Soares IC, Ribeiro TC, Fragoso MC, Marins LV, Wakamatsu A, et al. Steroidogenic factor 1 overexpression and gene amplification are more frequent in adrenocortical tumors from children than from adults. J Clin Endocrinol Metab. 2010;95:1458–62. doi: 10.1210/jc.2009-2040. [DOI] [PubMed] [Google Scholar]
- 7.Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagnere AM, et al. Mutations of beta-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005;65:7622–7. doi: 10.1158/0008-5472.CAN-05-0593. [DOI] [PubMed] [Google Scholar]
- 8.Teinturier C, Pauchard MS, Brugieres L, Landai P, Chaussain JL, Bougnere PF. Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Med Pediatr Oncol. 1999;32:106–11. doi: 10.1002/(sici)1096-911x(199902)32:2<106::aid-mpo7>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 9.Young WJ., Jr The Incidentally discovered adrenal mass. N Engl J Med. 2007;356:601–10. doi: 10.1056/NEJMcp065470. [DOI] [PubMed] [Google Scholar]
- 10.Ribeiro J, Ribeiro RC, Fletcher BD. Imaging findings in pediatric adrenocortical carcinoma. Pediatr Radiol. 2000;30:45–51. doi: 10.1007/s002470050013. [DOI] [PubMed] [Google Scholar]
- 11.Pacak K, Eisenhofer G, Goldstein DS. Functional imaging of endocrine tumors: Role of positron emission tomography. Endocr Rev. 2004;25:568–80. doi: 10.1210/er.2003-0032. [DOI] [PubMed] [Google Scholar]
- 12.Maurea S, Klain M, Mainolfi C, Ziviello M, Salvatore M. The diagnostic role of radionuclide imaging in evaluation of patients with nonhypersecreting adrenal masses. J Nucl Med. 2001;42:884–92. [PubMed] [Google Scholar]
- 13.Becherer A, Vierhapper H, Potzi C, Karanikas G, Kurtaran A, Schmaljohann J, et al. FDG-PET in adrenocortical carcinoma. Cancer Biother Radiopharm. 2001;16:289–95. doi: 10.1089/108497801753131363. [DOI] [PubMed] [Google Scholar]
- 14.Mackie GC, Shulkin BL, Ribeiro RC, Worden FP, Gauger PG, Mody RJ, et al. Use of [18F]fluorodeoxyglucose positron emission tomography in evaluating locally recurrent and metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2006;91:2665–71. doi: 10.1210/jc.2005-2612. [DOI] [PubMed] [Google Scholar]
- 15.Leboulleux S, Dromain C, Bonniaud G, Auperin A, Caillou B, Lumbroso J, et al. Diagnostic and prognostic value of 18-fluorodeoxyglucose positron emission tomography in adrenocortical carcinoma: A prospective comparison with computed tomography. J Clin Endocrinol Metab. 2006;91:920–5. doi: 10.1210/jc.2005-1540. [DOI] [PubMed] [Google Scholar]
- 16.Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C, et al. Weiss system revisited: A clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol. 2002;26:1612–9. doi: 10.1097/00000478-200212000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Schteingart DE, Doherty GM, Gauger PG, Giordano TJ, Hammer GD, Korobkin M, et al. Management of patients with adrenal cancer: Recommendations of an international consensus conference. Endocr Relat Cancer. 2005;12:667–80. doi: 10.1677/erc.1.01029. [DOI] [PubMed] [Google Scholar]
- 18.Tacon LJ, Prichard RS, Soon PS, Robinson BG, Clifton-Bligh RJ, Sidhu SB. Current and emerging therapies for advanced adrenocortical carcinoma. Oncologist. 2011;16:36–48. doi: 10.1634/theoncologist.2010-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]