

Abstract
We report a 17-year-old girl evaluated for primary amenorrhea. Cytogenetic analysis of the peripheral blood lymphocytes revealed normal autosomes with 46X inv (Y) confirming the diagnosis of Turner's syndrome with Y cell line. Treatment was initiated with conjugated estrogen while recommending bilateral prophylactic oophorectomy to the patient. One year later the patient presented with abdominal mass, biopsy of the specimen following resection confirmed dysgerminoma originating from right ovary with no invasion or metastasis. The literature is reviewed with regard to the various pathogenetic mechanisms proposed for the development of germ cell tumors in ovary, the cytogenetic findings and recommendations to handle such scenario.
Keywords: Dysgermonoma, gonadal dysgenesis, gonadoblastoma, mosaicism, Turner's syndrome, Y cell line
INTRODUCTION
Turner's syndrome (TS) is one of the most common chromosomal aneuploidy and is present in 1:2000 to 1:2500 live births.[1] It is a disorder caused by partial or complete X-chromosome monosomy and characterized by short stature, ovarian failure, and specific somatic abnormalities[2] Approximately 40-60% of patients with TS have 45XO karyotype, while remaining subjects have a chromosomal abnormality of X-chromosome including severe mosaicism.[3] Normal or a structurally abnormal Y-chromosome or Y-derived sequences in TS individuals is found in 6-9% cases.[4] Early detection of Y-derived sequences is of great importance because of the high risk (10-30%) of developing gonadal tumors.[5] Gonadoblastoma and dysgerminoma are the most common malignant tumors of the dysgenetic gonads.[6] At times the gonadoblastoma can undergo transformation into invasive dygerminoma in 60% cases and also into other malignant forms of germ cell tumors.[7] Prophylactic gonadectomy should be recommended in patients with TS and Y-chromosome mosaicism.[8]
CASE REPORT
A 17-year-old female was evaluated for primary amenorrhea. She was borne to parents of nonconsanguineous marriage, delivered at term with an uneventful birth history. Physical and mental milestones and developmental history throughout the infancy and childhood was normal. There was no history of headache, vomiting, seizures, chronic illness, any drug/radiation exposure. She was the second child of her parents with normal elder brother.
On examination patient appeared to be in good general health and a height of 155.5 cm, weight of 59 kg. She had no midline deformity or stigmata of TS. She had Tanner stage 1 breast and pubic hair development. On pelvic examination she had hypoplastic external genitalia. Systemic checkup including abdominal and cardiovascular examination was normal.
Routine hematology, urine analysis was normal. Bone age determined by Greulich and Pyle was 11 years. Hormonal evaluation revealed FSH- 46.5 IU/L (N-1.2-13.2), LH- 60.6 IU/L (N- 4.9-14.5), estrogen-4.0 pg/ml (N-19.0-111), prolactin 37 ng/ml (N-0.9-14.1). Hormonal tests for other anterior pituitary functions were within normal limits. Ultrasonography revealed a hypoplastic uterus (2.0 × 2.0 × 1.1 cm) with small ovaries (right 2 cm and left 1.9 cm). The chromosomal study of peripheral blood lymphocyte with G-T-G banding in 20 analyzed metaphases revealed normal autosomes and abnormal sex chromosomes in the form of 46X inv (Y) [Figure 1]. Treatment with conjugated estrogen (0.01 mg) was started. The patient was advised for prophylactic bilateral oophorectomy after explaining about the potential malignant risk.
Figure 1.
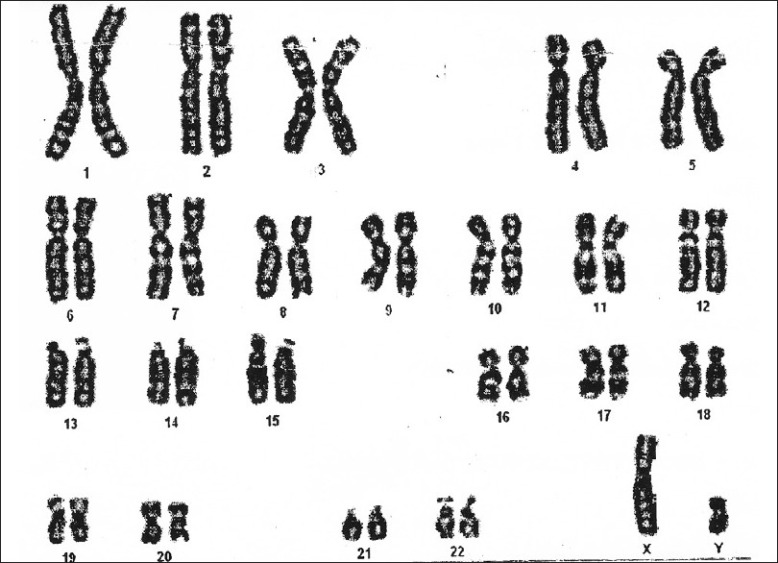
Karyotype of the patient - normal autosomes and abnormal sex chromosomes in the form of 46X inv (Y)
Examination at follow-up after 12 months revealed adequate breast development and growth of axillary and pubic hairs (B III, P III, A III). There was no menarche. On abdominal examination there was an non tender, ovoid 8 × 7 cm mass, solid in consistency and freely movable, in lower abdomen and pelvis. Ultrasonography revealed a large, solid, lobulated mass with irregular internal echogenicity emanating from right ovary [Figure 2]. The patient was subjected to laparotomy and a smooth and bosselated tumor of 11 × 10 × 7 cm was removed from right ovary. No metastases were evident. Histopathology of the biopsy specimen revealed uniformly appearing large, round cells with vesicular nuclei and clear or finely granular cytoplasm that is eosinophilic admixed with lymphocytes in the stroma, confirming the diagnosis of dysgerminoma [Figure 3]. Postoperative course was uneventful and the patient was discharged 4 days later.
Figure 2.
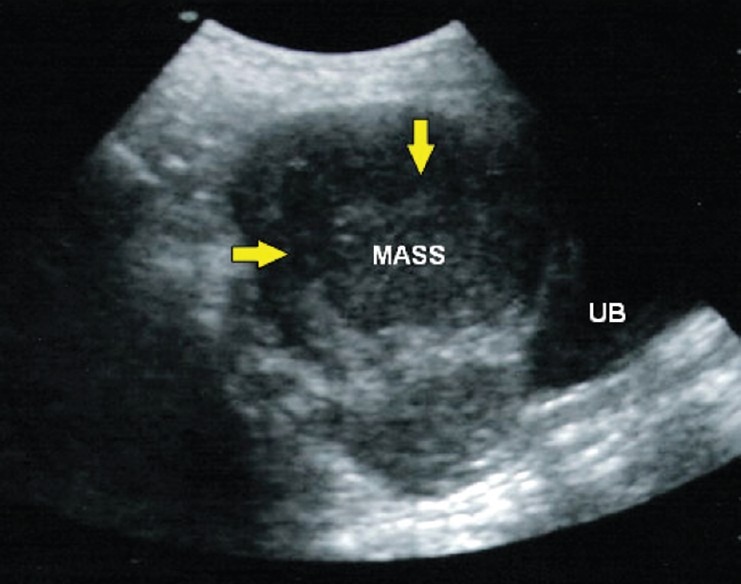
Ultrasonography of abdomen: large, solid, lobulated mass with irregular internal echogenicity emanating from the right ovary
Figure 3.
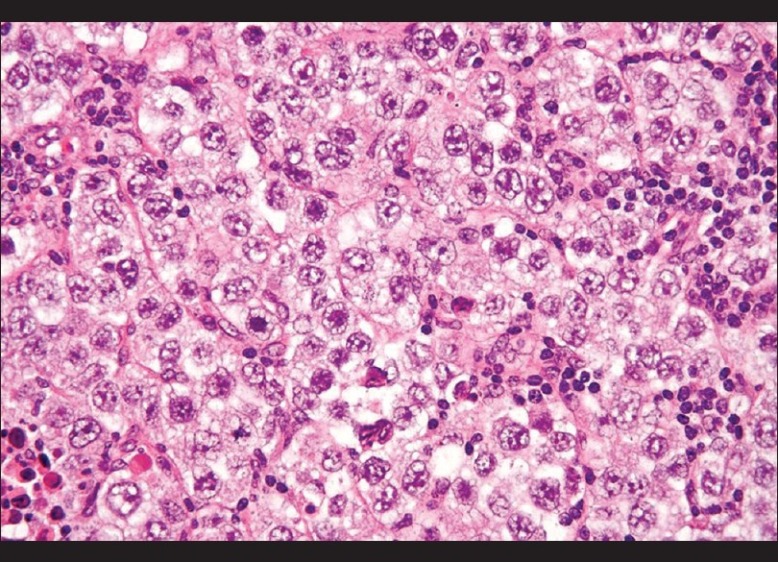
Histopathology of the biopsy specimen - large, round cells with vesicular nuclei and clear or finely granular cytoplasm that is eosinophilic admixed with lymphocytes in the stroma, confirming the diagnosis of dysgerminoma (H and E, ×300)
DISCUSSION
In the present case dysmorphic features of TS were not found. Guedes et al. also studied a girl who despite her 45X/46X, der (Y) karyotype displayed no features of TS except for decreased growth speed.[9] Our patient had only the features of delayed bone age. Except for short stature, all other findings are inconstant. Possible explanation might be undetected mosaicism, since diagnosis is usually made by analyzing between 5-30 peripheral blood lymphocytes to determine the karyotype and the second line is often present as a proportion of no more than 1-2% of the individual's cells.[10] The absence of SHOX (short stature homebox containing gene) haploinsufficiency or actual SHOX duplication could be the possible reason for lack of short stature in our patient.[11]
Hormonal evaluation of our patient revealed elevated gonadotropins with low estrogen levels. We found increase in prolactin as observed by other authors.[12] The frequency of Y-chromosome in TS as reported by various authors varies. It ranges from 4% to 61% depending on the methodological approach.[13] The use of molecular techniques such as fluorescence in situ hybridization (FISH) and polymerase chain reaction (PCR) has substantially improved the detection of low frequency cell lines and possible structural abnormalities.[14] In our patient inv Y was detected by the cytogenetic method.
Dysgenetic gonads serve as the risk factor for origin of germ cell tumors.[15] Patients with disorders of sexual development carry an additional risk. The precursor lesion in a dysgenetic gonad is gonadoblastom, which has the potential to progress toward invasive germ cell tumors, particularly dysgerminoma and less frequently embryonic carcinoma, teratoma, yolk sac tumor, and choriocarcinoma.[16] Hyperandrogenism is commonly associated with gonadoblastoma, especially in cases of coexistence with dysgerminoma.[17] Our patient had no such features. GBY (gonadoblastoma locus on the Y chromosome) is assumed to be related to the origin of tumors located in the pericentromeric region of Yp.[18] It is different than the testis determining region (SRY). SRY is located proximally while GBY is located distally. Germ cell tumors (GCT) are more common in whom the Y-chromosome retains the normally intense florescence of distal Yq. Gonadal dysgenesis is due to the presence of Y-chromosome lacking the male determining region (SRY).[19] SRY is suggested to be functional in spermatogenesis[20] and is the structural gene for H-Y antigen. H-Y antigen is an oncogene, and seems to be closely associated with development of GCTs.[21] Low occurrence of GCT in Turner females with Y-chromosome material might be related to the lower level of H-Y antigen found in Turner syndrome.[22] The lack of a regulatory Y-chromosome gene or genes on the X-chromosome, which may influence the level of H-Y antigen or other potential carcinogenic oncogenes might be the reason why Y-chromosome material in TS seems to cause the development of GCT to a much lesser degree than in XY females.[6]
Salo et al. have identified TSPY (tissue-specific protein Y encoded) as a candidate gene for GBY locus related to development of GCT.[23] Furthermore immune reactivity of POU5F1 (OCT 4) gene at 6p 21.31 is found to be positive for GCTs. Other studies have established the presence of Y-chromosome sequences like PABY (Y-chromosome pseudoautosomal bound region 1), SRY, ZFY (Y-chromosome-specific zinc finger gene), Yc (Y-chromosome centromeric alphoid region), and Yq (Y chromosome long-arm heterochromatic region) as risk factors for GCT.[24] Germ cells persist for longer time in the dysgenetic gonads of XY female than in gonads from 45X patients. Germ cells obviously have increased potential for malignancy in certain environment.[25]
It has traditionally been recommended that a search for Y-chromosome fragments in TS should only be performed under two circumstances: when there are signs of virilization and/or when there is a marker chromosome not identified by classical cytogenetics.[26] Nevertheless, when Canto et al.[27] used PCR to study 107 Turner syndrome patients with a 45, X karyotype, they identified Y-chromosome material in 10 (9.3%) of them. Prophylactic gonadectomy was indicated, and two of the six patients who agreed to undergo the surgery presented gonado-blastoma, thus indicating an incidence of 33%.
Similarly, Bianco et al.[14] studied different tissue samples from 20 TS patients by means of PCR and found that seven (35%) of the 45, X patients presented Y-chromosome-specific sequences in at least one of the tissues studied. Four (14%) of these patients underwent prophylactic gonadectomy, and bilateral gonadoblastoma was found in a 16-year-old girl. In this case, the presence of Y-chromosome sequences was not associated with virilization, thus reinforcing the idea that the absence of this characteristic does not rule out the possibility of the presence of hidden Y-chromosome fragments.
Prevalence of GCT in Y-chromosome varies among different studies pegged at 10-30%.[5] So prophylactic gonadectomy is recommended in such patients.[8] But Gravholt et al. have questioned this consensus on basis of their finding of frequency of Y-chromosome material in TS as 12.2% with occurrence of gonadoblastoma among Y-positive patients as 7-10%.[6] They recommended detailed vaginal sonography with color doppler sonography of gonads at regular intervals. But they go on to conclude that gonadectomy is still the procedure of choice to exclude malignancy with absolute certainty. However, as most of the studies have found higher incidence of GCT to the tune of 30-35%,[5] we thought it suitable to recommend prophylactic gonadectomy to our patient. The male genotype can exist as XY, XYY, a partial Y-chromosome such as dicentric Y, a ring Y or even a Y translocated to another chromosome.[28]
The mean age of gonadal tumor diagnosis as per previous reports is 18 years.[29] Our patient was 17 years old. Virilization in patients with TS indicates the presence of Y cell line within the gonads, even if Y cell is not identified in the peripheral blood. So virilization is an indication for the presence of Y mosaicism.[30] Our patient had no virilizing features. A few times, the gonadal tumor has been reported without evidence of Y-chromosome.[28] The basis was dysgenetic gonad itself giving rise to GCT. It leads to the suggestion that laparoscopic and ovarian biopsy might be considered in any patient suspected of dysgenetic or dysplastic ovaries.
CONCLUSION
Early detection of Y-chromosome sequence in TS is of great importance because of high risk of gonadal tumor development. Though the occurrence of Y-chromosome material in TS is low, it should be searched meticulously by molecular techniques like FISH, PCR. Since most studies have indicated 30-35% incidence of GCTs, prophylactic gonadectomy should be offered to TS patients with Y-chromosome.
ACKNOWLEDGMENTS
All the authors would extend their heartfelt thanks to Mrs Sruti Jammula, M Pharm, PhD, Dr Jagadeesh Tangudu, M Tech, MS, PhD and Mrs Sowmya Jammula, M Tech for their immense and selfless contribution towards manuscript preparation, language editing and final approval of text.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Lippe B. Turner syndrome. Endocrinol Metab Clin North Am. 1991;20:121–52. [PubMed] [Google Scholar]
- 2.Zinn AR, Page DC, Fisher EM. Turner syndrome: The case of the missing sex chromosome. Trends Genet. 1993;9:90–3. doi: 10.1016/0168-9525(93)90230-f. [DOI] [PubMed] [Google Scholar]
- 3.Mathur A, Stekol L, Schatz D, McLaren NK, Scott ML, Lippe B. The parental origin of the single X chromosome in Turner syndrome: Lack of correlation with parental age or clinical phenotype. Am J Hum Genet. 1991;48:682–6. [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobs P, Dalton P, James R, Mosse K, Power M, Robinson D, et al. Turner syndrome: A cytogenetic and molecular study. Ann Hum Genet. 1992;61:471–83. doi: 10.1046/j.1469-1809.1997.6160471.x. [DOI] [PubMed] [Google Scholar]
- 5.Verp MS, Simpson JL. Abnormal sexual differential and neoplasia. Cancer Genet Cytogenet. 1987;25:191–218. doi: 10.1016/0165-4608(87)90180-4. [DOI] [PubMed] [Google Scholar]
- 6.Gravholt CH, Fedder J, Naeraa RW, Muller J. Occurrence of gonadoblastoma in females with Turner syndrome and Y chromosome material: A population study. J Clin Endocrinol Metab. 2000;85:3199–202. doi: 10.1210/jcem.85.9.6800. [DOI] [PubMed] [Google Scholar]
- 7.Saenger P. Clinical review 48: The current status of diagnosis and therapeutic intervention in Turner syndrome. J Clin Endocrinol Metab. 1993;77:297–301. doi: 10.1210/jcem.77.2.8345029. [DOI] [PubMed] [Google Scholar]
- 8.Bianco B, Lipay MV, Melaragno MI, Guedes AD, Verreschi IT. Detection of hidden Y mosaicism in Turner's syndrome: Importance in the prevention of gonadoblastoma. J Pediatr Endocrinol Metab. 2006;19:1113–7. doi: 10.1515/jpem.2006.19.9.1113. [DOI] [PubMed] [Google Scholar]
- 9.Guedes AD, Bianco B, Lipay MV, Brunoni D, de Lourdes Chauffaille M, Verreschi IT. Determination of the sexual phenotype in a child with 45 X / 46 X, Idic (Yp) mosaicism: Importance of the relative proportion of the 45 X line in gonadal tissue. Am J Med Genet A. 2006;140A:1871–5. doi: 10.1002/ajmg.a.31363. [DOI] [PubMed] [Google Scholar]
- 10.Yorifuji T, Muroi J, Kawai M, Sasaki H, Momoi T, Furusho K. PCR-based detection of mosaicism in Turner syndrome patients. Hum Genet. 1997;99:62–5. doi: 10.1007/s004390050312. [DOI] [PubMed] [Google Scholar]
- 11.Ogata T, Kosho T, Wakui K, Fukushima Y, Yoshimoto M, Miharu N. Short stature homebox containing gene duplication on the der (X) chromosome in female with 45 X/ 46 X der (X), gonadal dysgenesis and tall stature. J Clin Endocrinol Metab. 2000;85:2927–30. doi: 10.1210/jcem.85.8.6745. [DOI] [PubMed] [Google Scholar]
- 12.Bohnet HG. Gonadotropins, prolactin and sex steroid secretion in pubertal maturation of normal girls and agonadal subjects. In: Flamigni S, Venturoli S, Givens JR, editors. Adolescence in females. Chicago: Year book Medical Publishes; 1985. pp. 153–65. [Google Scholar]
- 13.Quliter CR, Taylor K, Conway GS, Nathwani N, Delhanty JD. Cytogenetic and molecular investigations of Y chromosome sequences and their role in Turner syndrome. Ann Hum Genet. 1998;25:191–218. doi: 10.1046/j.1469-1809.1998.6220099.x. [DOI] [PubMed] [Google Scholar]
- 14.Patsalis PC, Sismani C, Hadjimarcou MI, Kitsjou-Tzeli S, Tzezou A, Hadjiathanasiou CG, et al. Detection and incidence of cryptic Y chromosome sequences in Turner syndrome patients. Clin Genet. 1998;53:249–57. doi: 10.1111/j.1399-0004.1998.tb02691.x. [DOI] [PubMed] [Google Scholar]
- 15.Shakkebaek NE, Holm M, Hoel-Hansen C, Jorgensen N, Rajpert-De Meyts E. Association between testicular dysgenesis syndrome (TDS) and testicular neoplasia: Evidence from 20 adult patients with signs of maldevelopment of the testis. APMIS. 2003;111:1–9. doi: 10.1034/j.1600-0463.2003.11101031.x. [DOI] [PubMed] [Google Scholar]
- 16.Pauls K, Franke FE, Büttner R, Zhou H. Gonadoblastoma: Evidence for a stepwise progression to dysgerminoma in a dysgenetic ovary. Virchows Arch. 2005;447:603–9. doi: 10.1007/s00428-005-1272-9. [DOI] [PubMed] [Google Scholar]
- 17.Mancilla EE, Poggi H, Repetto G, Rumié H, García H, Ugarte F, et al. Y chromosome sequences in Turner's syndrome: Association with virilization and gonadoblastoma. J Pediatr Endocrinol Metab. 2003;16:1157–63. doi: 10.1515/jpem.2003.16.8.1157. [DOI] [PubMed] [Google Scholar]
- 18.Tsuchiya K, Reijo R, Page DC, Diesteche CM. Gonadoblastoma: Molecular definition of the susceptibility region on the Y chromosome. Am J Hum Genet. 1995;57:1400–7. [PMC free article] [PubMed] [Google Scholar]
- 19.Diesteche CM, Casanova M, Saal H, Friedman C, Sybert V, Graham J, et al. Small deletions of the short arm of the Y chromosome in 46 XY females. Proc Natl Acad Sci U S A. 1986;83:7841–4. doi: 10.1073/pnas.83.20.7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fedder J, Hjort T. Evidence for more than one male-specific antigen in the human. In: Boutaleb Y, Gzouli A, editors. New concepts in reproduction. Carnforth: Parthenon; 1991. pp. 117–20. [Google Scholar]
- 21.Warner BA, Monsaert RP, Stumpf PG, Kulin HE, Wachtel SS. 46, XY gonadal dysgenesis: Is oncogenesis related to H-Y phenotype or breast development? Hum Genet. 1985;69:79–85. doi: 10.1007/BF00295534. [DOI] [PubMed] [Google Scholar]
- 22.Wolf U, Fraccaro M, Mayerova A, Hecht T, Zuffardi O, Hameister H. Turner syndrome patients are H-Y positive. Hum Genet. 1980;54:315–8. doi: 10.1007/BF00291575. [DOI] [PubMed] [Google Scholar]
- 23.Salo P, Kaariainen H, Petrovic V, Peltomaki P, Page DC, de la Chapelle A. Molecular mapping of the putative gonadoblastoma locus on the Y chromosome. Genes Chromosomes Cancer. 1995;14:210–4. doi: 10.1002/gcc.2870140309. [DOI] [PubMed] [Google Scholar]
- 24.Canto P, Alfaro SK, Jimenez AL, Soderlund D, Barron C, Reyes E, et al. Gonadoblastoma in Turner syndrome patients with non mosaic 45 X karyotype and Y chromosome sequences. Cancer Genet Cytogenet. 2004;150:70–2. doi: 10.1016/j.cancergencyto.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Cussen J, MacMohan RA. Germ cells and ova in dysgenetic gonads of a 46-XY female dizygotic twin. Am J Dis Child. 1979;133:373–5. doi: 10.1001/archpedi.1979.02130040027005. [DOI] [PubMed] [Google Scholar]
- 26.Frías JL, Davenport ML. Committee on Genetics and Section on Endocrinology.Health supervision for children with Turner syndrome. Pediatrics. 2003;111:692–702. doi: 10.1542/peds.111.3.692. [DOI] [PubMed] [Google Scholar]
- 27.Canto P, Kofman-Alfaro S, Jiménez AL, Söderlund D, Barrón C, Reyes E, et al. Gonadoblastoma in Turner syndrome patients with nonmosaic 45, X karyotype and Y chromosome sequences. Cancer Genet Cytogenet. 2004;150:70–2. doi: 10.1016/j.cancergencyto.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Alonso RP, Nieto K, Alvarez R, Palma I, Nájera N, Eraña L, et al. Distribution of Y-chromosome-bearing cells in gonadoblastoma and dysgenetic testis in 45, X/46, XY infants. Mod Pathol. 2005;18:439–45. doi: 10.1038/modpathol.3800293. [DOI] [PubMed] [Google Scholar]
- 29.Brant WO, Rajimwale A, Lovell MA, Travers SH, Furness PD, 3rd, Sorensen M, et al. Gonadoblastoma and Turner syndrome. J Urol. 2006;175:1858–60. doi: 10.1016/S0022-5347(05)00932-8. [DOI] [PubMed] [Google Scholar]
- 30.Modi D, Bhartiya D. Y chromosome mosaicism and occurrence of gonadoblastoma in cases of Turner syndrome and amenorrhoea. Reprod Biomed Online. 2007;15:547–53. doi: 10.1016/s1472-6483(10)60387-2. [DOI] [PubMed] [Google Scholar]