

Abstract
A 7-year-old boy presented with umbilical hernia and short stature. Growth retardation, recurrent upper respiratory tract infections and delayed developmental milestones were present from infancy. Umbilical hernia was diagnosed at the age of 5 years. On examination, he had short-trunk dwarfism, large head circumference, coarse facial features, joint stiffness, hepatosplenomegaly, and mild mental retardation. He had normal biochemical parameters, thyroid function tests and arterial blood gas analysis. Radiological evaluation showed that the child had Hunter syndrome with findings of J-shaped sellaturcica, proximal bulleting of metacarpals, spatulated ribs and anterior beaking of lumbar vertebrae. The second case was a 6-year-old girl with umbilical hernia, short stature, normal biochemistry and radiological findings of mucopolysaccharidosis. However, she also had corneal opacity; confirmed by slit-lamp examination, which led to the diagnosis of Hurler–Scheie syndrome. Enzymatic studies could not be done in both the cases, as they are not available at most centers.
Keywords: Mucopolysaccharidosis, short stature, umbilical hernia
INTRODUCTION
Children with short stature and umbilical hernia are often seen in endocrine clinics and are diagnosed to have congenital hypothyroidism. However, there are exceptions. Mucopolysaccharidosis should be considered in the differential diagnoses. Mucopolysaccharidosis is rare (incidence: 3.53 per 100,000 live births[1]). Mucopolysaccharidosis type I and mucopolysaccharidosis type II have incidences of 0.69 per 100,000 live births[1] and 0.64 per 100,000 live births, respectively. The Mucopolysaccharidosis Registry[2] has 76 cases of Hurler–Scheie syndrome enrolled (mucopolysaccharidosis IH-S) from 26 countries. International Hunter Syndrome Registry has 263 patients enrolled from 16 countries. There are a very few case reports from India [Tatapudi et al.[3] (mucopolysaccharidosis I), Chaturvedi et al.[4] (mucopolysaccharidosis II), Wander et al.[5] (mucopolysaccharidosis II)]. We present the first case report of mucopolysaccharidosis from northeastern region of India.
CASE REPORTS
Case 1
A 7-year-old boy presented to the Pediatric Surgery Out-patient department with umbilical hernia. He had growth retardation and was referred to us for endocrine evaluation. The child was born post term at 10 months of gestation (birth weight: 3.5 kg). There was no h/o prolonged neonatal jaundice, hoarse cry, feeding or sucking difficulty. Growth retardation, recurrent upper respiratory tract infections, and delayed developmental milestones were present from infancy. Umbilical hernia was diagnosed when the child was evaluated at the age of 5 years.
On examination, the child was short (height 99 cm; < fifth percentile; –4.02 Standard Deviation, height age 4 years). Upper segment to lower segment ratio was 0.811. He had a large head circumference (53 cm), coarse facial features, depressed nasal bridge, delayed eruption of permanent teeth, joint stiffness with flexion deformity of elbow and distal interphalangeal joints. There was hepatosplenomegaly, umbilical hernia, mild mental retardation (Intelligence Quotient: 58.22), normal fundus examination, bilateral normal hearing and no corneal clouding [Figures 1 and 2].
Figure 1.
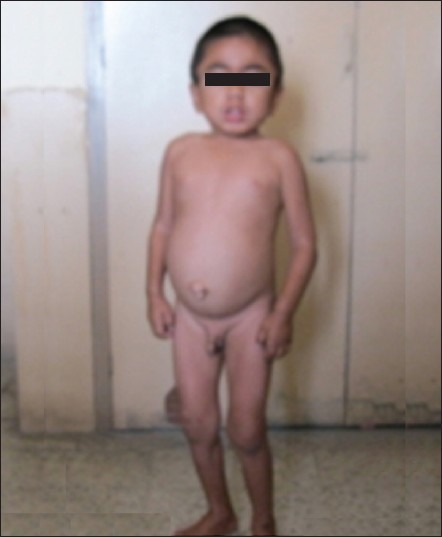
Child with Hunter syndrome
Figure 2.
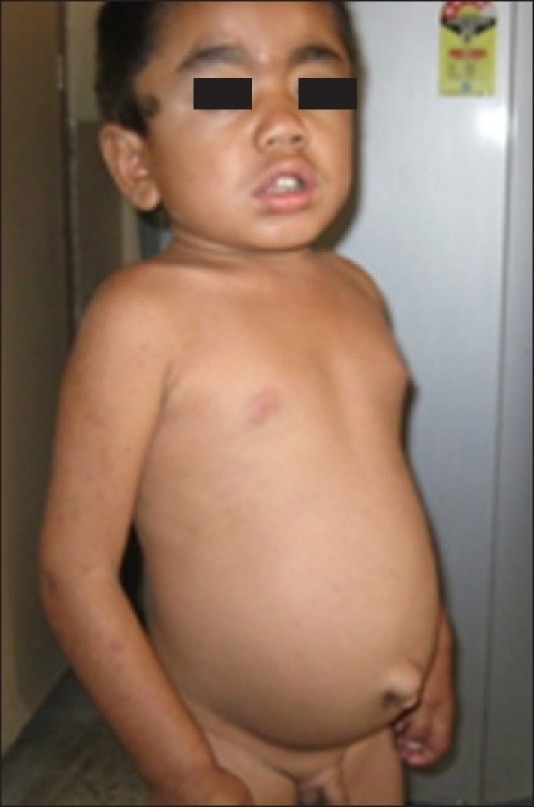
Child with Hunter syndrome
On investigation, he was found to be normoglycemic and had normal renal function (S. creatinine 0.4 mg/dl). The other findings were as follows: hemoglobin (Hb) 12.1 gm %; S. calcium 9.3 mg/dl; S. albumin 4.2 g/dl; S. phosphorus 4.4 mg/dl; S. alkaline phosphatase 172 U/l. He had no evidence of metabolic acidosis.The thyroid function tests were normal [thyroid stimulating hormone (TSH) 0.931 mIU/l; T4 level 134 nmol/l] [Figures 3–7].
Figure 3.

X-ray left hand: Proximal ends of metacarpals are tapering (bullet-shaped metacarpals)
Figure 7.
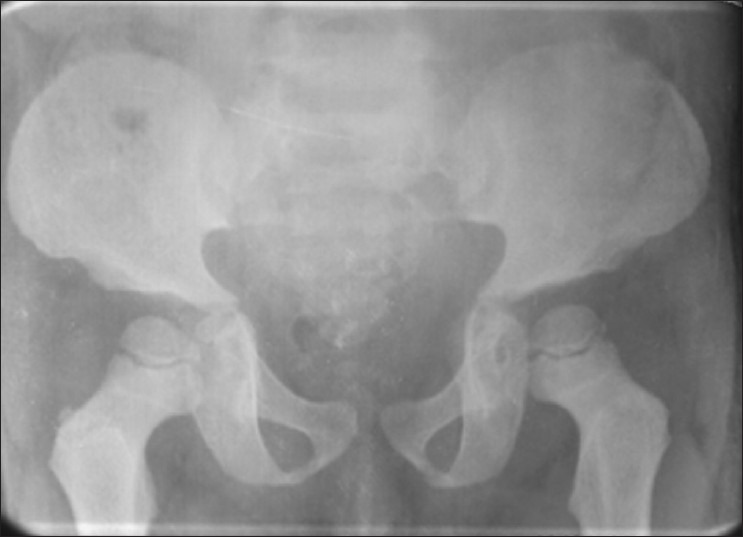
X-ray pelvis shows bilateral iliac flaring and acetabular roof is horizontal
Figure 4.
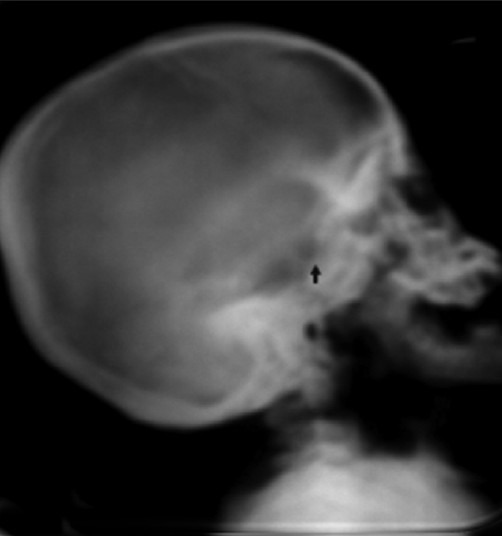
X-ray skull shows J-shaped sellaturcica
Figure 5.

X-ray chest shows spatulated ribs
Figure 6.

X-ray dorsolumbar spine shows beaking of anterior part of L3 vertebrae
USG abdomen revealed hepatosplenomegaly with umbilical hernia. Echocardiography (ECHO) study revealed normal findings. We could neither measure urinary glycosaminoglycan (GAG) levels nor perform iduronate sulfatase enzyme activity assay, as they are not done at our center and are also not available at most other centers. Our diagnosis of mucopolysaccharidosis II (Hunter syndrome) in this case was confirmed from his history, clinical examination and skeletal survey.
Case 2
A 6-year-old girl presented to the Pediatric Surgery department with umbilical hernia. It was found that she also had corneal opacity. The child had growth retardation and was referred to us for endocrine evaluation. She was born at term (birth weight: 2.9 kg). There was no h/o prolonged neonatal jaundice, hoarse cry, or feeding difficulty. Umbilical hernia and recurrent rhinorrhea were present since neonatal period. Growth retardation, delayed developmental milestones, and facial dysmorphism were present from infancy. Eruption of teeth started after 1 year of age. Opacities in eye were noticed by parents in the second year. She had difficulty in speech and could speak short sentences but not clearly.
On examination, the child was short (height 85 cm; < fifth percentile; –5.15 Standard Deviation; height age 2.5 years) and had upper segment to lower segment ratio of 41:46 (short trunk dwarfism). She had a large head circumference (54 cm), coarse facial features, depressed nasal bridge, cloudy cornea, joint stiffness with flexion deformity of elbow. There was hepatosplenomegaly, umbilical hernia and mild mental retardation (Intelligence Quotient: 56.11). Slit-lamp examination confirmed corneal clouding. Hearing assessment revealed bilateral normal hearing. Other examinations revealed normal findings.
On investigation, she was found to be normoglycemic and had normal renal function (S. creatinine 0.7 mg/dl). Her other findings were as follows: Hb 11.1 gm %; S. calcium 9.1 mg/dl; S. albumin 4.0 g/dl; S. phosphorus 4.4 mg/dl; S. alkaline phosphatase 177 U/l. Arterial blood gas analysis and thyroid function tests were normal [Figures 8 and 9].
Figure 8.
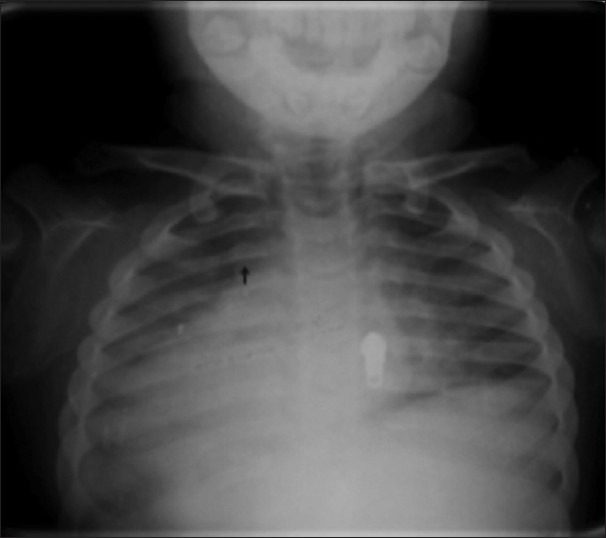
X-ray chest shows spatulated ribs
Figure 9.
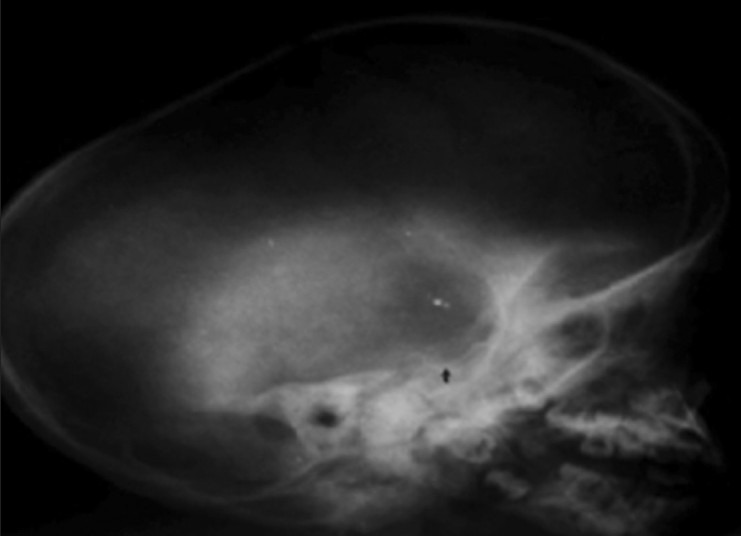
X-ray skull shows J-shaped sellaturcica
We could neither measure urinary GAG levels nor assay for α-l- iduronidase (IDUA) enzyme activity, as they are not performed at our center and are also not available at most other centers. Our diagnosis of mucopolysaccharidosis I (Hurler–Schei disease) in this case was confirmed from her history, clinical examination and skeletal survey.
DISCUSSION
Mucopolysaccharidosis was first described by Charles Hunter, a Canadian physician.[6,7] Mucopolysaccharidoses are a rare group of metabolic disorders (incidence: 0.69 cases per 100,000 live births).[1] Mucopolysaccharidosis I (incidence: 0.69 cases per 100,000 live births)[1] is caused by a deficiency of IDUA enzyme. Its clinical features range from severe mental retardation with hepatomegaly, corneal clouding, cardiac involvement and death in early childhood, to milder symptoms such as mild visceral involvement with normal intelligence and a normal life span. Hurler syndrome (mucopolysaccharidosis IH) is the severe form, while Scheie syndrome (MPS IS) is the milder form, and Hurler-Scheie syndrome (mucopolysaccharidosis IH-S) is the intermediate phenotype. Our patient (Case 2) had Hurler-Scheie syndrome (mucopolysaccharidosis IH-S). The Mucopolysaccharidosis Registry[2] has 302 cases of mucopolysaccharidosis I enrolled from 26 countries, of which 76 (~25%) are affected with Hurler–Scheie syndrome (mucopolysaccharidosis IH-S). Umbilical hernia and corneal opacity were the presenting features in most of the cases, as was in our case. Vijay et al.[8] have described the course of disease in 29 patients with Hurler–Schei syndrome who attended the mucopolysaccharidosis clinic in Manchester, United Kingdom. However, there have been a very few case reports from India. Tatapudi et al.[3] have reported a case of Hurler-Scheie syndrome from India.
Mucopolysaccharidosis type II or Hunter syndrome is rare and caused by a deficiency of iduronate-2-sulfatase. The common clinical presentations are a large head (dolichocephalic), short stature, mental retardation, coarse facial features, umbilical hernia, a broad nose with flared nostrils, and upper respiratory tract infection. Our patient (Case 1) had Hunter syndrome. The Hunter's Outcome Survey[9] is maintaining an International Registry of cases of Hunter syndrome. Two hundred and sixty-three patients from 16 countries have enrolled in Hunter's Outcome Survey.[9] The median age at enrollment was 12.2 years. The median age of onset of symptoms and diagnosis of Hunter syndrome were 1.5 and 3.5 years, respectively;[9] while in our case the symptoms were present since the first year of life and the case was diagnosed at the age of 7 years. Abdominal hernia was the earliest presenting symptom, as was in our case. Facial dysmorphism and hepatosplenomegaly were present in 95% and 89% of patients, respectively;[9] both these features were present in our case. The typical radiological findings in a case of Hunter syndrome help differentiate it from other causes of child presenting with short stature and umbilical hernia, such as congenital hypothyroidism. There have been only a few case reports of Hunter syndrome from India. Chaturvedi et al.[4] have described a case of Hunter syndrome from western India. Wander et al.[5] have described a case of Hunter syndrome from North India.
Thus, to conclude, mucopolysaccharidosis, although rare, should be considered in the differential diagnoses of children with short stature and umbilical hernia, and can be diagnosed based on clinical and radiological features, as enzymatic studies are not available at most centers.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Baehner F, Schmiedeskamp C, Krummenaver F, Miebach E, Baiboui M. Cumulative incidence rate of mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28:1011–7. doi: 10.1007/s10545-005-0112-z. [DOI] [PubMed] [Google Scholar]
- 2.Pastores G, Arn P, Beck M, Clarke JT, Guffon N, Kaplan P, et al. The MPS I registry: Design, methodology, and early findings of a global disease registry for monitoring patients with mucopolysaccharidosis type I. Mol Genet Metab. 2007;91:37–47. doi: 10.1016/j.ymgme.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 3.Tatapudi R, Gunashekhar M, Raju PS. Mucopolysaccharidosis type I Hurler-Scheie syndrome: A rare case report. Contemp Clin Dent. 2011;2:66–8. doi: 10.4103/0976-237X.79287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaturvedi P, Jain VK. Hunter's Syndrome: A case report. Indian J Pediatr. 1978;45:89–90. doi: 10.1007/BF02807303. [DOI] [PubMed] [Google Scholar]
- 5.Wander GS, Sandha GS, Chawla AS, Khurana SB. Multivalvular thickening in a case of Hunter's syndrome. J Assoc Physicians India. 1994;42:161–2. [PubMed] [Google Scholar]
- 6.Wraith JE, Scarpa M, Beck M, Bodamer OA, Meirleir LD, Guffon N, et al. Mucopolysaccharidosis Type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267–77. doi: 10.1007/s00431-007-0635-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Mufioz V, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome) Pediatrics. 2008;121:377–86. doi: 10.1542/peds.2007-1350. [DOI] [PubMed] [Google Scholar]
- 8.Vijay S, Wraith JE. Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr. 2005;94:872–7. doi: 10.1111/j.1651-2227.2005.tb02004.x. [DOI] [PubMed] [Google Scholar]
- 9.Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. Initial report from the Hunter Outcome Survey. Genet Med. 2008;10:508–16. doi: 10.1097/gim.0b013e31817701e6. [DOI] [PubMed] [Google Scholar]