

Abstract
Neuroblastoma is a childhood tumor in which transient therapeutic responses are typically followed by recurrence with lethal chemoresistant disease. In this study, we characterized the apoptotic responses in diverse neuroblastomas using an unbiased mitochondrial functional assay. We defined the apoptotic set-point of neuroblastomas using responses to distinct BH3 death domains providing a BH3 response profile, and directly confirmed survival dependencies. We found that viable neuroblastoma cells and primary tumors are primed for death with tonic sequestration of Bim, a direct activator of apoptosis, by either Bcl-2 or Mcl-1, providing a survival dependency that predicts the activity of Bcl-2 antagonists. The Bcl-2/Bcl-xL/Bcl-w inhibitor ABT-737 showed single agent activity against only Bim:Bcl-2 primed tumor xenografts. Durable complete regressions were achieved in combination with non-curative chemotherapy even for highest-risk molecular subtypes with MYCN amplification and activating ALK mutations. Furthermore, the use of unique isogenic cell lines from patients at diagnosis and at the time of relapse showed that therapy resistance was not mediated by upregulation of Bcl-2 homologues or loss of Bim priming, but by repressed Bak/Bax activation. Together, our findings provide a classification system that identifies tumors with clinical responses to Bcl-2 antagonists, defines Mcl-1 as the principal mediator of Bcl-2 antagonist resistance at diagnosis, and isolates the therapy resistant phenotype to the mitochondria.
Keywords: Bcl-2 homology proteins, mitochondrial profiling, animal models, Bcl-2 antagonist
INTRODUCTION
Cancer remains a major cause of mortality and the emergence of therapy resistance is the principal barrier to cure. Nascent cancer cells must bypass numerous apoptotic checkpoints that safeguard against oncogenesis (1, 2), while established cancers encounter therapeutics that interfere with vital functions to activate mitochondrial apoptosis (3, 4). Not surprisingly, alterations that attenuate these pathways are selected for during tumor initiation and progression and “apoptosis evasion” is considered an essential hallmark of cancer (5). Understanding how specific cancers circumvent the apoptotic program is critical to identifying rational therapeutic approaches. Indeed, numerous agents that target apoptotic mediators are under development and their effective integration into clinical use requires such knowledge.
Neuroblastoma (NB) is a highly lethal pediatric solid tumor. Most patients present with high-risk features (6) and despite intensive multimodal therapy the majority die from progression of therapy resistant disease (7). The Bcl-2 family proteins govern mitochondrial apoptotic responses and are candidate mediators of therapy response (8). They are classified by their highly conserved Bcl-2-homology (BH) domains into the pro-apoptotic BH3-only proteins (containing only the BH3 death-domain), pro-apoptotic multi-domain Bak and Bax (containing BH1, -2 and-3 domains), or anti-apoptotic multi-domain homologues (Bcl-2, Bcl-w, Bcl-xl, Mcl-1, and A1; sharing BH1, -2, -3 and -4 domains). BH3 proteins act as stress sentinels activated by a host of tumor-related insults that include radiation and chemotherapy.
A subset of BH3 proteins can directly engage Bak or Bax at the mitochondrial membrane, leading to their homo-oligomerization, mitochondrial outer membrane permeabilization (MOMP), release of apoptogenic factors such as cytochrome c, and death commitment. Such “direct-activator” BH3 proteins include Bim, Bid, and Puma (9). Alternatively, anti-apoptotic Bcl-2 proteins (e.g., Bcl-2, Mcl-1) can bind activator BH3’s, preventing Bak or Bax activation and providing a buffer against cell death. These tonically neutralized activator BH3 proteins can then be displaced by additional “enabler” BH3 proteins (e.g., Bik, Noxa, Bad) that have a higher affinity for anti-apoptotic docking sites. The state whereby activator BH3 proteins are sequestered by anti-apoptotic Bcl-2 proteins has been termed “primed for death” since such cells are paradoxically sensitized to select BH3 proteins (10). Of note, most non-transformed cells lack BH3 priming, providing a conceptual therapeutic index for agents that antagonize the anti-apoptotic Bcl-2 proteins (11).
We performed mitochondrial profiling on NB cell lines that were genetically diverse to identify their set point for apoptotic signal transduction following exposure to distinct BH3 death-domains, providing a “BH3 response profile” (12). We identified three classes of apoptosis resistance that accurately predicted sensitivity to the Bcl-2 antagonists in vitro. Most NB robustly and reproducibly responded to specific enabler peptides, confirming intact Bak/Bax signaling and inferring that endogenous activator BH3 proteins were tonically suppressed but displaceable (cells were primed). One third of high-risk NBs were most sensitive to BikBH3, suggesting a primary Bcl-2/Bcl-xL/Bcl-w dependence, and were exquisitely sensitive to the Bcl-2/-xL/-w inhibitor, ABT-737, in vitro (IC50 <50 nM). A second subset had a dominant NoxaBH3 response (indicating Mcl-1 survival dependence) and were relatively ABT-737 resistant. The third subset, consisting solely of cell lines derived at relapse following therapy, had absent enabler and markedly blunted activator BH3 peptide responses, a profile consistent with the profound therapy resistance seen clinically at relapse. We hypothesized that relapsed NBs either lose their primed for death status through repressed Bid, Bim or Puma activation, or repress Bak/Bax-mediated pore formation.
We now directly demonstrate that NBs are primed for death through sequestration of Bim, and that the pattern of Bim binding is predicted by the BH3 response profile. Further, the dominant anti-apoptotic site of Bim binding predicts the in vivo response to small molecule Bcl-2 antagonists. Xenografts from NB with Bim sequestered by Bcl-2 are exquisitely sensitive to ABT-737, and cures to a single course of therapy were obtained. Importantly this includes tumors with MYCN amplification and ALK mutations (both R1275Q and F1174L) that are associated with an extremely poor prognosis. Conversely, tumors with Bim sequestered to Mcl-1 are resistant to Bcl-2 antagonists. Using matched tumor cell line pairs obtained at diagnosis and following relapse after therapy, we unequivocally show that relapsed NBs retain Bim priming and anti-apoptotic Bcl-2 dependence patterns indistinguishable from pre-therapy cells. That acquired therapy resistance is associated with repression of Bak and/or Bax mediated apoptotic signal transduction implicates the mitochondria as a major contributor to the post-relapse therapy resistant phenotype.
MATERIALS AND METHODS
Cell Lines
Neuroblastoma cell lines with MYCN amplification [IMR5 (13), NLF, LA-N-5 (30), NGP (14), CHP134, NB-1643 (15), SMS-SAN, SMS-KCN and SMS-KCNR, SMS-KAN and KANR, SK-N-BE(1) and SK-N-BE(2), BE2C, CHLA-15 and CHLA-20, CHLA122 and CHLA136 (16)] and without [NB69 (17), SK-N-SH and SK-N-AS (18)] were grown in RPMI-1640 supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1% OPI, 100 U/ml of penicillin. Tissue culture was at 37° C in a humidified atmosphere of 5% CO2. All cell lines and isogenic pairs were confirmed using short tandem repeat (STR)-based genotyping (AmpFISTR, Applied Biosciences) and matched to the COG cell line genotype database (www.cogcell.org).
Co-immunoprecipitation
Cells were lysed in CHAPS buffer (10 mM HEPES, 150 mM NaCl, 2% CHAPS (Sigma-Aldrich) and added to antibody-matrix complex (ExactCruz Immunoprecipitation Matrix C (Santa Cruz, CA) plus 1-5mg IP antibody) for 24 hours, 4 degrees. Immunoprecipitated proteins were released from the matrix complex using 2X RIPA buffer, run on Nu-PAGE 10% BisTris gels (Invitrogen), transferred to PDVF membranes and detected for Bcl-2 family proteins as described (10). Primary frozen tumor samples were disassociated through a 0.45 μm sterile filter, washed twice with Red Blood Cell Lysing Buffer (Sigma), lysed and immunoprecipitated as above.
Antibodies
Anti-Mcl-1 (BD Pharmingen), anti-Bcl-2 (Dako; and Santa Cruz Biotechnology: sc-492), and anti-Bcl-xL (clone 7B2.5; gift of L.Boise, Emory University), anti-Bak, anti-Bax (#2772), anti-Puma (#4976) and anti-BID (#2002; Cell Signaling Technology), anti-Bim (Millipore Corporation: AB17003), anti-PARP (Cell Signaling #9542) and anti-Casp3 (Cell Signaling #9664) were used.
Mitochondrial profiling
Heavy membrane fractions enriched for functional mitochondria were obtained from NB cells during logarithmic growth, or from tumor xenografts, as described (19). Functional studies were performed with freshly isolated mitochondria suspended to a final concentration of 1 ug/uL in mitochondrial buffer, as described (20). BimBH3 peptide, recombinant tBid protein (R&D Systems; Minneapolis, MN) or 1% DMSO were incubated with mitochondria for 30 minutes at 30 C and cytochrome c release measured in duplicate by ELISA (R&D systems, Minneapolis, MN). Peptide synthesis and assay details are as in (19).
Whole cell JC-1 assay
Cells were plated at 2×104 NB cells/well into 384-well plates in 300 mM Trehalose, 10 mM Hepes-KOH, 80 mM KCl, 1 mM EGTA, 1 mM EDTA, 0.1% BSA, 5 mM succinate; T-EB, and incubated at RT with 100 uM BimBH3 peptide, 2 mM JC-1, 20 mg/ml oligomycin, 0.01% digitonin, and 10 mM b-mercaptoethanol, followed by continuous monitoring for JC1 fluorescence (BioTek Synergy Mx plate reader, Vermont) at 545 +/- 20 nm Ex and 590 +/- nm Em as described (21).
Bak and Bax oligomerization
Mitochondria from NB cells (1 ug/uL/treatment) were treated with BimBH3 peptide (0.0125-50uM) or DMSO for 30 minutes at 30° C followed by a 30 minute incubation with 0.9 mM of 1,6-bismaleimidohexane (BMH, 10mM stock in DMSO, Pierce, #22330) at RT to crosslink oligomers. Treatments were centrifuged to pellet mitochondria, the pellet was dissolved in 1X NuPage loading buffer (Invitrogen) and immunoblotted for Bak and Bax.
Cytotoxicity assays
2-5 × 104 NB cells/well were treated with ABT-737, cytotoxic agent or vehicle controls in RPMI-based media in triplicate. After 48hrs, WST-1 (Roche; Mannheim, Germany) was added and incubated at 37° C for 1 hour. Absorbance was recorded at 450 nm and 620 nm. For log-transformed nonlinear regression curve fit and determination of IC50, Graph Pad Prism software was used (Graph Pad Software, La Jolla, CA). For siRNA, 2×105 cells/mL were transfected with 50 nM Bim or Gapdh siRNA (siGENOME SMARTPOOL, Dharmacon). Etoposide was added 24 hours later and viability was measured by WST-1.
Annexin/PI apoptosis assay
NB cells were treated with ABT-737, melphalan, both agents or DMSO vehicle control. Following a 24 hour incubation, cells were harvested, washed twice with PBS and re-suspended in Binding Buffer (8g NaCl, 0.2g KCl, 1.44g Na2HPO4•7H20, 0.24 g KH2PO4) at a concentration of 1 × 106 cells/mL. Cells were incubated with Annexin V-FITC and Propidium Iodide (BD Biosciences, CA) for 15 minutes and then analyzed immediately by flow cytometry using the FACSCanto II flow cytometer (BD Biosciences, CA).
Murine xenograft studies
Xenografts were established in the flank of nu/nu athymic mice (Jackson Laboratories) as described (19). When tumors volume reached 200-400 mm3, mice (n=10 per arm) were treated by intraperitoneal injections of (1) vehicle control (normal saline, twice weekly for two weeks), (2) cyclophosphamide (75 mg/kg, twice weekly for two weeks), (3) ABT-737 (100 mg/kg daily × two weeks, as in (22)), or (4) the combination of cyclophosphamide with ABT-737 (according to their monotherapy schedule). Animals were sacrificed when tumor volumes exceeded 2000 mm3 and all animal work was performed under a protocol approved by the CHOP and Emory Institutional Animal Care and Use Committee.
Statistical analyses
Survival analyses were performed according to the method of Kaplan and Meier (23) with standard errors according to Peto (24). Comparisons of outcome between subgroups were performed by a 2-sided log-rank test.
RESULTS
Bim is sequestered at the mitochondria of viable NB, inducing a primed for death state that defines its anti-apoptotic dependence
We previously used an isolated mitochondrial cytochrome c release assay and inferred that most NB were primed for death as shown by robust responses to select enabler BH3 peptides that are incapable of directly activating Bak or Bax [(12) and Supplemental Fig. 1]. Using co-immunoprecipitation assays and cell lines from different BH3 response classes we confirmed that Bim is the principal BH3-only death activator in NB (Fig. 1A). All NBs that had a dominant cytochrome c release to the BikBH3 peptide had high Bcl-2 expression, and endogenous Bim bound to Bcl-2. We predicted these cells would have an activator BH3 protein bound to Bcl-xl or Bcl-w, rather than Bcl-2, as Bik is more avid for these hydrophobic pockets (25-27). However, Bik was discovered in association with Bcl-2 (“Bcl-2 interactive killer”) and its pro-apoptotic function can be abrogated by Bcl-2, supporting a functional interaction with Bik (28, 29). In support, BikBH3 peptide was capable of competitively displacing endogenous Bim from Bcl-2 in NB cells with Bim:Bcl-2 priming (Fig. 1B). In contrast, all NB cell lines that had a Noxa-dominant BH3 response profile had Bim neutralized almost exclusively by Mcl-1, despite the co-expression of Bcl-2 in many (Fig. 1A). The absence of ABT-737 responses for this subset reflects the inability of this small molecule to antagonize Bim:Mcl-1, as shown for other tumor models (8).
Figure 1. Bim is the primary death activator in NB.
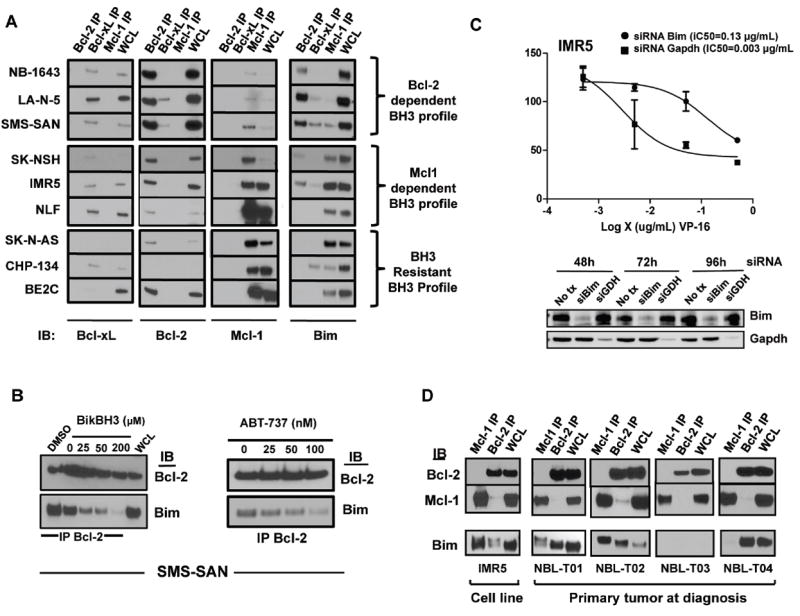
(A) Protein from NB cell lines of distinct BH3 response classes was immunoprecipitated using antibodies for Mcl-1, Bcl-2 or Bcl-xl and immunoblotted for Bim and the anti-apoptotic Bcl-2 family members. The identified dominant Bim interactions with the multidomain anti-apoptotic members are consistent with predictions inferred from BH3 profile results (12). (B) Co-immunoprecipitation (Co-IP) of Bcl-2 shows decreased Bim:Bcl-2 interactions following exposure of isolated mitochondrial lysates to a BikBH3 peptide for 30 minutes. Likewise Co-IP of Bcl-2 following treatment of intact NB cells with ABT-737 shows dose-dependent decreases in Bcl-2 bound Bim, supporting a competitive displacement model. (C) Bim or negative control GAPDH were inhibited using siRNA for 24 hours in IMR5 cells, followed by a 48-hour exposure to etoposide (VP-16). Survival was assessed by WST-1. Immunoblots confirmed target protein inhibition by siRNA. (D) Co-immunoprecipitations of fresh frozen NB tumor at diagnosis show similar Bcl-2 and Mcl-1 sequestration of Bim. WCL, whole cell lysate. All experiments were replicated at least twice and representative findings shown.
Bim was identified in complex with various anti-apoptotic partners in all NBs assessed, but neither alternative BH3 activators (Bid, Puma) nor multi-domain Bak or Bax proteins were sequestered by anti-apoptotic homologues (Supplemental Fig. 2). Bim-EL was the only Bim isoform detected in complex with Bcl-2 members and has been proposed as a necessary BH3-only protein downstream of chemotherapy-induced stress (30), and as a tumor suppressor that cooperates with MYC deregulation (30, 31). To test for a functional role for Bim in NB, we used siRNA-mediated knockdown and showed Bim loss attenuated etoposide-induced cytotoxicity in line with resistant cell line IC50s [Fig. 1C and (32)]. These data suggest Bim is the major mediator of chemoresponse in NB but that its activity is suppressed by anti-apoptotic Bcl-2 proteins. NB cells in which Bim is bound to Bcl-2 are extremely sensitive to ABT-737 in vitro (12). We therefore reasoned that ABT-737 derived its potency by displacing Bim from Bcl-2, and confirmed this by treating cells in culture with ABT-737 and showing a dose-responsive competitive displacement of Bim (Fig. 1B).
To interrogate whether these Bcl-2 family interactions were inherent to the primary tumor or secondary to cell culture adaptation, we performed Co-IPs using fresh frozen NB tumors of different risk-groups obtained at diagnosis (Supplemental Table 1). The same heterogeneous Bcl-2 family binding patterns were identified, even within a single clinical risk-group. Two tumors showed dominant Bcl-2:Bim priming (NBL-T01 and NBL-T04), one had Bim dominantly sequestered by Mcl-1 (NBL-T02), and one tumor that showed no evidence of Bim expression or priming (NBL-T03; Fig. 1D) failed to show other activator BH3’s, Bid or Puma, bound to Mcl-1 or Bcl-2, suggesting it lacks priming altogether (data not shown).
Post-therapy relapsed NBs retain Bim priming, but demonstrate repressed apoptotic signaling at the level of Bak/Bax
The third subset of NB cell lines identified by mitochondrial profiling were characterized by repressed enabler BH3 responses and blunted BidBH3 and BimBH3 responses (direct activators), suggesting they either had lost Bim priming or acquired apoptotic defects at or downstream of Bak and/or Bax (12). Notably, these NBs were derived at relapse following therapy. SK-N-AS, CHP-134, and BE2C had high Mcl-1 expression and sequestered Bim in patterns similar to NBs in the Mcl-1 dependent group, supporting no loss in Bim priming at relapse (Fig. 1A and 2B). Only CHP134 cells appeared qualitatively to have reduced priming when comparing whole cell Bim levels with the Bim pulled down with Mcl-1.
Figure 2. NB cell lines retain Bim expression and priming patterns following relapse after therapy.
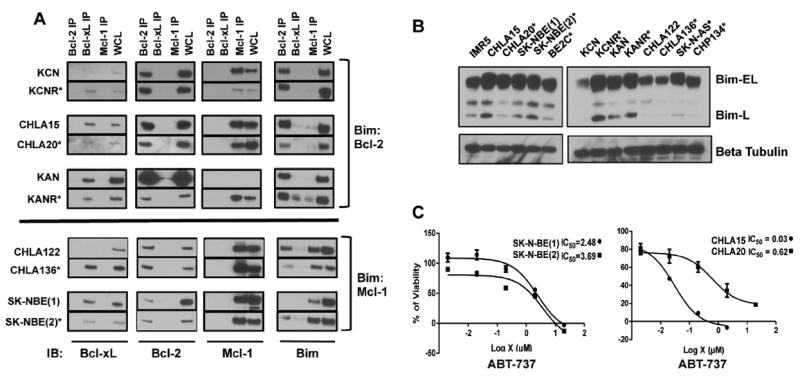
(A) Protein lysates from untreated NB cells derived from matched pre- and post-therapy relapse cell lines were immunoprecipitated with antibodies to Bcl-2 family members, and immunoblotted for Bim and the anti-apoptotic family members. *, denotes the post-relapse member of each pair. The dominant Bim binding protein did not change between pre- and post-relapse specimens. (B) Immunoblot for Bim using whole cell lysates show no loss of Bim-EL expression in post-relapse NB cells (C) Paired NB cell lines (pre- and post-relapse) were exposed to increasing concentrations of ABT-737 and assessed for viability using the WST-1 at 48 hours. As predicted, Bim:Mcl-1 primed SK-N-BE(1) and SK-N-BE(2) cells are ABT-737-resistant, whereas Bim:Bcl-2 primed CHLA-20 cells show increased resistance compared with isogenic pre-therapy CHLA-15 cells. All experiments were replicated at least twice and representative findings shown; error bars in panel (C) are representative of two or more independent biological replicates.
We next assayed isogenic matched tumor cell line pairs from the same patients at the time of initial diagnosis and at the time of relapse following therapy (Supplemental Table 2). Co-immunoprecipitation again demonstrated that most pre- and post-relapse pairs retained consistent anti-apoptotic Bcl-2 family expression as well as indistinguishable Bim priming patterns, suggesting that anti-apoptotic Bcl-2 dependence patterns are not substantially altered in response to therapy (Fig. 2A). Indeed, only KANR showed a gain in expression of an anti-apoptotic Bcl-2 homologue (Mcl-1) not expressed at diagnosis (in KAN) yet this acquired Mcl-1 expression did not neutralize Bim appreciably as minimal Bim:Mcl-1 was seen.
Immunoblots of Bim from whole cell lysates confirmed consistent Bim-EL expression between pre- and post-relapse cells (Fig. 2B). Although the initial cohort of post-relapse cell lines we investigated had Bim:Mcl-1 priming, the additional pre- and post-relapse pair studies identified NBs with Bim:Bcl-2 priming as well. This suggests that cells in either class could develop therapy resistance associated with maintained Bim priming but a reduction in BH3 responsiveness. We therefore assessed ABT-737 responsiveness before and after relapse in the context of both Bcl-2 and Mcl-1 dependence. SK-N-BE(1) (pre-therapy) and SK-N-BE(2) (post-relapse) demonstrate Bim:Mcl-1 priming, and as predicted, neither was sensitive to ABT-737 (IC50s >2uM; Fig. 2C). In contrast, both CHLA-15 (pre-therapy) and CHLA-20 (post-relapse, same patient as CHLA-15) had Bim:Bcl-2 priming that predicts ABT-737 sensitivity, yet only the pre-therapy CHLA-15 cells were sensitive (IC50=30 nM) while CHLA-20 cells were relatively resistant (IC50=620 nM; Fig. 2C). This suggests that diminished apoptotic responses seen in relapsed NB are not primarily due to loss of Bim expression or priming at anti-apoptotic docking sites, but arise downstream of Bim displacement. We reasoned that the defective response to death stimuli in relapsed NB may reside at the level of Bak and/or Bax activation or further downstream.
To explore this we assessed the dose-responsiveness of isolated NB mitochondria to the direct-activator BimBH3. In all cases the pre-therapy cells demonstrated a heightened responsiveness to BimBH3 than did their post-relapse counterparts even at concentrations that saturate anti-apoptotic binding sites (100uM) (Fig. 3A). To ensure responses reflected functional signaling we also assessed cytochrome c release in response to recombinant tBid with similar findings. To confirm this we used a complementary assay in which mitochondrial membrane potential is measured in intact cells using the JC-1 dye (21). Again, post-relapse cells were less sensitive to direct Bak/Bax activation by BimBH3 as gauged by JC-1 fluorescence as a measure of mitochondrial membrane depolarization. As a positive control, FCCP (p-trifluoromethoxy carbonyl cyanide phenyl hydrazone), a mitochondrial oxidative phosphorylation uncoupler that induces MOMP via a non-Bak/Bax mechanism, was capable of fully inducing JC-1 redistribution (Fig. 3B).
Figure 3. NB cell lines demonstrate repressed Bax/Bak activation at the time of relapse.
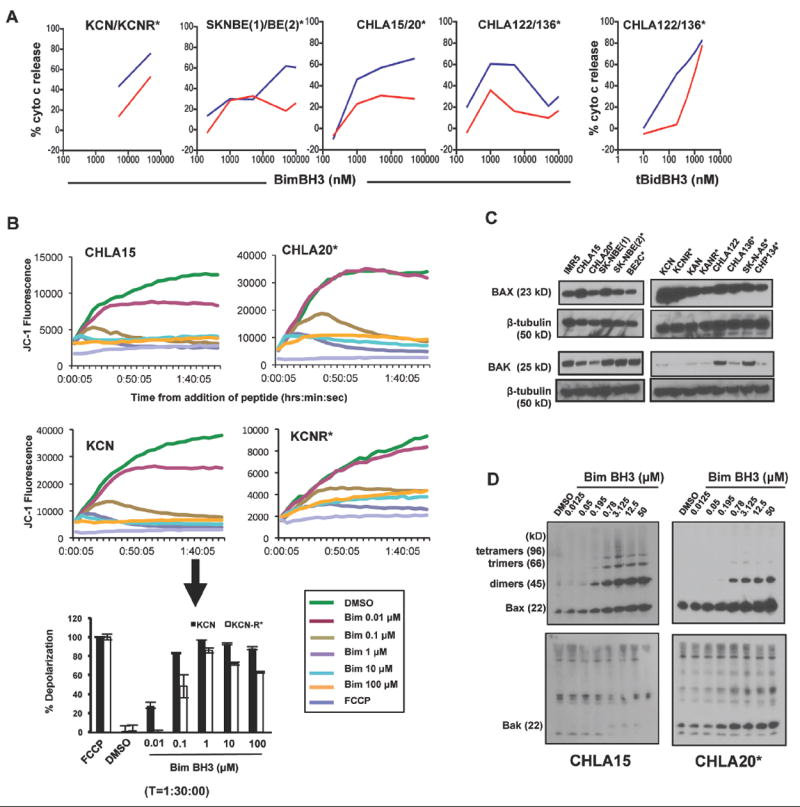
(A) Isolated mitochondria from isogenic NB cell line pairs (*, post-therapy relapse cells) were challenged with increasing concentrations of BimBH3 peptide or recombinant tBid protein and cytochrome c release was measured as a surrogate for apoptosis commitment. Pre-therapy cells (blue) were markedly more responsive to Bim (and tBID, as shown for CHLA122/136) compared to post-relapse cells (red). (B) Differences in mitochondrial responses to Bim in intact whole cells permeabilized with digoxin to incorporate BimBH3. Whole cell responses mimic isolated mitochondrial responses to Bim and are measured by JC-1 release from mitochondria (decreased fluorescence coincides with MOMP). Upper panel, JC-1 emission over time in response to different BimBH3 concentrations; lower panel, JC-1 fluorescence at 90 minutes highlighting decreased depolarization in KCNR compared to KCN. FCCP, p-trifluoromethoxy carbonyl cyanide phenyl hydrazone (positive control for MOMP induction). (C) Isogenic paired cell lines derived from the same tumor at diagnosis and following relapse were immunoblotted for Bax and Bak. In all cell line pairs, the post-therapy relapse cells had evidence for reduced Bak and/or Bax expression. β-tubulin serves as a loading control. (D) Acquired loss of higher order Bax homo-oligomers in CHLA-20 compared to CHLA-15 following direct Bax/Bak activation with BimBH3. Location of higher order oligomers have been previously defined (20). Nonspecific bands higher than 22kDa in the Bak immunoblot are not consistent with functional higher order oligomers (20). Data are representative of at least two biological replicates. Panel A data points represent replicate values and at all points the standard error is <5% (error bars omitted).
We next investigated the pro-apoptotic Bak and Bax proteins that are required for Bim-induced mitochondrial apoptosis (33). In cell lines established at relapse post-chemotherapy, Bak and/or Bax were reduced in expression (Fig. 3C). SK-N-BE cells had reductions in Bax in the relapsed lines (SK-N-BE2 and BE2C) with no reduction in overall Bak expression [similar to KAN and KANR]. CHLA-122 and CHLA-136 had no difference in total Bax but Bak levels were reduced in the post-relapse cells [also with KCN and KCNR]. CHLA-20 cells, in contrast, showed reductions in both Bak and Bax expression compared with the at-diagnosis isogenic CHLA-15. The functional status of the Bak/Bax axis was assessed using oligomerization assays in which higher-order Bak or Bax homo-oligomers are detected in response to a pro-apoptotic signal, BimBH3. Indeed, BimBH3 responsive CHLA-15 cells showed higher order oligomerization of Bax in response to Bim that were absent in their matched post-relapse counterpart, CHLA-20 cells (Fig. 3D).
Bcl-2 antagonists sensitize NBs with Bim:Bcl-2 priming to chemotherapy
We next investigated whether the Bim:Bcl-2 priming patterns seen in a subset of NBs could be exploited therapeutically. We tested NB cell lines with a Bcl-2 dependent profile (SMS-SAN and LA-N-5), Mcl-1-dependent profile (IMR5) or BH3 resistant profile (SK-N-AS) for the effect of ABT-737 on chemotherapy-induced death in vitro. As predicted, in Bcl-2 dependent NBs, cell death from doxorubicin and melphalan was enhanced by 5 nM ABT-737 (Fig. 4A). This synergistic loss of viability was due to increased apoptosis, as confirmed by PARP and caspase 3 cleavage, and phosphatidylserine externalization (Fig. 4B and 4C). In contrast, ABT-737 did not synergize with chemotherapeutics for Mcl-1 dependent (IMR5) or BH3-resistant (SK-N-AS) cells, even at 1 uM concentrations. Similar findings were seen with agents from two additional cytotoxic classes with activity against NB, cisplatinum (DNA platinator) and etoposide (topoisomerase II inhibitor; data not shown).
Figure 4. ABT-737 augments chemotherapy effects against NB in vitro.
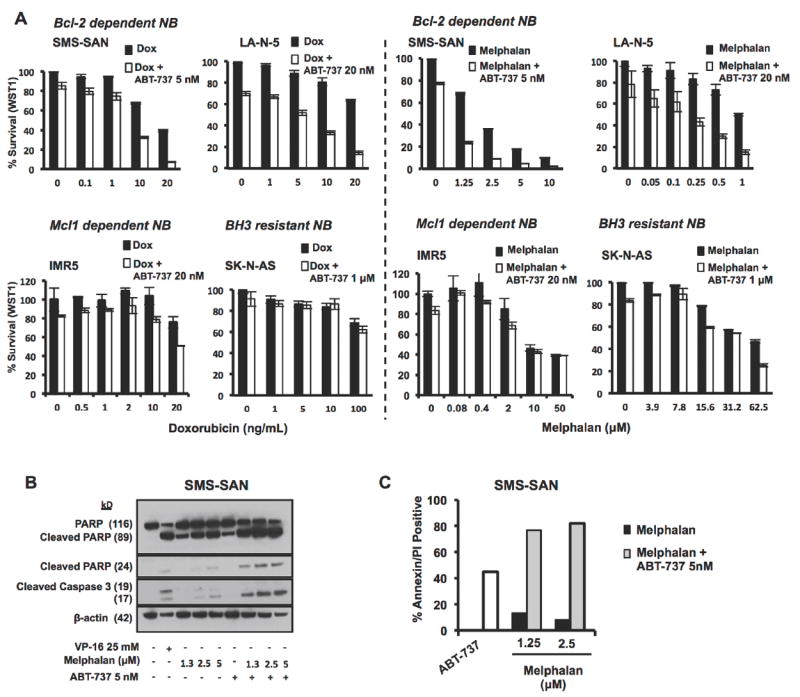
(A) Bcl-2 dependent NB cells, SMS-SAN, (5 × 104cells/well) were added to a 96 well plate and allowed to settle for 24h then treated with ABT-737, cytotoxic agent, combination or vehicle control in triplicate for 48 hours when they were assessed for changes in proliferation - values plotted are the viable cell number (biomass by WST-1 assay) normalized to vehicle-treated cells. ABT-737 enhances cell death from cytotoxics by augmenting the apoptosis effects of melphalan as demonstrated by increased Caspase 3 and PARP cleavage (B) and increased Annexin V positive cells by FACS (C). VP-16 was used as positive control for caspase activation. Error bars represent mean and standard deviation of greater than two biologic replicates.
Bcl-2 antagonists have potent activity in vivo against Bcl-2 dependent NBs
These data support that BH3 response profiles obtained from NB cell lines predict responses to selective Bcl-2 family antagonists (12) as well as synergy with cytotoxics in vitro. However, mitochondrial signaling may be modified through by extrinsic survival cues from the microenvironment or additional stress stimuli (e.g., hypoxia, acidosis) that are not recapitulated in vitro. We have shown that NB cell line BH3 response profiles are preserved in xenografts from mice supporting that the defining characteristics of these responses are relatively cell autonomous (12). However, to determine the extent to which in vivo responses to Bcl-2 antagonists can be predicted, we treated mice harboring xenografts of NB cell lines from different BH3 response classes with ABT-737 alone and with chemotherapy.
ABT-737 monotherapy induced notable tumor regression and extended overall survival by ~50% in mice bearing Bcl-2 dependent SMS-SAN xenografts (p<0.005 by log-rank test), with a median survival of 20.5 ± 10.5 d for ABT-737 treated mice and 14±3.2 d for control mice. For NBs with Mcl-1 dependence (IMR5) or BH3 resistance profile (BE2C), survival was not significantly extended by ABT-737 (Fig. 5; median survival 17±5 d versus 12±3.5 d and 12±5.3 d versus 11±1.6 d, respectively). The response of Bcl-2 dependent NBs to ABT-737 as a single agent supports that even in the context of in situ survival signals the tonic repression of activated Bim is required to maintain cancer cell survival, and antagonism of this is sufficient for anti-tumor activity.
Figure 5. Bim priming patterns predict NB sensitivity to ABT-737 in vivo.
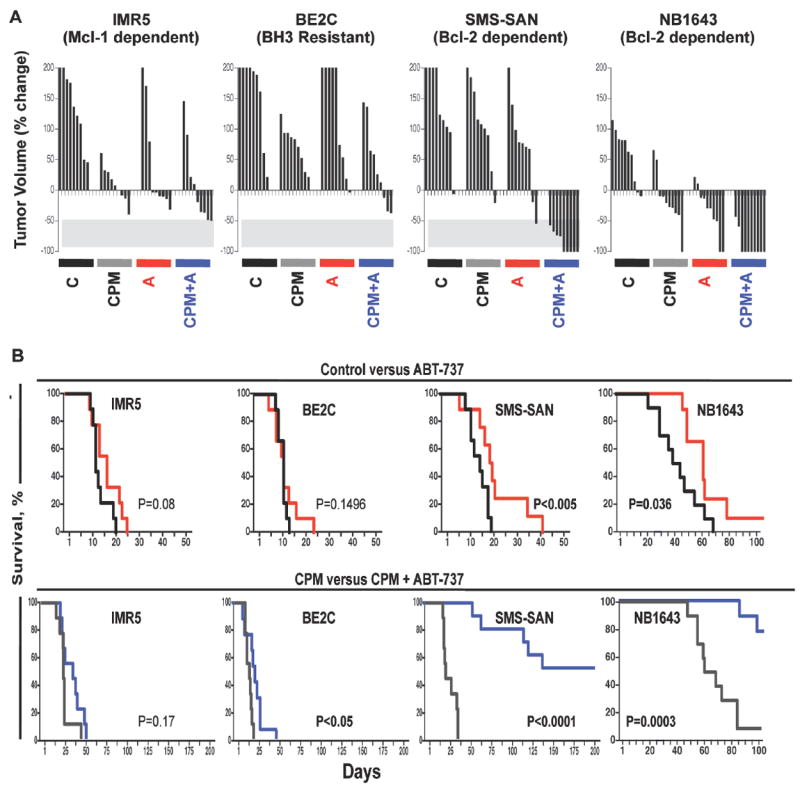
NB cell line xenografts representative of the three different BH3 response classes were established in the flank of nu/nu athymic mice. Mice with growing tumors >250 mm3 were randomized to receive either intraperitoneal cyclophosphamide (CPM), ABT-737 (A), Cyclophosphamide + ABT-737 (CPM +A), or vehicle control (C), as outlined in Methods. (A) Waterfall plot of individual tumor volume as % volume change from tumor volume pre-therapy. Negative values define best-response tumor shrinkage with -100% representing complete regression. (B) Kaplan-Meier curves comparing survival of control (black) versus ABT-737 (red), and CPM (gray) versus CPM+ABT-737 (blue) in representative cell lines with different Bcl-2 dependence patterns. p-values derived using the log-rank test. For NB-1643, n=10 mice per arm; for IMR5, BE2C and SMS-SAN, n=9 mice per arm.
Co-exposure to a genotoxic agent and a Bcl-2-antagonist was next assessed. We predicted that ABT-737, by saturating anti-apoptotic binding sites, would prevent sequestration of the excess BH3 death stimulus induced by chemotherapy (such as Bim) and increase anti-tumor activity for tumors functionally dependent on Bcl-2. A non-curative single course of cyclophosphamide was used. For each xenograft model, survival was extended in cyclophosphamide-treated mice compared with control-treated mice (p<0.0001 by log-rank test) consistent with the known anti-tumor activity of this drug in NB, yet all mice had progressive tumor after a transient regression (Fig. 5B, median survival of 22±7 d for SMS-SAN; 25±7.6 d for IMR5; and 18±3.6 d for BE2C in the respective cyclophosphamide-treated arms). Notably, the impact on survival time for SMS-SAN xenografts treated either with cyclophosphamide alone or ABT-737 alone were similar, whereas in IMR5 and BE2C the effect of cyclophosphamide was significantly greater than that of ABT-737.
The addition of ABT-737 to cyclophosphamide induced striking tumor responses in the SMS-SAN (Bcl-2 dependent) model, with complete regression obtained in 5/9 mice (Fig. 5A). At the time the last cyclophosphamide treated mouse was sacrificed for tumor progression (day 35), all cyclophosphamide-ABT-737 treated mice had tumors below pre-treatment volume, and median survival was extended to 126 days. Indeed, 4 of 9 mice showed no tumor re-growth at the time of sacrifice (>220 days) despite receiving only a single cycle of therapy (no tumor was identified at necropsy). The addition of ABT-737 to cyclophosphamide treatment did not significantly increase survival in the IMR5 Mcl-1 dependent model (median survival of 35±10.9 d versus 25±7.6 d), but did increase survival in the BE2C BH3 resistant model by log rank analysis, although the magnitude of this response was modest (median survival 21±6.4 d versus 18±3.6 d) and all BE2C tumor bearing mice succumbed to tumor progression by day 50 (Fig. 5B).
We assessed a second Bcl-2 dependent NB cell line, NB-1643, and confirmed robust activity. Tumors regressed and survival was extended for both cyclophosphamide (median survival 64 ± 12.5 d versus 42.5 ± 14.4 d) or ABT-737 (median survival 59±10.4 d versus 42.5±14.4 d; p<0.04 by log-rank) treated mice (Fig. 5). Compared to those treated with cyclophosphamide alone, all mice receiving both cyclophosphamide and ABT-737 had significant tumor regression (p<0.001 by log-rank) with 8/10 having complete regression and two mice sacrificed for tumor progression on days 83 and 95. These findings underscore the utility of this classifier to identify tumors with a marked sensitivity to ABT-737.
To elucidate the mechanisms of Bcl-2 antagonist resistance that might arise in response to ABT-737 selective pressure, we exposed the ABT-737-sensitive SMS-SAN NB cell line to ABT-737 at 100 nM in vitro and derived a cell line capable of growth under these conditions. SAN-ABTR (SMS-SAN-derived and ABT-737 Resistant) cells expressed most Bcl-2 family proteins at levels similar to parent cells, except Mcl-1, which was overexpressed (Fig. 6A). Co-immunoprecipitation revealed that Mcl-1 also became the dominant site for Bim sequestration (Fig. 6B). SAN-ABTR cells were confirmed to have an IC50 of ~1 log greater than parental SMS-SAN cells based on this switch to Mcl-1 for its survival bias (Fig. 6C). CHLA-15 cells were similarly exposed in vitro to obtain CHLA15-ABTR that switched from Bim:Bcl-2 priming to Bim:Mcl-1 priming, coincident with acquired ABT-737 resistance (Fig. 6B).
Figure 6. Bcl-2 dependent NB cell lines adopt an Mcl-1 dependence in association with ABT-737 resistance in vitro but not in vivo.
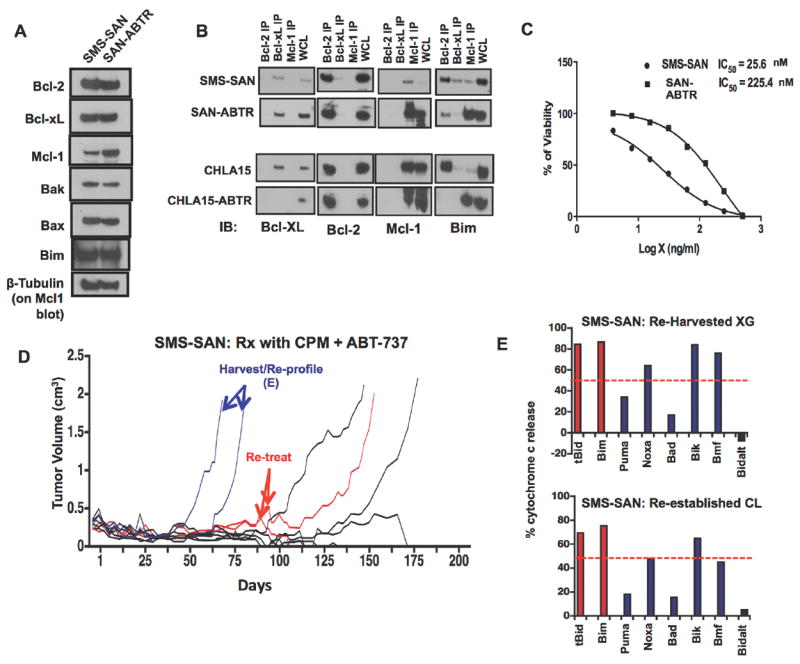
(A) Protein lysates from SMS-SAN and its ABT-737 resistant derivative, SAN-ABTR, were immunoblotted for expression of Bcl-2 family members. Increased Mcl-1 protein expression is seen in SAN-ABTR. (B) Co-immunoprecipitation of Bcl-2, Mcl-1 and Bcl-xL from parent NB cell lines and their derived ABT-737 resistant lines demonstrate a switch from Bim:Bcl-2 binding to Bim:Mcl-1 binding (for both SAN-ABTR and CHLA15-ABTR, an ABT-737-resistant variant of CHLA-15). (C) Dose response curves (using WST-1 assay) confirm increased resistance of SAN-ABTR to ABT-737 by >1 log. (D) Plot of tumor volume over time for SMS-SAN xenografts treated with ABT-737 and cyclophosphamide (same experiment as in Fig. 5). The first two tumors to re-grow (blue) were harvested and subjected to mitochondrial BH3 profiling directly as a xenograft and after re-establishment as a cell line. Both maintained a Bik-dominant (presumed Bcl-2 dependent) profile, (E). Two subsequent tumors that recurred (red) were retreated with ABT-737/Cyclophosphamide combination and showed regression, with durable complete regression obtained for one of two tumors. WCL, whole cell lysate. XG, xenograft, CL, cell line. Panels A-C, and E are representative data of replicate experiments; panel D shows 9 mice in a single experiment (as in Figure 5). Data in panel E histogram represents replicates with a standard error <5%; (error bars not shown).
Although standard cytotoxics used to treat NB did not appear to select for alterations in the dominant anti-apoptotic protein operative (see Fig. 2A), exposure to a Bcl-2 antagonist in vitro did. We therefore sought to determine whether SMS-SAN xenografts treated with ABT-737 and cyclophosphamide that re-grew had acquired a similar resistance mechanism. The first two SMS-SAN xenografts to recur and progress were harvested, and both the xenograft and the re-derived cell lines were shown to maintain a Bik-dominant BH3 response profile, nearly identical to the parent cells, and with no change in priming dependency or BH3 profile (Fig. 6D and 6E). We then re-treated the next two SMS-SAN xenografts that recurred with a second cycle of ABT-737 and cyclophosphamide. Both xenografts again completely regressed, with one mouse sacrificed at >200 days tumor free and the other succumbing to tumor at >50 days from the second treatment (Fig. 6D). Thus, over short term exposures to ABT-737 in combination with an alkylating agent there was no selection for ABT-737 resistance despite our in vitro selective pressure findings with single-agent ABT-737.
DISCUSSION
Cancers are extremely heterogeneous, even within a single histiotype. This is apparent in the divergent clinical responses among similarly treated patients, and underscored by deep sequencing results showing few highly recurrent oncogenic lesions but scores of low prevalence mutations. Such heterogeneity provides a practical challenge in an era of targeted therapeutics. Though the hope remains that this process will lead to more personalized and effective therapies, providing such agents for large numbers of distinct tumor genotypes remains daunting. An alternative approach to improve cancer outcomes is to target common downstream pathways that mediate therapy response and resistance, agnostic to the genetic heterogeneity that initiated or drives the tumor.
Targeting apoptotic programs to re-engage death signaling downstream of endogenous or therapeutic stressors provides one such opportunity. We hypothesized that identifying the apoptotic signaling set-point of neuroblastomas, downstream of their diverse oncogenic drivers and selective adaptations, would inform our understanding of their survival dependencies and provide insight into therapeutic approaches. Importantly, understanding the survival biases (and associated heterogeneity) operative in this often highly lethal solid tumor ought to have increasing therapeutic relevance as newer agents targeting the apoptotic pathway enter the clinic (Figure 7).
Figure 7. Functional mitochondrial assays identify mechanisms of therapy response and relapse in NB.
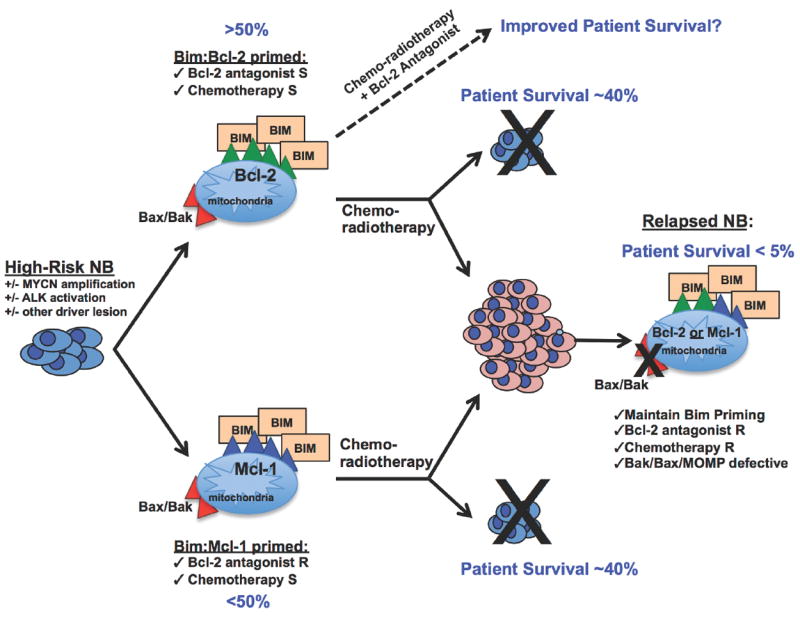
NB cell lines derived from the highest risk tumors are primed to die with activated Bim sequestered to pro-survival Bcl-2 or Mcl-1, regardless of concomitant genetic aberrations. Primary human tumors express Bcl-2 and/or Mcl-1 that confer poor prognosis, and diagnostic samples tested thus far show similar Bim sequestration, inferring these findings likely translate to primary disease (41). Bcl-2 sequestration of Bim occurred in a large subset of NB cell lines (> 50%), suggesting Bcl-2 inhibitors may have utility in treating NB in the upfront setting. NB mitochondrial responses to BH3 death-domains also identified, for the first time, a novel mechanism of acquired therapy resistance in relapsed NB isolated to the mitochondria through Bax/Bak repression. S, sensitive, R, resistant.
Mitochondrial profiling identified heterogeneous response patterns in neuroblastoma cell lines that are independent of common driver lesions, as neither MYCN status, 11q LOH, nor ALK aberration segregated with a single response profile. Viable neuroblastomas have activated but tonically neutralized Bim, providing a dependency that can be exploited therapeutically. Although Bim activation could arise as a tissue culture artifact, primary tumor Co-IPs from fresh frozen tissue demonstrate similar Bim priming states as cell lines for the majority, confirming Bim activation and tonic sequestration exist in situ. Bim was confirmed to be a principal mediator of chemotherapy response, suggesting its tonic neutralization provides a barrier to chemotherapy induced apoptosis. Such a role is supported by functional-genetic screens in which Bim knock-down induces resistance across multiple chemotherapy drug classes (34) and is consistent with findings from the Letai group correlating mitochondrial priming with chemotherapy response in multiple hematopoietic tumors and ovarian cancer (11). Subsets of neuroblastomas have Bim neutralized by either Bcl-2 or Mcl-1, although we found no tumors in which multiple anti-apoptotic homologues bound Bim. Bcl-xL was only modestly expressed and not a site of Bim binding, and Bcl-w was not evaluable as pull-down of this family member was not reliably achieved. The determinants of Bim binding when multiple anti-apoptotic Bcl-2 homologues are present remain to be elucidated.
Bcl-2 dependent neuroblastomas were exquisitely sensitive to ABT-737, a small molecule antagonist of Bcl-2/-xL/-w (22) and a homologue of ABT-263 (Navitoclax, Abbott Laboratories) that is in clinical trials (35, 36). We correlated a Bik-dominant mitochondrial response profile with ABT-737 sensitivity in vitro, confirmed a mechanism through competitive displacement of endogenous Bim, and demonstrated remarkable in vivo activity in a highly lethal xenograft model. Single agent activity for ABT-737 was recurrently shown for Bcl-2 dependent tumors, and combined with non-curative chemotherapy led to durable complete regressions not previously seen in this model. This includes tumors with MYCN genomic amplification (NB1643, SMS-SAN) and/or ALK mutation (NB1643, R1275Q mutation; SMS-SAN, F1174L mutation), lesions independently associated with poor clinical outcome (37-39). These data validate that phenotype-driven predictors may be important for triaging novel therapeutics for clinical use. The Pediatric Preclinical Testing Program (an NCI-supported initiative to accelerate the integration of new agents for pediatric cancer use) tested ABT-263 across a panel of neuroblastoma cell lines and found 1 of 6 to be sensitive (40). This was NB-1643 that we show to be Bcl-2-dependent with Bim:Bcl-2 binding, predicting this response.
Importantly, Bcl-2 family expression and interactions in cell lines are consistent with those we identified in primary tumors [(41) and Figure 1D]. Greater than 80% of high-risk neuroblastomas express IHC-detectable Bcl-2 or Mcl-1 and over a third of primary tumors express Bcl-2 alone, supporting that Bcl-2 antagonists may have clinical utility for a large subset of patients (41). Furthermore, Co-immunoprecipitations confirm Bim:Bcl-2 and Bim:Mcl-1 priming in distinct primary tumors, suggesting Bim sequestration by Bcl-2 or Mcl-1 plays a part in the pathogenesis of untreated tumors and infers a predictive response to BH3 mimetics. For NB cell lines demonstrating Bim:Mcl-1 priming, ABT-737 had little impact. However, Mcl-1 is unique among Bcl-2 anti-apoptotic homologues as it has a short half-life and is highly regulated, providing opportunities to resensitize Mcl-1-dependent tumors to Bcl-2 antagonists through targeting regulators of Mcl-1 expression.
BH3 profiles also defined an apoptosis resistant pattern exclusive to chemoresistant, post-therapy relapsed neuroblastoma (12). Using near-isogenic pre- and post-therapy cell line pairs we demonstrate that this reflects attenuation of Bak and/or Bax activation rather than a loss of direct-activator BH3 priming or gain-of-function of anti-apoptotic Bcl-2 family members. Though the mechanisms of this repression require further investigation, these findings from mitochondrial assays (and confirmed by a complimentary whole cell assay) isolate the resistance phenotype to a single organelle, the mitochondria, and provide a new conceptual target for reverting chemoresistance. Understanding the broader functional attributes of therapy resistant mitochondria may uncover vulnerabilities that are distinct from both normal cells and pre-therapy tumor cells. Importantly, tumors with Bim:Bcl-2 priming (predicted to be ABT-263 responders) are likely to have reduced sensitivity to a Bcl-2 antagonist at the time of relapse based on our findings. This has implications for how ABT-263 and similar agents are studied in the clinic. Our data support integrating such an agent into therapy prior to relapse, although clinical development standards call for initial testing to be done in patients at relapse, a patient population (based on our data) that are likely to have tumors less sensitive to BH3 mimetics as single agents. However, the data we present here, and preclinical studies combining ABT-737 with the fenretinide (42), suggest that recurrent neuroblastomas may be responsive to drug combinations that include BH3-mimetic agents.
The functional mitochondria profiling approach used here was initially applied to the characterization of hematological neoplasms (10, 43-46) and only more recently to select solid tumors that arise in the context of an organ with dependence on heterotypic cell types, contacts, and signaling (11). Our work demonstrates that despite this complexity, mitochondrial responses from isolated neuroblastoma cells recapitulate in vivo responses to similar stressors, and mitochondrial responses to stressors that promote apoptosis are therefore more cell intrinsic than might have been predicted. A more definitive test of this concept will require assessing the profile of more solid tumor samples harvested directly from their autochthonous primary and metastatic sites, and the subsequent demonstration that these profiles indeed predict response to Bcl-2 directed therapeutics in the clinic.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the advice and assistance of Tony Letai and Jeremy Ryan (Dana Farber Cancer Institute) in optimizing BH3 profiling and JC-1 assays for solid tumors; Lawrence Boise (Winship Cancer Institute, Emory University) for insightful review of our work; the Children’s Oncology Group (COG) Cell Culture and Xenograft Repository (www.cogcell.org) for providing select cell lines; The Atlanta Pediatric Biorepository and Children’s Healthcare of Atlanta Pathology Department for providing primary tumor specimens; Steven Elmore and Abbott Laboratories for ABT-737; and the assistance of the CHOP Pathology Core.
GRANT SUPPORT
This work was supported by NIH CA97323, the King Family, the Richard and Sheila Sanford Chair in Pediatric Oncology, Alex’s Lemonade Stand Foundation (to M.D.H.), NIH K08-CA128925, The Cure Childhood Cancer Foundation (to K.C.G), and NIH CA82830 (to C.P.R.).
Footnotes
The authors have no conflicting financial interests.
References
- 1.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 2.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407:810–6. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- 4.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–12. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 7.Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. Journal of Clinical Oncology. 2009;27:1007–13. doi: 10.1200/JCO.2007.13.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–32. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–3. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 11.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–33. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldsmith KC, Lestini BJ, Gross M, Ip L, Bhumbla A, Zhang X, et al. BH3 response profiles from neuroblastoma mitochondria predict activity of small molecule Bcl-2 family antagonists. Cell Death Differ. 2010;17:872–82. doi: 10.1038/cdd.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tumilowicz JJ, Nichols WW, Cholon JJ, Greene AE. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res. 1970;30:2110–8. [PubMed] [Google Scholar]
- 14.Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983;305:245–8. doi: 10.1038/305245a0. [DOI] [PubMed] [Google Scholar]
- 15.Schlesinger HR, Gerson JM, Moorhead PS, Maguire H, Hummeler K. Establishment and characterization of human neuroblastoma cell lines. Cancer Res. 1976;36:3094–100. [PubMed] [Google Scholar]
- 16.Keshelava N, Zuo JJ, Chen P, Waidyaratne SN, Luna MC, Gomer CJ, et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001;61:6185–93. [PubMed] [Google Scholar]
- 17.Feder MK, Gilbert F. Clonal evolution in a human neuroblastoma. J Natl Cancer Inst. 1983;70:1051–6. [PubMed] [Google Scholar]
- 18.Schmechel D, Marangos PJ, Brightman M. Neuron-specific enolase is a molecular marker for peripheral and central neuroendocrine cells. Nature. 1978;276:834–6. doi: 10.1038/276834a0. [DOI] [PubMed] [Google Scholar]
- 19.Goldsmith KC, Liu X, Dam V, Morgan BT, Shabbout M, Cnaan A, et al. BH3 peptidomimetics potently activate apoptosis and demonstrate single agent efficacy in neuroblastoma. Oncogene. 2006;25:4525–33. doi: 10.1038/sj.onc.1209489. [DOI] [PubMed] [Google Scholar]
- 20.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 21.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci U S A. 2010;107:12895–900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association. 1958;53:457–81. [Google Scholar]
- 24.Peto R, Lee PN, Paige WS. Statistical analysis of the bioassay of continuous carcinogens. Br J Cancer. 1972;26:258–61. doi: 10.1038/bjc.1972.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 26.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 Domains of BH3-Only Proteins Differentially Regulate Bax-Mediated Mitochondrial Membrane Permeabilization Both Directly and Indirectly. Mol Cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–9. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 28.Gillissen B, Essmann F, Graupner V, Starck L, Radetzki S, Dorken B, et al. Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. The EMBO journal. 2003;22:3580–90. doi: 10.1093/emboj/cdg343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, et al. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene. 1995;11:1921–8. [PubMed] [Google Scholar]
- 30.Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, et al. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7:227–38. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 31.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101:6164–9. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keshelava N, Seeger RC, Groshen S, Reynolds CP. Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res. 1998;58:5396–405. [PubMed] [Google Scholar]
- 33.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang H, Pritchard JR, Williams RT, Lauffenburger DA, Hemann MT. A mammalian functional-genetic approach to characterizing cancer therapeutics. Nat Chem Biol. 2011;7:92–100. doi: 10.1038/nchembio.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–59. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clinical Cancer Research. 2010;16:4353–62. doi: 10.1158/1078-0432.CCR-09-2660. [DOI] [PubMed] [Google Scholar]
- 38.Martinsson T, Eriksson T, Abrahamsson J, Caren H, Hansson M, Kogner P, et al. Appearance of the novel activating F1174S ALK mutation in neuroblastoma correlates with aggressive tumor progression and unresponsiveness to therapy. Cancer Res. 2011;71:98–105. doi: 10.1158/0008-5472.CAN-10-2366. [DOI] [PubMed] [Google Scholar]
- 39.Passoni L, Longo L, Collini P, Coluccia AM, Bozzi F, Podda M, et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009;69:7338–46. doi: 10.1158/0008-5472.CAN-08-4419. [DOI] [PubMed] [Google Scholar]
- 40.Lock R, Carol H, Houghton PJ, Morton CL, Kolb EA, Gorlick R, et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1181–9. doi: 10.1002/pbc.21433. [DOI] [PubMed] [Google Scholar]
- 41.Lestini BJ, Goldsmith KC, Fluchel MN, Liu X, Chen NL, Goyal B, et al. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther. 2009;8 doi: 10.4161/cbt.8.16.8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang H, Harned TM, Kalous O, Maldonado V, DeClerck YA, Reynolds CP. Synergistic activity of fenretinide and the Bcl-2 family protein inhibitor ABT-737 against human neuroblastoma. Clin Cancer Res. 2011;17:7093–104. doi: 10.1158/1078-0432.CCR-11-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–32. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 44.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111:2300–9. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.