

Abstract
Chronic kidney disease (CKD) increases cardiovascular risk and mortality. However, traditional cardiovascular risk factors do not adequately account for the substantial increase in mortality observed in CKD. The aim of this study was to examine the relative contributions of novel cardiovascular risk factors to the risk between CKD and mortality. The study population included 4,680 consecutive new patients from a tertiary care preventive cardiology program from 1996 to 2005. Estimated glomerular filtration rate was calculated using the Modification of Diet in Renal Disease (MDRD) method. Baseline levels of traditional (low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, hypertension, triglycerides, total cholesterol, and fasting glucose) and emerging (apolipoproteins A-I and B, lipoprotein[a], fibrinogen, homocysteine, and high-sensitivity C-reactive protein) risk factors were examined. All-cause mortality was obtained from the Social Security Death Index. There were 278 deaths over a median follow-up period of 22 months. CKD (estimated glomerular filtration rate ≤60 ml/min/1.73 m2) was strongly associated with mortality after adjusting for traditional cardiovascular risk factors (hazard ratio 2.31, 95% confidence interval 1.77 to 3.11, p <0.001) and with the addition of propensity score (hazard ratio 2.33, 95% confidence interval 1.75 to 3.10, p <0.001). Of all the traditional and emerging risk factors monitored, only the addition of homocysteine and fibrinogen significantly attenuated the association between CKD and mortality (adjusted hazard ratio 1.73, 95% confidence interval 1.23 to 2.34, p <0.001), explaining 38% of the attributable mortality risk from CKD. A significant interaction (p = 0.004) between homocysteine and estimated glomerular filtration rate was observed whereby the annual mortality rate in subjects with CKD with homocysteine <10 μmol/L (the bottom tertile) was similar to those with normal renal function (1% per year), whereas homocysteine levels ≥12.5 μmol/L (the top tertile) were associated with a sevenfold greater mortality risk. In conclusion, homocysteine and fibrinogen levels explain nearly 40% of the attributable mortality risk from CKD.
Previous work has suggested that patients with chronic kidney disease (CKD) have significant lipoprotein derangements.1 In addition, studies have also demonstrated heightened levels of oxidative stress, endothelial damage, thrombosis, inflammation, insulin resistance, and glucose intolerance.2,3 Whether a comprehensive panel of markers with mechanistic links to these pathophysiologic processes can help explain the elevated mortality risk in subjects with CKD has not been fully explored. Therefore, we hypothesized that markers of lipids and lipoproteins (low-density lipoprotein [LDL] cholesterol, high-density lipoprotein cholesterol, triglycerides, and total cholesterol), apolipoproteins (apolipoprotein [apo] A-I and apo B), inflammation (highsensitivity C-reactive protein), thrombosis (lipoprotein[a] and fibrinogen), oxidative and endothelial damage (homocysteine), and glucose metabolism (fasting glucose) might help explain the link between CKD and mortality.
Methods
The population consisted of consecutive new patients seen at a tertiary preventive cardiology program from 1996 to 2005. This population constituted primary and secondary cardiovascular prevention. Generally, all staff members within the preventive cardiology section follow a similar algorithm when treating risk factors such as hypertension, hyperlipidemia, obesity, glucose intolerance, and others. Patients were excluded if they lacked a United States Social Security number. In addition, we excluded individuals with end-stage renal disease requiring maintenance hemodialysis at baseline. Of a total of 5,636 new subjects seen, 4,680 had baseline laboratory data available. The Institutional Review Board of the Cleveland Clinic approved this study.
All patients underwent detailed interviews by professional nursing staff members to obtain information regarding the presence of cardiovascular risk factors, previous cardiac procedures, other medical diagnoses, and medication use, as outlined previously.2 In addition, blood samples were drawn to assess a number of traditional and novel cardiovascular risk factors, including fasting levels of lipids and lipoproteins, apolipoproteins, inflammation, thrombosis, oxidative and endothelial damage, and glucose metabolism. Death was ascertained using the Social Security Death Index.
Estimated glomerular filtration rate (eGFR) was calculated using the Modification of Diet in Renal Disease (MDRD) equation, which incorporates serum creatinine, body weight, age, and race. On the basis of the recommendation of the National Kidney Foundation,4 CKD was defined by an eGFR ≤60 ml/min/1.73 m2. Therefore, for descriptive purposes, we divided the group into those with CKD (eGFR ≤0 ml/min/1.73 m2) and those without CKD (eGFR >60 ml/min/1.73 m2).
All measurements were performed at baseline. Standard methods were used to measure fasting lipid profiles. Apo A-I and apo B were measured by immunonephelometry using a Beckman Immage Nephelometer and Beckman reagents (Beckman Coulter, Brea, California), and high-sensitivity C-reactive protein was quantified using an immunoturbidometric method, the Roche CRP Latex HS assay (Roche Diagnostics, Indianapolis, Indiana). Baseline serum creatinine was measured by colorimetric assay (Roche Diagnostics). Lipoprotein(a) was measured using a monoclonal antibody–based enzyme-linked immunosorbent assay (Genentech, South San Francisco, California), fibrinogen was measured using a Sysmex CA 7000 (Dade Behring, Deerfield, Illinois) by the Claus method, homocysteine was measured using immunochemiluminescence on a Bayer Centaur (Bayer Diagnostics, Tarrytown, New York), and glucose was measured by hexokinase assay (Roche Diagnostics).
Wilcoxon’s rank-sum test for continuous variables and chi-square tests for categorical variables were used to examine differences between 2 groups. We used Cox proportional-hazards modeling to examine the association between CKD and all-cause mortality. In the first model, we examined the association between CKD and mortality after accounting only for the differences in baseline demographic characteristics, medical history, and medication use. In the second model, in addition to the baseline demographics, we adjusted for propensity score. Propensity score was calculated using nonparsimonious logistic regression models for predicting CKD for each subject. In the subsequent full model, in addition to these confounders and the propensity score, we adjusted for novel cardiovascular risk factors. The proportional-hazards assumption was confirmed by testing the weighted Schoenfeld residuals and by plotting hazard ratios (HRs) against time plots for selected variables. In addition to using CKD as a dichotomous variable in these models, similar models were created using eGFR as a continuous variables.
Our goal in this study was to identify and quantify the relative contributions of novel cardiovascular risk factors to the observed association between CKD and mortality. To address this, we used the following formula5,6: 1 — (log HRadjusted/log HRunadjusted), where HRunadjusted is the HR for mortality adjusted for all patient characteristics other than specific novel cardiovascular risk factors, and HRadjusted reflects the HR for mortality including specific novel cardiovascular risk factors. For example, if we were interested in the relative contribution of homocysteine, HRunadjusted would include all confounding variables listed in Tables 1 and 2 except homocysteine, and HRadjusted would include all variables included in HRunadjusted plus homocysteine. In addition, we tested all potential interactions and selected those with clinical relevance for further testing. Analyses were performed using SAS version 9.1 (SAS Institute Inc., Cary, North Carolina) and S-plus version 6.2 (Insightful, Inc., Seattle, Washington).
Table 1.
Baseline characteristics according to estimated glomerular filtration rate cut points
| Variable | eGFR (ml/min/1.73 m2) |
P Value | |
|---|---|---|---|
| ≤60 (n = 524) |
>60 (n = 4,156) |
||
| Age (yrs) | 64 ± 11 | 54 ± 12 | <0.001 |
| Men | 307 (59%) | 2,678 (64%) | 0.009 |
| Black race | 61 (12%) | 331 (8%) | 0.004 |
| Body mass index (kg/m2) | 29 ±5 | 29 ± 6 | 0.174 |
| Diabetes mellitus | 178 (34%) | 599 (14%) | <0.001 |
| Systolic blood pressure (mm Hg) | 132 ± 22 | 122 ± 19 | <0.001 |
| Current smoker | 39 (7%) | 582 (14%) | <0.001 |
| Known coronary artery disease | 308 (59%) | 1,729 (42%) | <0.001 |
| Previous myocardial infarction | 193 (37%) | 896 (22%) | <0.001 |
| Previous peripheral vascular disease | 64 (12%) | 152 (4%) | <0.001 |
| Aspirin use | 340 (65%) | 2,705 (65%) | 0.93 |
| Lipid lowering treatment | 392 (75%) | 2,769 (67%) | <0.001 |
| Less than high school education | 31 (6%) | 137 (3%) | <0.001 |
| High school education | 202 (38%) | 1,097 (26%) | <0.001 |
| Some college | 291 (56%) | 2,922 (70%) | <0.001 |
Table 2.
Cardiovascular risk factors according to estimated glomerular filtration rate cut points
| Cardiovascular Risk Factor | eGFR (ml/min/1.73 m2) |
p Value | |
|---|---|---|---|
| ≤60 (n = 524) |
>60 (n = 4,156) |
||
| Total cholesterol (mg/dl) | 218 (180–275) | 216 (179–256) | 0.03 |
| LDL (mg/dl) | 126 (96–157) | 126 (99–154) | 0.99 |
| High-density lipoprotein (mg/dl) | 44 (35–54) | 44 (37–54) | 0.31 |
| Triglycerides (mg/dl) | 193 (133–321) | 167 (111–266) | <0.001 |
| Apo A-I/apo B | 0.89 (0.78–1.0) | 0.88 (0.73–1.0) | <0.001 |
| Lipoprotein(a) (mg/dl) | 30 (10–72) | 24 (9–56) | <0.001 |
| Fibrinogen (mg/dl) | 363 (317–424) | 322 (284–367) | <0.001 |
| Homocysteine (μmol/L) | 16 (13–19) | 11 (9–13) | <0.001 |
| Fasting glucose (mg/dl) | 94 (86–112) | 90 (81–99) | <0.001 |
| High-sensitivity C-reactive protein (mg/L) | 3.0 (1.3–6.6) | 2.2 (1–4.7) | <0.001 |
Data are expressed as median (interquartile range).
Results
A total of 4,680 patients met our study criteria, of whom 524 met the criterion for CKD. The baseline characteristics of all patients are listed in Table 1. Patients with CKD were older, in general had a significantly higher prevalence of traditional cardiovascular risk factors, and had a lower overall education level. However, they were less likely to be current smokers.
Baseline levels of novel cardiovascular risk factors are listed in Table 2. Patients with CKD had significantly higher levels of high-sensitivity C-reactive protein, lipoprotein(a), fibrinogen, homocysteine, triglycerides, and glucose. In contrast, CKD was associated with lower total cholesterol, LDL cholesterol, and high-density lipoprotein cholesterol.
Of the novel markers that were added to a model that contained age, gender, body mass index, diabetes, hypertension, current smoking, history of coronary artery disease, triglycerides, LDL cholesterol, lipid-lowering medications, education level, and aspirin use, the ratio of apo A-I to apo B (adjusted HR 1.71, 95% confidence interval [CI] 1.15 to 2.54, p = 0.008), fibrinogen (adjusted HR 3.88, 95% CI 2.25 to 6.68, p <0.001), and homocysteine (adjusted HR 2.50, 95% CI 1.77 to 3.55, p <0.001) were significantly associated with all-cause mortality, whereas lipoprotein(a) (adjusted HR 1.10, 95% CI 0.98 to 1.23, p = 0.105) and glucose (adjusted HR 0.77, 95% CI 0.49 to 1.21, p = 0.250) were not. In a subsequent model in which all established and novel cardiovascular risk factors were collectively examined, only homocysteine (adjusted HR 2.12, 95% CI 1.47 to 3.04, p <0.001) and fibrinogen (adjusted HR 2.80, 95% CI 1.60 to 4.89, p <0.001) predicted death.
CKD was significantly associated with all-cause mortality in unadjusted, multivariate-adjusted, and propensity score–adjusted models (Table 3, Figure 1). When eGFR was included in the models, only homocysteine (adjusted HR 1.92, 95% CI 1.28 to 2.87, p <0.001) and fibrinogen (adjusted HR 2.99, 95% CI 1.69 to 5.30, p <0.001) predicted death, whereas the ratio of apo A to apo B, lipoprotein(a), triglycerides, and glucose failed to predict all-cause mortality. Moreover, 38% of the association between CKD and mortality was explained by the addition of homocysteine and fibrinogen to the full model, despite the inclusion of all traditional cardiovascular risk factors and laboratory values.
Table 3.
Association between chronic kidney disease and all-cause mortality
| Variable | eGFR (ml/min/1.73 m2) |
HR (95% CI) | p Value | |
|---|---|---|---|---|
| ≤60 (n = 524) |
>60 (n = 4,156) |
|||
| Model 1: unadjusted | 96 (18%) | 182 (4%) | 4.10 (3.17–5.30) | <0.001 |
| Model 2: partially adjusted* | 96 (18%) | 182 (4%) | 2.35 (1.77–3.11) | <0.001 |
| Model 3: propensity adjusted† | 96 (18%) | 182 (4%) | 2.33 (1.75–3.10) | <0.001 |
| Model 4: fully adjusted‡ | 96 (18%) | 182 (4%) | 1.69 (1.23–2.32) | 0.001 |
| Model 5: fully adjusted plus high-sensitivity C-reactive protein (n = 2,427) |
21 (7%) | 40 (2%) | 1.25 (0.97–1.58) | 0.083 |
Adjusted for age, gender, diabetes, hypertension, current smoking, history of myocardial infarction, history of coronary artery disease, history of peripheral vascular disease, aspirin use, lipid-lowering medication use, educational status, triglycerides, LDL cholesterol, high-density lipoprotein cholesterol, and total cholesterol.
Adjusted for all factors in model 1 plus propensity score.
Adjusted for all factors in model 1, propensity score, and apo A-I, apo B, lipoprotein(a), fibrinogen, homocysteine, and fasting glucose.
Figure 1.
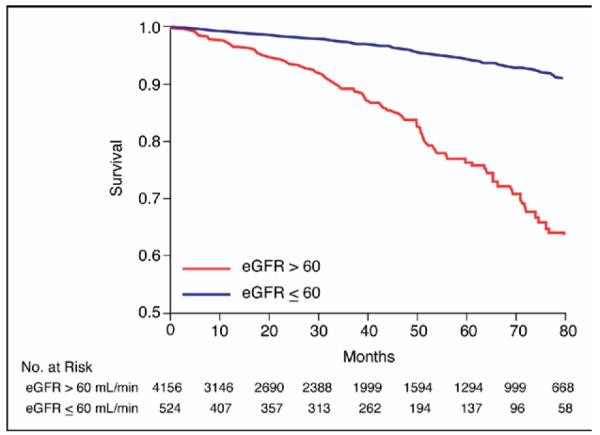
Kaplan-Meier curves according to CKD status.
Given that patients with CKD have elevated levels of homocysteine and fibrinogen, we tested potential interactions between eGFR and these markers. A strong interaction between homocysteine level and eGFR (adjusted p for interaction = 0.004) was noted whereby the probability of death was significantly higher for the highest tertile of homocysteine, especially for eGFR <60 ml/min/1.73 m2, as shown in Figure 2. Similarly, a trend toward a significant interaction was noted for the association between fibrinogen and eGFR, also shown in Figure 2 (adjusted p for interaction = 0.08).
Figure 2.

(A) Adjusted probability of mortality for tertiles of homocysteine (HCys) according to eGFR. EGFR is presented as a continuous measure on the x-axis and adjusted probability of mortality on the y-axis. Subjects were divided by homocysteine tertiles (cut points were ≤10, 10.1 to 12.4, and ≥12.5 μmol/L from lowest to highest tertile). Note the interaction between eGFR and homocysteine whereby as eGFR decreased, the highest tertile of homocysteine was significantly associated with mortality, in contrast to the lowest and intermediate tertiles. (B) Predictive value of fibrinogen according to CKD status. EGFR is presented as a continuous measure on the x-axis and adjusted probability of mortality on the y-axis. Subjects were divided by fibrinogen tertiles (cut points were ≤300, 301 to 350, and ≥351 mg/dl from lowest to highest tertile). Note that in general, there was an association between decreasing eGFR and increasing mortality risk that was more prominent in the highest compared with the lowest tertile of fibrinogen.
Given the prevalence of CKD, we tested whether homocysteine or fibrinogen could potentially identify those patients with CKD who are at increased risk for death. As shown in Figure 3, the annual mortality rate was very similar (1%) for patients in the lowest tertile of homocysteine regardless of eGFR. However, patients with CKD who were in the second and third tertiles of homocysteine had significantly higher annual mortality rates compared with those without CKD (6% to 8%). Fibrinogen also had significant predictive value when added to eGFR; however, unlike homocysteine, it did not have discriminative value to significantly predict low- versus high-risk patients.
Figure 3.
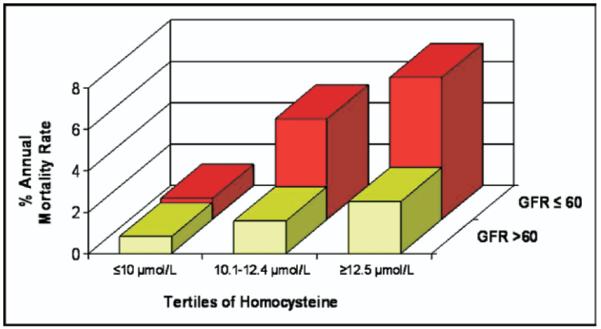
Subjects with CKD and low homocysteine had low mortality risk. Homocysteine is presented as tertiles on the x-axis, CKD (defined as eGFR <60 ml/min/1.73 m2) on the y-axis, and percentage annual mortality rate on the z-axis. Homocysteine tertile cut points were ≤10, 10.1 to 12.4, and ≥12.5 μmol/L from lowest to highest tertile. Note that subjects with eGFRs ≤60 ml/min/1.73 m2 had the same mortality (1% per year) as those with eGFRs <60 ml/min/1.73 m2 if in the lowest tertile of homocysteine.
Discussion
In this observational cohort study, homocysteine and fibrinogen levels explained 38% of the association between CKD and mortality despite multiple adjustments for traditional as well as novel cardiovascular risk factors. Although the association between CKD and cardiovascular morbidity and mortality is irrefutable, no specific therapeutic measures except addressing traditional cardiovascular risk factors are available. The present studies are hypothesis generating and thus suggest homocysteine and fibrinogen as 2 potential therapeutic targets that may affect CKD-associated mortality beyond addressing traditional cardiovascular risk factors. Furthermore, we found an interaction between eGFR and homocysteine, whereby patients with CKD who were in the lowest tertile of homocysteine had low mortality risk, similar in fact to those without CKD. These results thus indicate that homocysteine levels may be useful in risk stratifying patients with CKD, with a homocysteine level <10 μmol/L being an excellent prognostic indicator with respect to overall mortality.
Numerous previous studies have demonstrated elevated homocysteine levels in subjects with CKD.7,8 Although the exact mechanisms for the elevated homocysteine levels in CKD are unknown, a number of hypotheses exist. One obvious contributor is that the renal excretion of homocysteine is reduced in patients with CKD. However, additional alternative mechanisms may contribute as well. For example, alterations in remethylation pathways and disturbance in cysteine disposal might also be impaired in patients with CKD.9 The net consequences of elevated homocysteine levels may in turn be enhanced risk for the development of atherosclerosis. Although recent clinical trials aimed at lowering homocysteine levels have been disappointing, a preponderance of genetic and biochemical data suggest that elevated homocysteine levels promote a proatherogenic phenotype, with potential mechanisms including enhanced oxidative stress, inflammation, thrombosis, and endothelial dysfunction.10,11 Indeed, it has been suggested that homocysteine has an inhibitory effect on nitric oxide functional bioavailability, with resultant diminished endothelium-dependent and endothelium-independent vasodilatation in patients with hyperhomocysteinemia compared with healthy controls.12,13 The observed strong relation between homocysteine levels and mortality in CKD raises the intriguing hypothesis that therapies aimed at lowering homocysteine may have potential clinical benefit in patients with CKD.
Several large, prospective, randomized, placebo-controlled studies have examined the clinical impact of homocysteine-lowering therapies in the general population, with discouraging results. Although the impact of homocysteine reduction in end-stage renal disease has been examined,14,15 lowering homocysteine levels in patients with CKD has not yet been closely examined. The Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) examined the impact of folic acid supplementation in patients with chronic renal failure.16 In this multicenter, randomized controlled trial, homocysteine levels were significantly reduced, but no changes in fatal outcomes were observed. Although informative, this study had several limitations for the present discussion, including a heterogenous population, with most patients already receiving maintenance dialysis (of the 315 total subjects, 267 [85%] were receiving peritoneal dialysis or hemodialysis, and 48 [15%] had creatinine clearance <25 ml/min). Additionally, the most common cocktail of vitamin supplements used in trials aimed at lowering homocysteine levels have included folic acid and vitamin B12. It has been suggested, however, that in addition to lowering homocysteine levels, these vitamins may promote alternative adverse effects that might outweigh or mask any beneficial effect.10 For example, folic acid may promote cell proliferation through thymidylate synthesis, the mechanism proposed to account for the higher in-stent restenosis noted in subjects supplemented with folic acid, vitamin B12, and vitamin B6.17 Alterations in methylation reactions through the modulation of methylation potential (the ratio of S-adenosylmethionine to S-adenosylhomocysteine) has also been suggested to have a potential proatherogenic impact.10 For this reason, alternative methods for homocysteine reduction, such as enhancing urinary excretion or its conversion to cysteine in the liver, are potential new therapeutic strategies.10 Others have recommended parenteral cobalamin in addition to folic acid as an alternative to folic acid and vitamin B12 to enhance methionine synthase activity to decrease homocysteine levels.18 The present study suggests that future trials may wish to focus on patients with earlyonset CKD, as opposed to the general population, as a more likely cohort to benefit from homocysteine-lowering therapeutic strategies.
Fibrinogen also demonstrated strong association with mortality, even after adjustments for traditional and emerging risk factors. Fibrinogen is an acute-phase protein that affects rheology, platelet aggregation, and coagulation19 and has prognostic implications in patients with coronary artery disease.20 Elevated levels of fibrinogen have also been linked to cardiovascular end points in patients with CKD and end-stage renal disease.21 In chronic renal failure, the activation of interleukin-6 may lead to a heightened inflammatory state or acute phase response and, consequently, elevated fibrinogen levels.22,23 Shlipak et al24 proposed that patients with CKD may also have decreased fibrinogen clearance. Evidence shows that serum fibrinogen levels have been significantly correlated with the severity of chronic renal insufficiency, even after adjustments for multiple confounders.24–26
The present study had several limitations. First, given its observational nature, the present findings are hypothesis generating and do not establish causality. However, the contribution of homocysteine and fibrinogen to the link between CKD and mortality was tested in multiple models, including propensity-matched models, indicating that these nontraditional risk factors can help explain nearly 40% of the attributable mortality risk associated with CKD. Second, the glomerular filtration rate was estimated using the MDRD equation, which was originally intended for patients with impaired renal function and may not represent the true estimation of glomerular filtration rate. Additionally, MDRD is limited in its ability to diagnose CKD in younger age groups. As a result, the present study only identified a portion of the patients with CKD. Hence, this reduced the discriminatory capabilities of the model. However, compared with other measures of eGFR, such as the Cockcroft-Gault formula, it may be superior.27 Third, we did not have high-sensitivity C-reactive protein data for all patients, necessitating subgroup analyses on this cohort (n = 2,427) with only 61 deaths. Fourth, nontraditional markers examined in the present study were evaluated in real time as part of the initial patient assessment within an academic preventive cardiology clinic. Although many nontraditional markers were monitored, a number of others were not routinely available in subjects (e.g., LDL-associated phospholipase A2, lipoprotein subfractions, myeloperoxidase, and the urinary microalbumin/creatinine ratio) and thus were not included in the present analyses.
Acknowledgment
We are grateful to Dr. Donald W. Jacobsen (Department of Cell Biology and Cardiovascular Medicine, Cleveland Clinic, Cleveland, Ohio) and Dr. Emilio Poggio (Department of Nephrology, Cleveland Clinic) for their thoughtful comments and helpful criticisms of this report. Additionally, we thank Cindy Stevenson and Brian Hoar for their assistance with data collection and management.
This study was supported by Grants P01 HL076491, P01 HL77107, and P01 HL087018 from the National Institutes of Health, Bethesda, Maryland. Dr. Shishehbor is supported by Clinical and Translational Science Award 1KL2RR024990 from Case Western Reserve University/Cleveland Clinic, Cleveland, Ohio.
References
- 1.Farbakhsh K, Kasiske BL. Dyslipidemias in patients who have chronic kidney disease. Med Clin North Am. 2005;89:689–699. doi: 10.1016/j.mcna.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Acevedo M, Pearce GL, Kottke-Marchant K, Sprecher DL. Elevated fibrinogen and homocysteine levels enhance the risk of mortality in patients from a high-risk preventive cardiology clinic. Arterioscler Thromb Vasc Biol. 2002;22:1042–1045. doi: 10.1161/01.atv.0000020007.25154.62. [DOI] [PubMed] [Google Scholar]
- 3.Peppa M, Uribarri J, Cai W, Lu M, Vlassara H. Glycoxidation and inflammation in renal failure patients. Am J Kidney Dis. 2004;43:690–695. doi: 10.1053/j.ajkd.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 4.K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39:S1–S266. [PubMed] [Google Scholar]
- 5.Brotman DJ. Mediators of the association between mortality risk and socioeconomic status. JAMA. 2006;296:763–764. doi: 10.1001/jama.296.7.763-b. [DOI] [PubMed] [Google Scholar]
- 6.Shishehbor MH, Litaker D, Pothier CE, Lauer MS. Association of socioeconomic status with functional capacity, heart rate recovery, and all-cause mortality. JAMA. 2006;295:784–792. doi: 10.1001/jama.295.7.784. [DOI] [PubMed] [Google Scholar]
- 7.Muntner P, Hamm LL, Kusek JW, Chen J, Whelton PK, He J. The prevalence of nontraditional risk factors for coronary heart disease in patients with chronic kidney disease. Ann Intern Med. 2004;140:9–17. doi: 10.7326/0003-4819-140-1-200401060-00006. [DOI] [PubMed] [Google Scholar]
- 8.Muntner P, He J, Astor BC, Folsom AR, Coresh J. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: results from the atherosclerosis risk in communities study. J Am Soc Nephrol. 2005;16:529–538. doi: 10.1681/ASN.2004080656. [DOI] [PubMed] [Google Scholar]
- 9.van Guldener C. Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol Dial Transplant. 2006;21:1161–1166. doi: 10.1093/ndt/gfl044. [DOI] [PubMed] [Google Scholar]
- 10.Loscalzo J. Homocysteine trials—clear outcomes for complex reasons. N Engl J Med. 2006;354:1629–1632. doi: 10.1056/NEJMe068060. [DOI] [PubMed] [Google Scholar]
- 11.Weiss N, Keller C, Hoffmann U, Loscalzo J. Endothelial dysfunction and atherothrombosis in mild hyperhomocysteinemia. Vasc Med. 2002;7:227–239. doi: 10.1191/1358863x02vm428ra. [DOI] [PubMed] [Google Scholar]
- 12.Nakanishi T, Ishigami Y, Otaki Y, Izumi M, Hiraoka K, Inoue T, Takamitsu Y. Impairment of vascular responses to reactive hyperemia and nitric oxide in chronic renal failure. Nephron. 2002;92:529–535. doi: 10.1159/000064078. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Li H, Jin H, Ebin Z, Brodsky S, Goligorsky MS. Effects of homocysteine on endothelial nitric oxide production. Am J Physiol Renal Physiol. 2000;279:F671–F678. doi: 10.1152/ajprenal.2000.279.4.F671. [DOI] [PubMed] [Google Scholar]
- 14.Isbel NM, Haluska B, Johnson DW, Beller E, Hawley C, Marwick TH. Increased targeting of cardiovascular risk factors in patients with chronic kidney disease does not improve atheroma burden or cardiovascular function. Am Heart J. 2006;151:745–753. doi: 10.1016/j.ahj.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 15.Wrone EM, Hornberger JM, Zehnder JL, McCann LM, Coplon NS, Fortmann SP. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J Am Soc Nephrol. 2004;15:420–426. doi: 10.1097/01.asn.0000110181.64655.6c. [DOI] [PubMed] [Google Scholar]
- 16.Zoungas S, McGrath BP, Branley P, Kerr PG, Muske C, Wolfe R, Atkins RC, Nicholls K, Fraenkel M, Hutchison BG, Walker R, McNeil JJ. Cardiovascular morbidity and mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in chronic renal failure: a multicenter, randomized, controlled trial. J Am Coll Cardiol. 2006;47:1108–1116. doi: 10.1016/j.jacc.2005.10.064. [DOI] [PubMed] [Google Scholar]
- 17.Lange H, Suryapranata H, De Luca G, Borner C, Dille J, Kallmayer K, Pasalary MN, Scherer E, Dambrink JH. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med. 2004;350:2673–2681. doi: 10.1056/NEJMoa032845. [DOI] [PubMed] [Google Scholar]
- 18.Hoffer LJ. Testing the homocysteine hypothesis in end-stage renal disease: problems and a possible solution. Kidney Int. 2006;69:1507–1510. doi: 10.1038/sj.ki.5000279. [DOI] [PubMed] [Google Scholar]
- 19.Kannel WB. Overview of hemostatic factors involved in atherosclerotic cardiovascular disease. Lipids. 2005;40:1215–1220. doi: 10.1007/s11745-005-1488-8. [DOI] [PubMed] [Google Scholar]
- 20.Rosenson RS, Koenig W. Utility of inflammatory markers in the management of coronary artery disease. Am J Cardiol. 2003;92(suppl):10I–8I. doi: 10.1016/s0002-9149(03)00504-6. [DOI] [PubMed] [Google Scholar]
- 21.Zoccali C, Mallamaci F, Tripepi G, Cutrupi S, Parlongo S, Malatino LS, Bonanno G, Rapisarda F, Fatuzzo P, Seminara G, et al. Fibrinogen, mortality and incident cardiovascular complications in end-stage renal failure. J Intern Med. 2003;254:132–139. doi: 10.1046/j.1365-2796.2003.01180.x. [DOI] [PubMed] [Google Scholar]
- 22.Prinsen BH, Rabelink TJ, Beutler JJ, Kaysen GA, De Boer J, Boer WH, Hagen EC, Berger R, De Sain-Van Der Velden MG. Increased albumin and fibrinogen synthesis rate in patients with chronic renal failure. Kidney Int. 2003;64:1495–1504. doi: 10.1046/j.1523-1755.2003.00211.x. [DOI] [PubMed] [Google Scholar]
- 23.Raj DS, Oladipo A, Lim VS. Amino acid and protein kinetics in renal failure: an integrated approach. Semin Nephrol. 2006;26:158–166. doi: 10.1016/j.semnephrol.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 24.Shlipak MG, Fried LF, Crump C, Bleyer AJ, Manolio TA, Tracy RP, Furberg CD, Psaty BM. Elevations of inflammatory and procoagulant biomarkers in elderly persons with renal insufficiency. Circulation. 2003;107:87–92. doi: 10.1161/01.cir.0000042700.48769.59. [DOI] [PubMed] [Google Scholar]
- 25.Descamps-Latscha B, Witko-Sarsat V, Nguyen-Khoa T, Nguyen AT, Gausson V, Mothu N, London GM, Jungers P. Advanced oxidation protein products as risk factors for atherosclerotic cardiovascular events in nondiabetic predialysis patients. Am J Kidney Dis. 2005;45:39–47. doi: 10.1053/j.ajkd.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 26.Landray MJ, Wheeler DC, Lip GY, Newman DJ, Blann AD, McGlynn FJ, Ball S, Townend JN, Baigent C. Inflammation, endothelial dysfunction, and platelet activation in patients with chronic kidney disease: the Chronic Renal Impairment in Birmingham (CRIB) study. Am J Kidney Dis. 2004;43:244–253. doi: 10.1053/j.ajkd.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 27.Stevens LA, Coresh J, Greene T, Levey AS. Assessing kidney function—measured and estimated glomerular filtration rate. N Engl J Med. 2006;354:2473–2483. doi: 10.1056/NEJMra054415. [DOI] [PubMed] [Google Scholar]