

Abstract
One of the central problems in biochemistry in the post-genomic era is the elucidation of functions of proteins, including “orphan” human cytochromes P450 (P450s), when the substrates are unknown. A general strategy for identification of endogenous substrates of P450s in tissue extracts using metabolomic and isotopic labeling approaches is described, involving four main steps: (1) In vitro incubation of a P450 enzyme system with cofactor and tissue extract is done under a mixture of 18O2/16O2 (1:1). (2) LC-MS assay of an organic extract of the reaction mixture is performed. (3) The isotopic labeling products appearing as M/M+2 doublets can be directly identified using the program DoGEX (R. Sanchez-Ponce and F. P. Guengerich, Anal. Chem. 79, 3355-3362, 2007). (4) Characterization of potential candidates is done. Validation of the strategy was established using human P450 7A1 as an initial model to identify its known product, 7α-hydroxycholesterol, in liver extracts. The strategy was then applied to human P450s 1A2, 2C8, and 2C9 in untargeted substrate searches with human liver extracts. A total of seven fatty acids were identified and verified as substrates of these three hepatic P450s. The products were subsequently characterized as hydroxylation and epoxidation derivatives of fatty acids, using GC-MS analysis. Finally, kinetic studies were performed to confirm that the fatty acids are oxidized by P450s 1A2, 2C8, and 2C9. Thus, this strategy has been demonstrated to be useful in identifying reactions in tissue extracts with orphan human P450s.
Keywords: cytochrome P450, substrate searches, LC-MS, metabolomics, isotopic labeling
INTRODUCTION
Enzymes are critical for almost all cellular transduction systems and metabolic pathways. The successful completion of the human genome project allows various gene products, i.e. proteins, to become available rapidly. Thus, an important and challenging task in modern biochemistry is elucidation of functions of proteins, including establishing the catalytic activities of novel enzymes when their substrates remain unknown.1, 2
Cytochrome P450 (P450) enzymes play significant roles in the metabolism of a large number of compounds, including sterols, fatty acids, eicosanoids, vitamins, and xenobiotics.3 To date 57 apparently functional genes and at least 58 pseudogenes of human P450s have been identified.4 However, ~13 human P450 genes are termed “orphans” because of their unknown functions.4,5 Thus far, several strategies are available for identifying substrates and products of human P450s. Trial-and-error has been used as a historical approach to establish enzyme function but realistically is too time consuming and incomprehensive.5 Alternatively, the affinities of ligands (substrates) for the P450 enzymes can be used in some strategies but distinct issues (e.g., difficult isolation of candidates and false positive effects) limit the application.5 Recently global metabolic profiling (i.e. gathering as much information about metabolites in biological systems as possible) has been demonstrated to be a powerful approach to elucidation of the function of an enzyme.6 Isotopic labeling methods have also shown great potential in the elucidation of function, with the inherent advantages of high sensitivity and selectivity.7 However, regardless of whether global metabolic profiling or isotopic labeling methods are performed, equal sensitivity of all compounds in a sample and a wide dynamic range are issues.8-10
NMR and mass spectrometry (MS) are the most widely used analytical tools for assigning functions of enzymes by metabolomic analysis.8-10 1H-NMR provides unbiased and highly quantitative analysis of various chemical species, without the requirement for sample pretreatment, but relatively poor sensitivity and resolution (resulting in the signal loss of the trace but interesting compounds) limit the applications. Alternatively, MS analysis can be performed, either directly or coupled with chromatographic methods, and possesses high sensitivity, selectivity, and throughput capacity. In recent years, liquid chromatography (LC)-MS has proven to be of particular use in elucidation of enzyme function because of chromatographic separation, leading to less ion suppression, higher selectivity, and resolution with no or minimal chemical derivatization steps (which are generally indispensable for GC-MS).11-13 Particularly when the samples are complex mixtures (e.g., urine, plasma, tissue extracts, etc.), LC-MS enables direct analysis of the sample and provides a reasonable dynamic range and abundant structural information.11-13
With LC-MS as the experimental platform for elucidation of enzyme function, a significant issue is the vast amount of data produced. Appropriate bioinformatics tools are needed and have been developed.14 Many commercial solutions (e.g., BlueFuse, MetAlign, MarkerLynx) are not released for public use, and freely available tools have frequently been utilized for metabolomic data processing.14 E.g., MZmine has been demonstrated to be a useful tool in differential analysis of LC-MS data by performing noise filter, peak detection, and retention time (tR) alignment for corresponding peaks of different runs.15 XCMS, written in R statistical language, is implemented with nonlinear retention time correction of LC-MS data16 and has been successfully used for analysis of endogenous and exogenous metabolites in human serum.17 The Matlab-based computer program DoGEX was developed in this laboratory for untargeted analysis of MS data for elucidation of functions of enzymes.18 DoGEX processes LC-MS data using data alignment, noise removal, baseline correction, and peak detection.18 A major advantage of DoGEX is selective detection of the chromatographic peaks with a particular isotopic ratio, using spectral filtering. This feature can be exploited to identify the isotopic labeling products formed by an enzymatic reaction in complex biological systems.
The aim of the present study is to establish a general strategy for identification of endogenous substrates of human P450s in tissue extracts using LC-MS assays and the DoGEX program. Most of the P450 reactions involve the addition of oxygen (16 a.m.u.): if in vitro incubation of a P450 enzyme system with cofactor and tissue extract is done with a mixture of 18O2/16O2 (1:1), most stable oxidation products should appear as M/M+2 doublets. An automated peak search of the LC-MS profiling can be done to identify the products with characteristic isotopic signatures using the DoGEX program. In order to validate the strategy, both targeted and untargeted searches were performed to identify the potential substrates and products in human liver extracts. Human P450 7A1 was selected as an initial model to identify its known product, 7α-hydroxycholesterol, and subsequently untargeted substrate searches were done for human P450s 1A2, 2C8, and 2C9.
EXPERIMENTAL SECTION
Materials and Reagents
Human liver samples (from organ donors) were obtained from Tennessee Donor Services and stored at -70 °C. Human P450s 7A1,19 1A2,20 2C8,21, 22 and 2C923 and rat NADPH-P450 reductase24 and cytochrome b525 were expressed and purified as previously reported. L-α-1,2-Dilauroyl-sn-glycero-3-phosphocholine, pentafluorobenzyl (PFB) bromide, N,N-diisopropylethylamine, myristic acid (C14:0), palmitic acid (C16:0), palmitoleic acid (C16:1), oleic acid (C18:1), linoleic acid (C18:2), arachidonic acid (C20:4), and docosahexaenoic acid (C22:6) were purchased from Sigma-Aldrich (St. Louis, MO). [1-14C] C14:0 (specific activity 55 mCi mmol-1), [1-14C] C16:0 (specific activity 56 mCi mmol-1), [1-14C] C16:1 (specific activity 55 mCi mmol-1), [1-14C] C18:1 (specific activity 55 mCi mmol-1), [1-14C] C18:2 (specific activity 55 mCi mmol-1), [1-14C] C20:4 (specific activity 55 mCi mmol-1), and [1-14C] C22:6 (specific activity 55 mCi mmol-1) were purchased from American Radiolabeled Chemicals (St. Louis, MO). N,O-bis-(Trimethylsilyl)-trifluoroacetamide (BSTFA):trimethylchlorosilane (TMCS):trimethylsilylimidazole (TMSI):pyridine (3:2:3:10, v/v/v/v) was purchased from Regis Technologies (Morton Grove, IL). All other reagents and solvents were obtained from general commercial suppliers.
Human Liver Extracts
Human liver samples (0.6 g each) from four different individuals were homogenized in a CHCl3/CH3OH mixture (2:1, v/v) with a final volume 20-fold that of the liver sample. The mixture was stirred for 20 min at 23 °C and the homogenate was filtered through paper. The crude extract was mixed with 0.2 volume of H2O and the mixture was centrifuged at 7 × 103 × g for 20 min at 23 °C. The lower organic layer was carefully collected, dried under a stream of N2, stored at -70 °C, and dissolved in 2 mL of ethanol as a stock solution prior to incubations with P450.
In vitro P450 Incubations
Each in vitro enzyme reaction mixture included 1.0 mL of 100 mM phosphate buffer (pH 7.4) containing purified human P450 (1 μM), NADPH-P450 reductase (2 μM), L-α-1,2-dilauroyl-sn-glycero-3-phosphocholine (75 μM), and an aliquot of the ethanolic solution of human liver extract (2%, v/v). In the cases of P450s 2C8 and 2C9, cytochrome b5 (1 μM) was added to improve the enzyme activity.26 For isotopic labeling experiments, reactions were performed in modified 20-mL Thunberg tubes, which were subjected to 10 cycles of alternate vacuum and Ar purging to remove air, using a manifold device.27, 28 The 18O2/16O2 mixture (1:1, v/v) was added into the tube (under vacuum) from a premixed pressurized cylinder (Cambridge Isotopes, Andover, MA) via a short needle.18 The enzyme reaction was initiated by the addition of an NADPH-generating system including 100 μL of 100 mM glucose 6-phosphate, 50 μL of 10 mM NADP+, and 2 μL of a 1 mg mL-1 solution of yeast glucose 6-phosphate dehydrogenase. All incubations were carried out at 37 °C for 20 min and quenched by adding 2 mL of CH2Cl2, followed by extraction. After centrifugation at 2 × 103 × g for 40 min, the (lower) organic phase was carefully removed, transferred into another vial, and dried under an N2 stream for LC-MS assay. For the incubation of P450 7A1 and liver extract, the sample was further derivatized with succinic anhydride (2.5 mg) in pyridine (50 μL) at 65 °C for 6 h prior to LC-MS analysis.
LC-MS/MS Metabolomics
The LC separation system was a Waters Acquity UPLC system (Waters, Milford, MA) using an Acquity UPLC BEH octadecylsilane (C18) column (1.7 μm; 1.0 mm × 100 mm) at 50 °C. The analysis was carried out with a linear gradient from 95% mobile phase A, 10 mM NH4CH3CO2 in a 5:95 (v/v) CH3CN/H2O mixture, to 100% mobile phase B, 10 mM NH4CH3CO2 in a 95:5 (v/v) CH3CN/H2O mixture, over 20 min and held at 100% mobile phase B for 5 min, all at a flow rate of 0.1 mL min-1. The sample (10 μL, dissolved in CH3CN/H2O, 2:1, v/v) was injected using an autosampler system. MS/MS analysis was performed on a ThermoFinnigan LTQ ion trap mass spectrometer (ThermoFisher, Watham, MA) equipped with an electrospray ionization (ESI) source. Both negative and positive ion modes were used, with a full scan range of m/z 50 to 1000. The source spray voltage was set at 3.5 kV and the capillary voltage was set at -50 V, with the capillary temperature at 300 °C. The sheath gas flow rate was 24 and the auxiliary gas flow rate was 8. MS fragmentation analysis was done with normalized collision energy 35%, 1 m/z isolation width, activation Q 0.25, and activation time 30 ms. Data were acquired using a Finnigan Xcalibur software package.
Data Analysis by DoGEX
The analysis of the resulting LC-MS data was automatically done with the Matlab-based computer program DoGEX.18 DoGEX employs wavelets and morphological analysis to process LC-MS data and generates a matrix of integration areas versus retention time and molecular mass of all the detected chromatographic peaks.18 A major advantage of this program is selective detection of the chromatographic peaks with a particular isotopic ratio using spectral filtering. Therefore, in the present study the 18O2/16O2 oxidation products appearing as M and M+2 doublets can be easily identified using the DoGEX program.
Substrate Quantitation in Human Liver Extract by GC-MS
For quantitation of total free fatty acids in human liver extract, PFB ester derivatives were prepared. The ethanolic solution of human liver extract (20 μL) was dried under N2 stream and redissolved in 0.5 mL of CH2Cl2; diisopropylethylamine (40 μL) and PFB bromide (10 μL) were successively added to the above solution and the mixture was heated at 60 °C for 1 h. After cooling to room temperature, the organic phase was collected and evaporated under an N2 stream. The resulting PFB esters of the fatty acids in the liver extract were dissolved in 100 μL of hexane and analyzed on a ThermoFinnigan Trace GC 2000 gas chromatograph interfaced with a Finnigan Trace GC DSQ mass spectrometer (ThermoFinnigan, Austin, TX) using the electron impact (EI) negative ion mode. Samples (4 μL) were injected onto a HP-5 capillary column (30 m × 0.25 mm × 0.25 μm, Agilent Technologies, Santa Clara, CA). The temperature program started at 150 °C (held for 1 min) and then increased to 300 °C at 8 °C min-1, and held at 300 °C for 6 min more.
Spectral Binding Titrations
Binding assays of fatty acids and human P450s 2C8 and 2C9 were done29 using an Aminco DW-2a/OLIS spectrophotometer (On-line Instrument Systems, Bogart, GA). Purified P450 (2 μM) was titrated with fatty acid (in C2H5OH) in 1.0 mL of 100 mM phosphate buffer (pH 7.4), and the reference cuvette (containing the same concentration of enzyme, in buffer) was titrated with an equal volume of the vehicle solvent (C2H5OH). The final concentration of C2H5OH was < 1% (v/v). Visible spectra (350-500 nm) were recorded after each addition of fatty acid. The difference in absorbance between the wavelength maximum at 390 nm and minimum at 420 nm, ΔA390 – A420, was plotted versus the fatty acid concentration. The value of spectral dissociation constant, Ks, was estimated using a hyperbolic equation in GraphPad Prism software (GraphPad Software, San Diego, CA). In the case of high affinity binding (apparent Ks value within 3-fold of the P450 concentration), the quadratic equation
| (1) |
was used to correct for the bound enzyme concentration.
Characterization of Oxidation Products and Kinetic Analysis of P450 Reactions
14C-radiolabeled substrates were commonly used for analysis of rates of oxidation of fatty acids. In vitro incubations were performed using 14C radioactive fatty acid substrates and the recombinant P450 system under the same conditions as described above for the 18O2/16O2 assay (without a change of the atmosphere). For characterization of oxidation products, 50 nmol of fatty acid (typically containing 0.56 μCi of radioactivity) was used in each 1.0 mL reaction mixture. For steady-state kinetic studies, the concentration of the substrate containing radioactive fatty acid was varied, ranging from 0 to 100 μM. After drying the CH2Cl2 extract of the reaction mixture under N2 stream, the sample was dissolved in CH3CN/H2O (2:1, v/v) for HPLC analysis. HPLC separation was done using a 4.6 mm × 150 mm Prodigy octadecylsilane (C18) column (5 μm; Phenomenex, Torrance, CA) with the same mobile phases as described above for LC-MS assay. A linear gradient began with mobile phase A/B (20:80, v/v) increasing to A/B (0:100, v/v) over 25 min at a flow rate of 1.0 mL min-1 and was held at 100% B for 5 min for C14:0 and C16:0. In the cases of the other five fatty acids, the mobile phase program began with a 50-100% B linear gradient over 50 min and held at 100% B for 5 min, at a flow rate of 1.0 mL min-1. Radioactivity was determined in line with a β-RAM flow counter (IN/US Systems, Tampa, FL), with the scintillation cocktail pumped at a rate of 3.0 mL min-1.
Characterization of the oxidation products was performed by GC-MS after preparing the corresponding trimethylsilyl (TMS) ethers. The fractions of the products were collected separately from the HPLC column, the organic solvent was evaporated, and the products were extracted twice with ethyl acetate. After the extracts were dried under an N2 stream, silylation was done on each residue with 100 μL of silylation reagent (BSTFA: TMCS: TMSI: pyridine, 3:2:3:10, v/v/v/v) at 60 °C for 30 min. For the unsaturated fatty acids, hydrogenation of each sample was performed prior to silylation using 2 mg of Pd powder under an H2 stream for 3 min. The resulting TMS derivatives were directly analyzed by GC-MS in the electron impact mode using the same conditions as with the PFB esters for substrate quantitation.
RESULTS AND DISCUSSION
Validation of Metabolomic and Isotopic Labeling Approach
The strategy of identification of endogenous substrates of human P450s in tissue extracts using metabolomic and isotopic labeling approaches involved four main steps (Figure 1). The isotopic labeling incubation of a recombinant human P450 system with cofactor and tissue extract was first done under an atmosphere of an 18O2/16O2 mixture (1:1). The organic extract of the reaction mixture was then submitted to LC-MS analysis for metabolic profiling. An automated search was done to find the oxidation products appearing as M/M+2 doublets, using the DoGEX program. Finally, the potential candidates were further characterized using MS or other analytical tools. Human P450 7A1 was selected as an initial model to validate this strategy because this P450 specifically oxidizes cholesterol to form 7α-hydroxycholesterol.19,30 An organic extract of human liver tissue was employed as the substrate library because the liver is the major location of P450 7A1 and it contains a large variety of compounds, including the substrate cholesterol.
Figure 1.
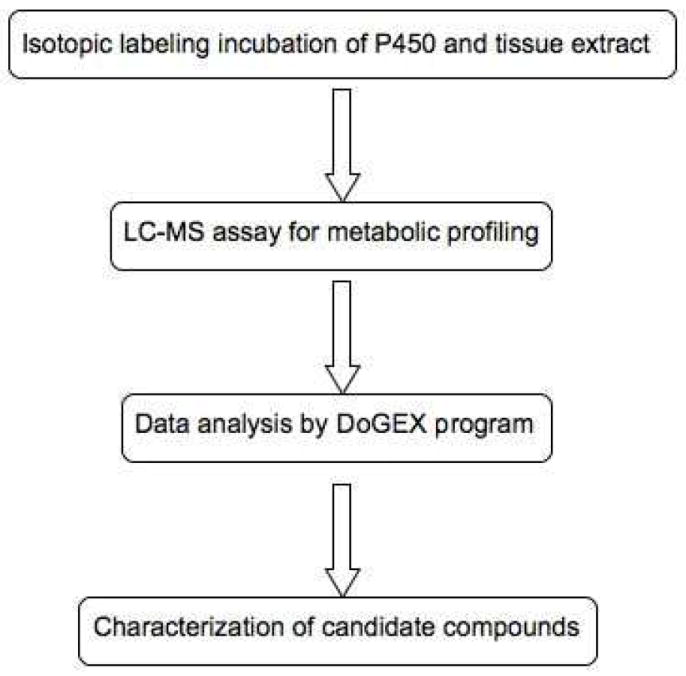
Strategy for identification of endogenous substrates of human P450s in tissue extracts using LC-MS metabolomics and isotopic labeling methods.
Because 7α-hydroxycholesterol easily loses the (7-) hydroxyl group during the MS assay,31,32 the sample prepared from incubation of human P450 7A1 and CHCl3-CH3OH liver extract was further derivatized by succinic anhydride prior to LC-MS analysis. The resulting succinate ester is readily analyzed in the ESI negative ion mode, with a molecular ion ([M-H]-) of m/z 601. The total ion chromatogram of the incubation of human P450 7A1 and liver extract is shown, along with the detected peaks (Figure 2A). An automated search was done using the DoGEX program, with an isotopic ratio range of 0.95-1.9 (designed for a 2-fold visual display range in Matlab), and the doublet of m/z 603 and 601 was identified (Figure 2B, 2C, and 2D). The identity of the 7α-hydroxycholesterol was subsequently confirmed by comparison (tR, MS spectrum) with an authentic sample. This result demonstrated that the strategy is simple but efficient and sensitive enough to identify substrates and products of P450 enzymes in tissue extracts.
Figure 2.
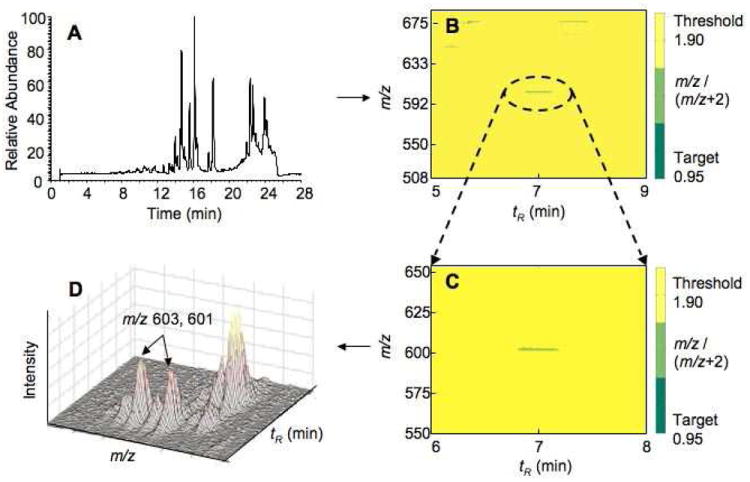
Total ion chromatogram of the incubation of human P450 7A1 and liver extract (A), data analysis for identifying the oxidation product 7α-hydroxycholesterol using the program DoGEX (B), expansion of part B (C), and three-dimensional profile of part C (D).
Searches for Human P450s 1A2, 2C8, and 2C9 Substrates in Liver Extracts
After successful validation with P450 7A1, we applied this strategy to more P450 enzymes and tried to find new substrates and products for some established hepatic enzymes, i.e. P450s 1A2, 2C8, and 2C9. Organic extracts were also used as the substrate library instead of the crude homogenates because the CHCl3-CH3OH extracts (resuspended in C2H5OH for in vitro incubations) contain most types of small molecules suitable for substrate/product searches (with the exception of a few compounds that may be poorly extracted or not soluble in C2H5OH), with the removal of proteins, nucleic acids, and carbohydrates that might hinder chromatography and detection of more relevant chemicals. Unlike the incubation of P450 7A1 with liver extract, this was a genuine untargeted search in human liver extract using the above strategy, i.e. without knowing the nature of the substrates and products.
In order to profile as many metabolites as possible, the reaction mixtures were analyzed using UPLC-MS in both the negative and positive ion modes. However, no peaks with the characteristic isotopic signatures were detected using the positive ion mode. The DoGEX results for the analysis of the P450 1A2/liver extract (negative ion mode) showed several potential products (Figure 3). A total of seven different doublets (m/z 245/243, 271/269, 273/271, 297/295, 299/297, 321/319, and 345/343) were identified. These seven doublets were also found in searches with human liver extract incubated with P450s 2C8 and 2C9. Verification of these results was done with Xcalibur, the Waters UPLC-MS software package, to determine if the identified peaks were really products of the P450 reactions. In comparisons of the samples of the incubations of P450 1A2 and liver extract with and without NADPH, the seven groups of peaks (marked with numbers) were found in the sample in which the cofactor NADPH was included, but none was found in the sample without NADPH (Figure 4). The results were further confirmed by comparisons to incubations done in the absence of added P450 enzymes (results not presented). Moreover, these peaks were consistent with the results generated with the DoGEX program, i.e. same tR and m/z values. Thus, all seven doublets identified by the DoGEX program were judged to be oxidation products formed in liver extracts with human P450s 1A2, 2C8, and 2C9.
Figure 3.
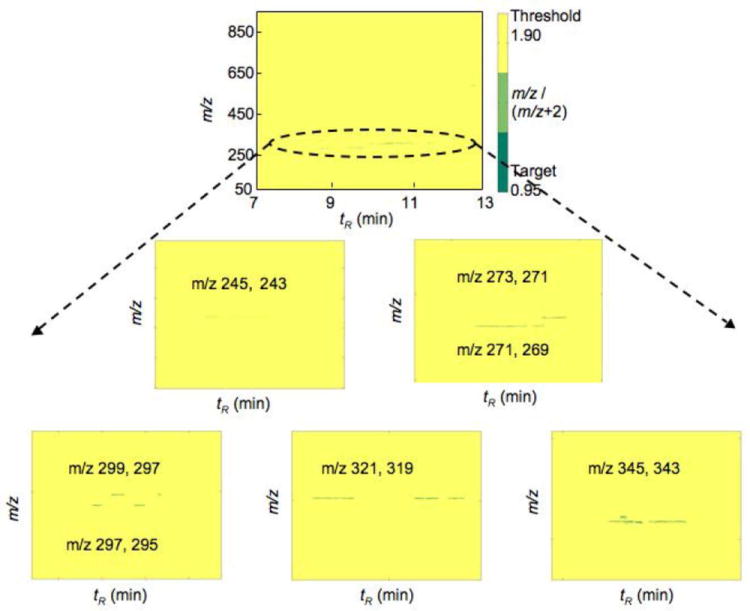
DoGEX results of substrate searches with liver extract and human P450 1A2.
Figure 4.
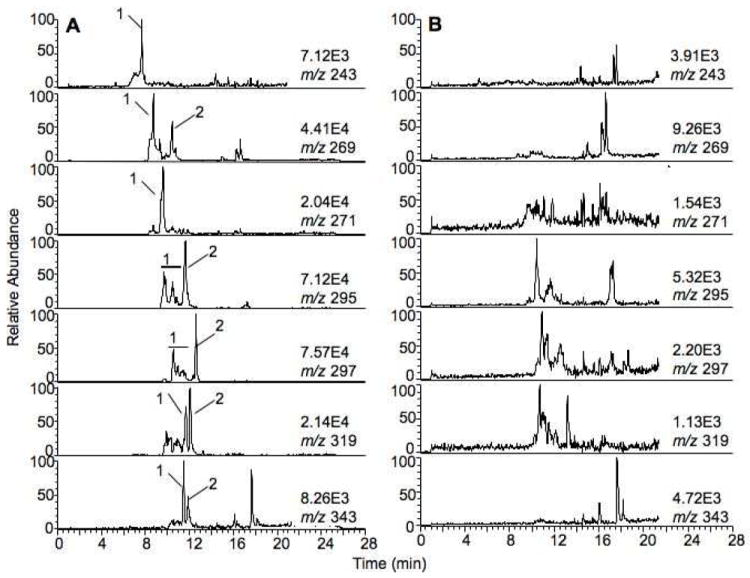
Comparison of LC-MS analysis for incubation of P450 1A2 and liver extract with (A) and without (B) NADPH-generating system.
In principle, the m/z values of the respective substrates can be deduced from those of the products by subtracting 16 a.m.u. (oxygen). Therefore, the molecular masses of the potential substrates corresponding to the doublets described above were 228, 254, 256, 280, 282, 304, and 328. MS fragmentation analysis was done for the seven identified products (Table S-1). The main ion fragments [M-H]-, [M-H-18]- (loss of H2O), [M-H-44]- (loss of CO2), and [M-H-18-44]- (loss of both H2O and CO2) were found for the product peaks. Based on these results and comparison with the database of LIPIDMAPS (http://www.lipidmaps.org), fatty acids were suspected as the potential substrates of the P450 reactions, including C14:0, C16:0, C16:1, C18:1, C18:2, C20:4, and C22:6, corresponding to the molecular ions described above. To test this hypothesis, these seven synthetic fatty acids were used in incubations with P450 1A2 and analyzed by LC-MS/MS under the same conditions. All of the product peaks formed in the incubations of the authentic fatty acids with P450 1A2 yielded exactly the same peaks identified in human liver extracts with the DoGEX program. These results imply that these fatty acids are substrates in human liver for P450s 1A2, 2C8, and 2C9 (Figure 5). Among these fatty acids, C20:4 is the most commonly studied fatty acid oxidized by P450s, including human P450s 1A2, 2C8, and 2C9.33 The other unsaturated fatty acids, i.e. C18:1,34,35 C18:2,36 and C22:6,37,38 and the saturated fatty acids, i.e. C14:035 and C16:0,35 have also been reported as substrates of the P450s in human liver. This appears to be the first report that C16:1 is a substrate of human P450s.39
Figure 5.
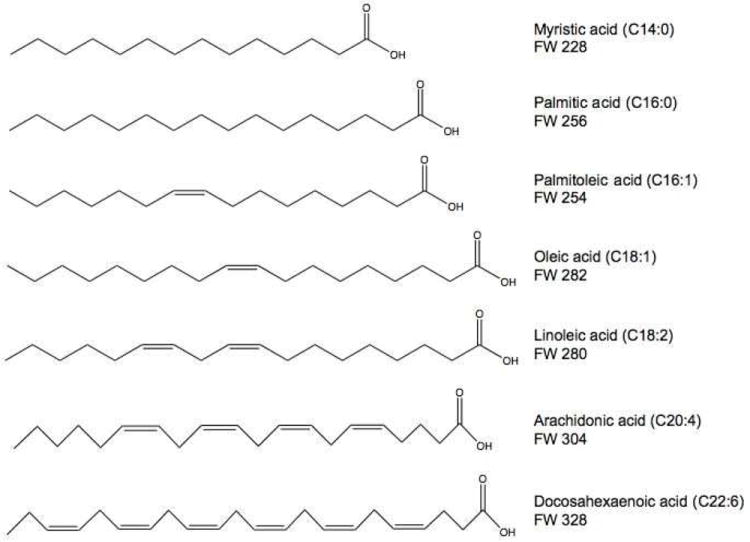
Structures of the fatty acids.
Fatty Acid Quantitation in Human Liver Extracts
In order to quantify the free fatty acids in human liver extract, PFB ester derivatives were prepared and analyzed by GC-MS, because the PFB esters of fatty acids show little variation of ionization response among different fatty acids, allowing direct comparison of ratios of the fatty acids in liver extract (electron impact negative ion mode, Figure S-1). The relative ratios of free fatty acids in human liver were then calculated from the peak areas (Figure S-1) and are summarized in Table 1. Of the seven fatty acids identified, C18:1, C18:2, and C16:0 were the main fatty acids in human liver and C14:0, C16:1, and the polyunsaturated fatty acids C20:4 and C22:6 were present at a level one order of magnitude lower (compared with the major fatty acids), consistent with earlier literature.40 Thus, the fatty acids are putative substrates of the P450 enzymes and are also found in human liver.
Table 1.
Quantitation of fatty acids in human liver extract by GC-MS analysisa
| Fatty acid | Relative ratio in human liver (% total) |
|---|---|
| C14:0 | 2 |
| C16:0 | 16 |
| C16:1 | 6 |
| C18:1 | 44 |
| C18:2 | 26 |
| C20:4 | 5 |
| C22:6 | 1 |
PFB esters of free fatty acids in human liver extract were prepared using the method described in the Experimental Section and analyzed by GC-MS (Figure S-1).
Spectral Binding Titrations
Because the fatty acids were verified as potential endogenous substrates for P450s 1A2, 2C8, and 2C9 in human liver, we investigated interactions between the fatty acids and P450 enzymes. Human P450 1A2 has an exclusively high-spin iron configuration,20,41 and therefore the spectral binding studies were done only for the low-spin iron P450s 2C8 and 2C9. The Ks values were estimated from the titration curves of ΔA390 – ΔA420 versus the concentrations of fatty acids (Table 2). All of the seven fatty acids exhibited micromolar affinity for P450s 2C8 and 2C9, except C22:6 (no real binding to P450 2C8, ΔA390 < 0.005 at a substrate concentration of 50 μM). The spectral changes induced by binding of C14:0 to P450 2C8 are shown as an example (estimated Ks 8.0 μM, Figure S-2). These binding results provide further evidence that the seven fatty acids that were identified are human liver ligands for human P450s 2C8 and 2C9.
Table 2.
Spectral titration of human P450 2C8, and 2C9 with fatty acids
| Fatty acid | P450 2C8 | P450 2C9 | ||
|---|---|---|---|---|
| ΔAmaxa | Ksb (μM) | ΔAmax | Ks (μM) | |
| C14:0 | 0.051 ± 0.001c | 8.0 ± 0.7c | 0.040 ± 0.002 | 13 ± 2 |
| C16:0 | 0.044 ± 0.002 | 1.5 ± 0.6 | 0.055 ± 0.004 | 11 ± 2 |
| C16:1 | 0.041 ± 0.003 | 7.9 ± 1.8 | 0.056 ± 0.005 | 8.7 ± 1.8 |
| C18:1 | 0.027 ± 0.002 | 1.1 ± 0.7 | 0.046 ± 0.005 | 6.8 ± 2.1 |
| C18:2 | 0.046 ± 0.003 | 0.5 ± 0.3 | 0.045 ± 0.002 | 0.8 ± 0.3 |
| C20:4 | 0.062 ± 0.005 | 13 ± 3.0 | 0.037 ± 0.002 | 0.5 ± 0.3 |
| C22:6 | –d | – | 0.042 ± 0.005 | 9.3 ± 2.6 |
Difference in absorbance between the wavelength maximum at 390 nm and minimum at 420 nm, ΔA390 – A420. The concentration range used was 0 to 50 μM fatty acid in each case.
Estimated from the hyperbolic fitting plot of ΔAmax versus the fatty acid concentration.
Data presented in Supporting Information Figure S-2.
–, no detectable binding (ΔA390 < 0.005 at 50 μM).
Characterization of Oxidation Products of Fatty Acids
P450s react with fatty acids to form a number of hydroxylation and epoxidation products.33 The identities of these products were established using the approach described in the Experimental Section. The radioactive HPLC peaks (Figure 4) were collected and the TMS derivatives were prepared for GC-MS assay. In the case of the unsaturated fatty acids, hydrogenation was done before silylation to simplify the analysis of the GC-MS fragments. Human P450 1A2 was employed as a prototype for incubations with the radioactive fatty acids for characterization of the oxidation products also formed by P450s 2C8 and 2C9.
For hydroxylated products, the base peak m/z 117 has been shown to be the characteristic ion fragment specific to ω-1 hydroxylated fatty acids, although the m/z 117 ion can also be observed for the cleavage of the carboxylate end, resulting in the fragment [COO-SiMe3+].42-44 For instance, the spectrum of the product of the incubation of P450 1A2 and C14:0 showed that the major ion fragment was m/z 117 (Figure S-3), resulting from cleavage of the ω-terminal group (CH3-CHO-SiMe3+). Moreover, the fragments at m/z 373 [M+-15] and m/z 283 [M+-90-15] were formed by the loss of methyl and TMS plus methyl groups, respectively. Another major ion fragment was m/z 73, belonging to the TMS moiety, [SiMe3+]. Therefore, the product of the reaction of C14:0 and P450 1A2 was ω-1 hydroxylation. Thus, the positions of the hydroxyl group of the metabolites could be characterized as ω, ω-1, ω-2, etc., based on the shifts of the m/z 117 base peaks by multiples of 14.45 For the epoxidation products, acid hydrolysis was done to convert the products to dihydrodiols for GC-MS analysis, and the positions of the epoxy groups were defined based on the two hydroxyl groups. The TMS derivatives of the products formed by human P450 1A2 (same for P450s 2C8 and 2C9) (Table 3) were hydroxylation and epoxidation products, as described in reported literature.33-36,38
Table 3.
EI GC-MS analysis of TMS derivatives of the oxidation products of fatty acids produced by human P450 1A2
| Substrate | Product numbera | [M+] | Major fragment ions | Product |
|---|---|---|---|---|
| C14:0 | 1 | 388 | 117, 283, 373b | (ω-1)-OH |
| C16:0 | 1 | 416 | 117, 311, 401 | (ω-1)-OH |
| C16:1 | 1 | 416 | 103/ 117/ 159/ 173 | ω/ ω-1/ ω-4/ ω-5-OH |
| 2 | 504 | 187, 317 | 9,10-epoxy | |
| C18:1 | 1 | 444 | 103/ 117/ 187/201 | ω/ ω-1/ ω-6/ ω-7-OH |
| 2 | 532 | 215, 317 | 9,10-epoxy | |
| C18:2 | 1 | 444 | 117/ 131/ 145/ 159/ 201 | ω-1/ ω-2/ ω-3/ ω-4/ ω-7-OH |
| 2 | 444 | 173/ 187/ 229/ 257 | ω-5/ ω-6/ ω-9/ ω-11-OH | |
| C20:4 | 1 | 560 | 215, 229, 345, 359/173 | 11, 12-epoxy/ 15-HETEc |
| 2 | 560 | 257, 271, 303, 317/229 | 8,9-epoxy/ 11-HETEc | |
| C22:6 | 1 | 500 | 117, 383 | (ω-1)-OH |
| 2 | 588 | 215, 229, 271/257, 271, 331, 345 | 10,11-epoxy/13, 14-epoxy |
Product number is the peak number in Figure 4.
The structure of the product was characterized by GC-MS (see Figure S-3).
HETE: hydroxyeicosatetraenoic acid.
Kinetic Analysis of P450 Fatty Acid Oxidation Reactions
Kinetic studies were performed for all groups of the products (marked by numbers in Figure 4), with concentrations of the fatty acids ranging from 0 to 100 μM and a reaction time of 20 min. The kinetic parameters kcat and Km were estimated based on Michaelis-Menten plots and non-linear regression analysis (Table 4). In most of the previous literature kcat and Km values have seldom been measured, and only the values for C20:4 (P450s 2C8 and 2C9) were available (but calculated only for the formation of epoxidation products).40,46 The catalytic efficiencies (kcat/Km) measured in the present study are slower than those measured for many common P450 drug oxidations (some catalytic efficiencies < 103 min-1 M-1), but the fatty acids were definitely oxidized by human P450s 1A2, 2C8, and 2C9. These results indicate that all seven fatty acids are present in human liver and are substrates for human P450s 1A2, 2C8, and 2C9, which generate the hydroxylation and epoxidation products with different catalytic efficiencies.
Table 4.
Steady-state kinetic analysis of oxidations of fatty acids by human P450s 1A2, 2C8, and 2C9.a
| Fatty acid | P450 | Product number | kcat(min-1) | Km(μM) | kcat / Km (min-1 μM-1) |
|---|---|---|---|---|---|
| C14:0 | 1A2 | 1 | 0.29 ± 0.04 | 34 ± 10 | 0.0085 ± 0.0040 |
| 2C8 | 1 | 0.052 ± 0.004 | 30 ± 5 | 0.0017 ± 0.0008 | |
| 2C9 | 1 | 0.053 ± 0.004 | 22 ± 5 | 0.0024 ± 0.0008 | |
| C16:0 | 1A2 | 1 | 0.027 ± 0.002 | 13 ± 3 | 0.0020 ± 0.0007 |
| 2C8 | 1 | 0.025 ± 0.002 | 29 ± 3 | 0.00086 ± 0.00067 | |
| 2C9 | 1 | 0.049 ± 0.014 | 49 ± 20 | 0.0010 ± 0.0007 | |
| C16:1 | 1A2 | 1 | 0.28 ± 0.01 | 14 ± 2 | 0.020 ± 0.005 |
| 2 | 0.070 ± 0.003 | 12 ± 2 | 0.0058 ± 0.0015 | ||
| 2C8 | 1 | 0.080 ± 0.006 | 41 ± 7 | 0.0020 ± 0.0009 | |
| 2 | 0.023 ± 0.001 | 18 ± 2 | 0.0013 ± 0.0005 | ||
| 2C9 | 1 | 0.074 ± 0.004 | 20 ± 3 | 0.0037 ± 0.0013 | |
| 2 | 0.14 ± 0.01 | 13 ± 3 | 0.011 ± 0.003 | ||
| C18:1 | 1A2 | 1 | 0.20 ± 0.01 | 29 ± 4 | 0.0069 ± 0.0025 |
| 2 | 0.21 ± 0.01 | 28 ± 4 | 0.0075 ± 0.0025 | ||
| 2C8 | 1 | 0.024 ± 0.002 | 23 ± 5 | 0.0010 ± 0.0004 | |
| 2 | 0.020 ± 0.001 | 29 ± 5 | 0.00069 ± 0.00020 | ||
| 2C9 | 1 | 0.085 ± 0.006 | 63 ± 8 | 0.0013 ± 0.0008 | |
| 2 | 0.16 ± 0.01 | 35 ± 6 | 0.0046 ± 0.0017 | ||
| C18:2 | 1A2 | 1 | 0.26 ± 0.01 | 14 ± 2 | 0.019 ± 0.005 |
| 2 | 0.13 ± 0.01 | 12 ±3 | 0.011 ± 0.003 | ||
| 2C8 | 1 | 0.17 ± 0.01 | 32 ± 3 | 0.0053 ± 0.0033 | |
| 2 | 0.094 ± 0.007 | 26 ± 5 | 0.0036 ± 0.0014 | ||
| 2C9 | 1 | 0.14 ± 0.02 | 32 ± 9 | 0.0044 ± 0.0022 | |
| 2 | 0.15 ± 0.01 | 25 ± 6 | 0.0060 ± 0.0017 | ||
| C20:4 | 1A2 | 1 | 0.064 ± 0.005 | 16 ± 3 | 0.0040 ± 0.0017 |
| 2 | 0.17 ± 0.02 | 17 ± 4 | 0.010 ± 0.005 | ||
| 2C8 | 1 | 0.027 ± 0.001 | 19 ± 3 | 0.0014 ± 0.0003 | |
| 2 | 0.023 ± 0.002 | 34 ± 5 | 0.00068 ± 0.00040 | ||
| 2C9 | 1 | 0.12 ± 0.01 | 21 ± 3 | 0.0057 ± 0.0033 | |
| 2 | 0.078 ± 0.005 | 24 ± 4 | 0.0033 ± 0.0013 | ||
| C22:6 | 1A2 | 1 | 0.15 ± 0.01 | 22 ± 3 | 0.0068 ± 0.0033 |
| 2 | 0.12 ± 0.01 | 33 ± 3 | 0.0036 ± 0.0033 | ||
| 2C8 | 1 | 0.065 ± 0.004 | 25 ± 5 | 0.0026 ± 0.0008 | |
| 2 | 0.061 ± 0.005 | 25 ± 6 | 0.0024 ± 0.0008 | ||
| 2C9 | 1 | 0.027 ± 0.002 | 25 ± 4 | 0.0011 ± 0.0005 | |
| 2 | 0.15 ± 0.01 | 26 ± 4 | 0.0058 ± 0.0025 | ||
kcat and Km values were determined using the method described in the Experimental Section. In some cases multiple products are grouped because of limited LC resolution.
CONCLUSIONS
In summary, we have described a general strategy for identification of endogenous substrates in tissue extracts, which can be used to elucidate functions of human P450s. The combination of LC-MS metabolomics and the program DoGEX enables considerable simplification of a procedure for substrate searches in tissue extracts by direct analysis of the sample and automated identification of the potential products. The strategy was successfully validated employing human P450 7A1 to identify its known product in a human liver extract. In genuine untargeted searches with human P450s 1A2, 2C8, and 2C9, seven fatty acids were identified as potential substrates. The studies on fatty acid quantitation and spectral binding titrations confirmed that these seven fatty acids are human liver ligands for P450s 1A2, 2C8, and 2C9. Moreover, characterization of the products and kinetic analysis of the P450 reactions demonstrated that these fatty acids are oxidized by P450s 1A2, 2C8, and 2C9 to form hydroxylation and epoxidation products, as some previous literature reported. The physiological relevance of most of these products remains to be established.33 This strategy has been shown to be efficient, sensitive, and robust. It provides an example of application of the program DoGEX and also demonstrates the potential for identification of reaction candidates with true orphan human P450s in tissue extracts.
Supplementary Material
Acknowledgments
We thank D. L. Hachey, M. W. Calcutt, and A. Harlan for mass spectrometry assistance and discussions, Y. Xiao for helping with some of the studies, and A. R. Brash for use of a hydrogenation system. This work was supported in part by National Institutes of Health grants R37 CA090426 and P30 ES000267.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Additional information as noted in text (Table S-1 and Figures S-1, S-2, and S-3). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Carpenter AE, Sabatini DM. Nat Rev Genet. 2004;5:11–22. doi: 10.1038/nrg1248. [DOI] [PubMed] [Google Scholar]
- 2.Hughes TR, Robinson MD, Mitsakakis N, Johnston M. Curr Opin Microbiol. 2004;7:546–554. doi: 10.1016/j.mib.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 3.Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. KluwerAcademic/Plenum Publishers; New York: 2005. [Google Scholar]
- 4.Guengerich FP. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. Ortiz de Montellano PR, editor. Kluwer Academic/Plenum Press; New York: 2005. pp. 377–530. [Google Scholar]
- 5.Guengerich FP, Wu Z-L, Bartleson CJ. Biochem Biophys Res Commun. 2005;338:465–469. doi: 10.1016/j.bbrc.2005.08.079. [DOI] [PubMed] [Google Scholar]
- 6.Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. Biochemistry. 2004;43:14332–14339. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 7.Mutlib AE. Chem Res Toxicol. 2008;21:1672–1689. doi: 10.1021/tx800139z. [DOI] [PubMed] [Google Scholar]
- 8.Saghatelian A, Cravatt BF. Curr Opin Chem Biol. 2005;9:62–68. doi: 10.1016/j.cbpa.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Schlotterbeck G, Ross A, Dieterle F, Senn H. Pharmacogenomics. 2006;7:1055–1075. doi: 10.2217/14622416.7.7.1055. [DOI] [PubMed] [Google Scholar]
- 10.Pan Z, Raftery D. Anal Bioanal Chem. 2007;387:525–527. doi: 10.1007/s00216-006-0687-8. [DOI] [PubMed] [Google Scholar]
- 11.Want EJ, Cravatt BF, Siuzdak G. Chembiochem. 2005;6:1941–1951. doi: 10.1002/cbic.200500151. [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Gonzalez FJ, Idle JR. Drug Metab Rev. 2007;39:581–597. doi: 10.1080/03602530701497804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dettmer K, Aronov PA, Hammock BD. Mass Spectrom Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katajamaa M, Oresic M. J Chromatogr A. 2007;1158:318–328. doi: 10.1016/j.chroma.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 15.Katajamaa M, Oresic M. BMC Bioinformatics. 2005;6:179. doi: 10.1186/1471-2105-6-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 17.Nordstrom A, O’Maille G, Qin C, Siuzdak G. Anal Chem. 2006;78:3289–3295. doi: 10.1021/ac060245f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez-Ponce R, Guengerich FP. Anal Chem. 2007;79:3355–3362. doi: 10.1021/ac0622781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mast N, Graham SE, Andersson U, Bjorkhem I, Hill C, Peterson J, Pikuleva IA. Biochemistry. 2005;44:3259–3271. doi: 10.1021/bi047566a. [DOI] [PubMed] [Google Scholar]
- 20.Sandhu P, Guo Z, Baba T, Martin MV, Tukey RH, Guengerich FP. Arch Biochem Biophys. 1994;309:168–177. doi: 10.1006/abbi.1994.1099. [DOI] [PubMed] [Google Scholar]
- 21.Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:9497–9503. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 22.Schoch GA, Yano JK, Sansen S, Dansette PM, Stout CD, Johnson EF. J Biol Chem. 2008;283:17227–17237. doi: 10.1074/jbc.M802180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandhu P, Baba T, Guengerich FP. Arch Biochem Biophys. 1993;306:443–450. doi: 10.1006/abbi.1993.1536. [DOI] [PubMed] [Google Scholar]
- 24.Hanna IH, Teiber JF, Kokones KL, Hollenberg PF. Arch Biochem Biophys. 1998;350:324–332. doi: 10.1006/abbi.1997.0534. [DOI] [PubMed] [Google Scholar]
- 25.Yun C-H, Kim K-H, Calcutt MW, Guengerich FP. J Biol Chem. 2005;280:12279–12291. doi: 10.1074/jbc.M411019200. [DOI] [PubMed] [Google Scholar]
- 26.Yamazaki H, Komatsu T, Ohyama K, Nakamura M, Asahi S, Shimada N, Guengerich FP, Nakajima A, Yokoi T. Protein Expr Purif. 2002;24:329–337. doi: 10.1006/prep.2001.1578. [DOI] [PubMed] [Google Scholar]
- 27.Burleigh BD, Jr, Foust GP, Williams CH., Jr Anal Biochem. 1969;27:536–544. doi: 10.1016/0003-2697(69)90067-0. [DOI] [PubMed] [Google Scholar]
- 28.Guengerich FP, Krauser JA, Johnson WW. Biochemistry. 2004;43:10775–10788. doi: 10.1021/bi0491393. [DOI] [PubMed] [Google Scholar]
- 29.Schenkman JB, Remmer H, Estabrook RW. Mol Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]
- 30.Pikuleva IA. Pharmacol Ther. 2006;112:761–773. doi: 10.1016/j.pharmthera.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 31.Manini P, Andreoli R, Careri M, Elviri L, Musci M. Rapid Commun Mass Spectrom. 1998;12:883–889. [Google Scholar]
- 32.Razzazi-Fazeli E, Kleineisen S, Luf W. J Chromatogr A. 2000;896:321–334. doi: 10.1016/s0021-9673(00)00719-6. [DOI] [PubMed] [Google Scholar]
- 33.Capdevila JH, Falck JR, Harris RC. J Lipid Res. 2000;41:163–181. [PubMed] [Google Scholar]
- 34.Adas F, Berthou F, Picart D, Lozac’h P, Beauge F, Amet Y. J Lipid Res. 1998;39:1210–1219. [PubMed] [Google Scholar]
- 35.Adas F, Salaun JP, Berthou F, Picart D, Simon B, Amet Y. J Lipid Res. 1999;40:1990–1997. [PubMed] [Google Scholar]
- 36.Bylund J, Kunz T, Valmsen K, Oliw EH. J Pharmacol Exp Ther. 1998;284:51–60. [PubMed] [Google Scholar]
- 37.Fer M, Dreano Y, Lucas D, Corcos L, Salaun JP, Berthou F, Amet Y. Arch Biochem Biophys. 2008;471:116–125. doi: 10.1016/j.abb.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Fer M, Corcos L, Dreano Y, Plee-Gautier E, Salaun JP, Berthou F, Amet Y. J Lipid Res. 2008;49:2379–2389. doi: 10.1194/jlr.M800199-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Niwa T, Murayama N, Yamazaki H. Curr Drug Metab. 2008;9:453–462. doi: 10.2174/138920008784746364. [DOI] [PubMed] [Google Scholar]
- 40.Hagenfeldt L, Wahren J, Pernow B, Raf L. J Clin Invest. 1972;51:2324–2330. doi: 10.1172/JCI107043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sansen S, Hsu MH, Stout CD, Johnson EF. Arch Biochem Biophy s. 2007;464:197–206. doi: 10.1016/j.abb.2007.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perkins EG, Argoudelis CJ. Lipids. 1969;4:619–621. doi: 10.1007/BF02531051. [DOI] [PubMed] [Google Scholar]
- 43.Wood KV, Bonham CC, Jenks MA. Rapid Commun Mass Spectrom. 2001;15:873–877. doi: 10.1002/rcm.285. [DOI] [PubMed] [Google Scholar]
- 44.Wheelan P, Zirrolli JA, Murphy RC. Am Soc Mass Spectrom. 1995;6:40–51. doi: 10.1016/1044-0305(94)00090-M. [DOI] [PubMed] [Google Scholar]
- 45.Chun Y-J, Shimada T, Sanchez-Ponce R, Martin MV, Lee L, Zhao B, Kelly SL, Waterman MR, Lamb DC, Guengerich FP. J Biol Chem. 2007;282:17486–17500. doi: 10.1074/jbc.M700863200. [DOI] [PubMed] [Google Scholar]
- 46.Barbosa-Sicard E, Markovic M, Honeck H, Christ B, Muller DN, Schunck WH. Biochem Biophys Res Commun. 2005;329:1275–1281. doi: 10.1016/j.bbrc.2005.02.103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.