

Abstract
Acute liver injury causes massive hepatocyte apoptosis and/or fatal liver damage. Fibronectin, an extracellular matrix glycoprotein, is prominently expressed during adult tissue repair. However, the extent of fibronectin dependence on the hepatocyte response to acute liver damage remains to be defined. As identification of hepatic survival factors is critical for successful therapeutic intervention in liver failure, this relationship has been investigated using a fibronectin-deficient mouse model of acute liver injury. Here we show that the lack of fibronectin induces significantly increased hepatocyte apoptosis, which is accompanied by significant downregulation of the anti-apoptotic protein Bcl-xL. Furthermore, fibronectin deficiency leads to a significantly elevated production of hepatocyte growth factor in hepatic stellate cells post injury, which in turn results in an earlier onset and acceleration of hepatocyte regeneration. Primary hepatocytes on fibronectin are protected from reactive oxygen species (ROS)-induced cellular damage, retaining expression of Bcl-xL, whereas those on type I collagen are not. This retained expression of Bcl-xL is inhibited by PI3-kinase inhibitor LY294002.
Conclusion
We provide evidence that the fibronectin-mediated matrix survival signals for hepatocytes are transduced through the PI3-kinase-Bcl-xL signaling axis in response to injury. This work defines fibronectin as a novel anti-apoptotic factor for hepatocytes following acute liver injury but demonstrates that fibronectin is not essential for subsequent hepatocyte proliferation.
Keywords: fibronectin, extracellular matrix, Bcl-xL, PI3-kinase, hepatocyte growth factor, hepatocyte survival
Introduction
Acute liver injury caused by the hepatitis virus or hepatotoxins induces massive hepatocyte apoptosis and/or fatal liver damage, and eventually liver transplantation is required (1–2). Identification of hepatic survival factors is critical for successful therapeutic intervention in liver failure. Following acute injury, the liver undergoes a wound healing response to restore tissue architecture and maintain homeostasis (3). Liver regeneration requires both a well-orchestrated proliferation of hepatocytes and the reconstruction of the extracellular matrix (ECM). In response to injury, the “provisional matrix” formation between plasma fibrin(ogen) and plasma-type fibronectin during an initial stage stabilizes wound areas and provides a road map for cell migration (4). The components of this provisional matrix, plasma fibrinogen and plasma-type fibronectin, are produced by hepatocytes in the liver (4–6).
Fibronectin is a large dimeric glycoprotein that exists in soluble form as a plasma component and in insoluble form as a part of the ECM of almost every tissue (5–6). Both isoforms are generated from a single gene by alternative splicing (7). Extensive in vitro functional studies have indicated that fibronectin plays key roles in a wide variety of cellular behaviors (5–6). Prominent expression of fibronectin is observed during tissue repair, including liver injury (8–9). To explore the function of fibronectin in adult tissues, we generated plasma-type fibronectin conditional-knockout mice and demonstrated that plasma-type fibronectin supports neuronal cell survival following brain damage but is not essential for skin-wound healing (10). To further define the functional identity of fibronectin in the initial stage of adult tissue remodeling, a null-condition for both plasma and cellular fibronectin isoforms from adult liver was recently established in our group. We showed that, in contrast to the long-standing concept that collagen fibril assembly requires fibronectin matrix in culture (11–13), the lack of fibronectin does not actually interfere with reconstruction of collagen organization in the initial fibrogenic response to liver damage (14).
The functional role of fibronectin in parenchymal cells (hepatocytes) following liver injury remains obscure, but regulation of hepatocyte proliferation has been suggested. The level of fibronectin mRNA during liver regeneration after partial hepatectomy is upregulated 3-fold in the liver (15), and fibronectin can promote epidermal growth factor (EGF)-initiated DNA synthesis in primary hepatocyte cultures (16). However, the extent of fibronectin dependence on the hepatocyte response to acute liver damage has not yet been addressed. Here, using a mouse model lacking fibronectin in the adult liver, the suitability of fibronectin as a target for promoting hepatocyte survival and proliferation following acute liver injury has been investigated.
Materials and Methods
Mutant mice generation and mouse maintenance
A mouse strain carrying the fibronectin-floxed gene (Fn[fl/fl]) was generated. Mice lacking both plasma and cellular type fibronectin (‘Liver fibronectin-null mice’) were established by intraperitoneal injections of polyinosinic-polycytidic acid (pI-pC) in the Fn(fl/fl)/Mx-Cre+ strain (C57BL6/129sV mixed background) as described previously (14). Fibronectin-null livers showed normal liver morphology, and no obvious inflammation or fibrosis was noted after pI-pC injections (14). All mice were maintained and bred at the Cleveland Clinic’s Biological Resources Unit in accordance with institutional guidelines and National Institutes of Health standards. The study was approved by the Institutional Animal Care and Use Committee. Mice were regularly monitored and had free access to standard mouse chow and water. Mice received humane care in accordance with institutional guidelines and National Institutes of Health recommendations outlined in the “Guide for the Care and Use of Laboratory Animals.”
Induction of acute liver injury by carbon tetrachloride (CCl4)
Acute liver injury was induced by a single intraperitoneal dose administration of CCl4 solution in olive oil (Fluka) (1.0 ml/kg body weight as 50% [vol/vol]) in sex-matched, 12- to 15-week-old mice as described previously (14, 17). Control and liver Fn-null mice were derived from the same litters.
Antibodies, cytokines and reagents
The following antibodies were used for the analyses: rabbit polyclonal antibody (pAb) against mouse fibronectin (Chemicon); goat pAb against fibrinogen (Nordic Immuno. Lab.); rabbit mAb (E184, Epitomics) against alpha smooth muscle actin (α-SMA); rabbit mAb against Ki67 (clone SP6, Lab Vision); rabbit pAb against cleaved caspase 3 (Cell Signaling); mouse mAb against phospho-extracellular signal-regulated kinase (Erk)1/2 (pT202/pY204) and rabbit pAb against total Erk (Cell Signaling); rabbit pAbs against phospho-Akt (pS473) and total Akt (Cell Signaling); rabbit pAb against Bcl-xL (Cell Signaling); rabbit pAb against phosho-Histone H3 (pS10) (Cell Signaling); mouse mAb against heat shock cognate protein 70 (HSC70, Santa Cruz); and mouse mAb against β-actin (clone AC15, Sigma). Fluorescein isothiocyanate (FITC)- and Cy3-conjugated donkey anti-rabbit and anti-goat IgG, and peroxidase-conjugated donkey anti-mouse, anti-rabbit and anti-goat IgG were from Jackson ImmunoRes Lab. 4′6-diamidino-2-phenylindole (DAPI) was from Molecular Probes. EnvisionTM+ System-HRP labeled polymer was from DAKO.
Histological analysis, immunohistochemistry and immunofluorescence
Histological analysis, immunohistochemistry and immunofluorescence were performed as described previously (14, 18). To quantify damaged areas in response to acute liver injury, images were captured with the same gain, offset, magnitude and exposure time. A minimum of 5 different images from each mouse were randomly selected and the intensities were quantified using Image-Pro Plus software (version 6.1, Media Cybernetics) as described previously (17). For quantification of Bcl-xL signal intensity in primary hepatocytes, images were captured with the same gain, offset, magnitude and exposure time. Then 6 areas per image were randomly selected, and the mean intensity in each cell (intensities/areas, 50 cells) was measured using Image-Pro Plus. The average intensity per cell (relative fluorescent units) was calculated as described elsewhere (19).
Western-blot analysis
Western blot analyses were performed as described elsewhere (14). In some immunoblotting analyses, samples were transferred onto Immobilon-FL polyvinylidene fluoride (PVDF) membrane (Millipore Corp.) and probed with primary and IRDye 800CW- or IRDye 680-conjugated secondary antibodies (LI-COR Biosci.). Immunoreactive bands were detected using the Odyssey Infrared Imaging System (LI-COR Biosci.).
Enzyme-linked immunosorbent assay (Elisa) for hepatocyte growth factor (HGF)
The amount of HGF in liver samples was determined using an Elisa kit (Institute of Immunology, Japan) according to the manufacturer’s instructions.
Serum biochemical analysis
Serum alanine aminotransferase (ALT) levels were determined using a standard kit (Genzyme Diagnostics P.E.I. Inc., Canada).
In vitro assay using mouse primary hepatocytes
Primary hepatocytes were isolated from adult fibronectin-null mouse livers using collagenase perfusion and suspended in Williams’ E medium supplemented with 5μg/ml insulin, 5μg/ml transferrin, 10ng/ml EGF, 10−5M aprotinin, 10−5M dexamethasone, 10−3M nicotinamide, and 10% fibronectin-depleted FBS as described elsewhere (14, 18). For analysis of reactive oxygen species (ROS), hydrogen peroxide (H2O2) (Fisher Scientific) was used as a ROS inducer. To examine the effects of H2O2 on primary hepatocytes, H2O2 was applied at a final concentration of 1.0mM. Primary hepatocytes were subsequently plated on plasma fibronectin (Sigma) and on type I collagen (Upstate) coated substrata (10μg/ml), incubated for 15hrs, then analyzed. Where indicated, cultures contained 5μM LY294002 (IC50=1.4μM) to inhibit phosphatidylinositol 3 (PI3)-kinase, 20μM PD98059 (IC50=2μM) to inhibit mitogen-activated protein kinase kinase (MEK)1/2, 1μM 420119 (IC50=40nM) to inhibit c-Jun N-terminal kinase (JNK), and 5μM SB203580 (IC50=34nM) to inhibit p38 MAP kinase (all inhibitors from Calbiochem).
Data presentation and statistical analysis
All experiments were performed in triplicate as a minimum, on separate occasions, andthe data shown were chosen as representative of results consistently observed. Results are presented as means ± standard deviation (S.D.). Differences between groups were analyzed using the two-sided Student’s t test on raw data. A P value of <0.05 was considered significant.
Results
Fibrin(ogen)-matrix formation without fibronectin in response to acute liver injury
The initial responses to tissue damage are 1) an exudation of plasma proteins and 2) formation of a provisional matrix composed of plasma fibrin(ogen) and fibronectin. Therefore, it was first examined whether the deposition and tissue distribution pattern of the provisional matrix were altered in fibronectin-null livers following acute injury. The expression of fibrin(ogen) showed similar levels between control and mutant livers before and after liver injury (up to 72hrs) by western analysis (Fig. 1A). In control livers, intense deposition of fibronectin and fibrin(ogen) was observed following injury, and was often co-localized as evidenced by yellow fluorescence in merged images (Fig. 1B). However in mutant livers the deposition of was remarkably decreased (<8% compared with control untreated liver as shown by western analysis; Fig. 1A,B). Thus, this indicates that fibrin(ogen) alone can form the initial matrix in response to acute liver damage. An earlier report reveals a small remnant amount of fibronectin associated with platelets in plasma type fibronectin-null mice (10). Very low level deposition of fibronectin protein was occasionally observed in the injured regions of the fibronectin-null liver post injury (Fig. 1B). No obvious extensive hemorrhage was observed in either fibronectin-null or control liver throughout the periods (data not shown).
Fig. 1. Fibrin(ogen) alone forms the initial matrix without fibronectin in response to CCl4-induced acute liver injury.

(A) Western blot analysis of fibronectin and fibrinogen in control and fibronectin-null livers at 0, 6, 12, 36 and 72hrs after injury under reducing conditions. Pooled samples from 3 mouse livers from each strain were used for the analysis. Band intensity was measured by densitometry and normalized to HSC70 (loading control), then the intensity of the control liver at 0hr was set to 1. Each intensity is shown relative to the control value.
(B) Time-course of fibronectin and fibrin(ogen) deposition in control and Fn-null livers at 12, 36, and 72hrs after injury. Double immunofluorescence staining for fibronectin (in red) and fibrin(ogen) (in green), and the merged images. CV, central vein. Bar=50μm.
Lack of fibronectin induces a significant increase in apoptosis in hepatocytes, which is accompanied by a significant downregulation of Bcl-xL expression
It has been previously demonstrated that plasma-type fibronectin can support cell survival following adult tissue damage and an association with anti-apoptotic pathways suggested (10). Since many in vitro studies indicate that fibronectin acts as a survival factor in a variety of cell types (20–23), we next examined whether fibronectin actually prevents hepatocyte apoptosis or whether the initial matrix formed only with fibrin(ogen) is sufficient for hepatocyte survival after CCl4-induced acute liver injury. A significant increase in the number of cleaved caspase 3 (a marker for apoptosis)-positive cells was observed in fibronectin-null livers at 24, 36 and 48hrs post injury (34.8±14.0 cells/field in mutant vs. 1.3±0.4 in control livers at 36hrs post injury [n=5; field=0.24mm2, P<0.01], Fig. 2A,B).
Fig. 2. The lack of fibronectin significantly diminishes survival signals for hepatocytes, mediated by Bcl-xL.

(A) Immunostaining for cleaved caspase 3 (in brown) at 36hrs after injury. Sections were counterstained with hematoxylin. CV, central vein. Bar=50μm.
(B) Analysis of cleaved caspase 3-positive cells. Data are means±S.D. (cells/field; field=0.14mm2 [n=5 for each group]). *, P<0.05; **, P<0.01.
(C) Western blot analysis of Bcl-xL in control and fibronectin-null livers post injury under reducing conditions. Pooled samples from 3 mouse livers from each strain were used for the analysis. Band intensity was measured by densitometry, and each Bcl-xL intensity was normalized to HSC70 (loading control). Then the intensity at 0hr in control livers was set to 1. Each intensity is shown relative to the control value.
(D) Upper panels: Immunostaining for Bcl-xL (in red) and DAPI (to visualize cell nuclei, in blue) at 36hrs after injury (at lower magnification). CV, central vein. Bar=50μm. Lower panels: Triple immunofluorescence staining for Bcl-xL (in red), hepatocyte marker albumin (in green), and DAPI (in blue) at 36hr post injury (at higher magnification). Arrowheads indicate Bcl-xL and albumin double- positive hepatocytes (orange to yellow color). CV, central vein. Bar=25μm.
(E) Analysis of Bcl-xL-positive hepatocytes. Data are means±S.D. (cells/field; field=0.14mm2 [n=5 for each group]). *, P<0.05.
Bcl-xL is an anti-apoptotic member of the Bcl-2 (B-cell lympohoma-2) family of proteins and is the guardian of the mitochondrial pathway for cell death (24). Since Bcl-xL is up-modulated during liver regeneration (25), the expression of Bcl-xL in fibronectin-null livers following injury was examined. The expression level of Bcl-xL protein was remarkably downregulated by ~55.6% and ~50.0% in fibronectin-null livers at 24hr and 36hr post injury, respectively, by western analysis (Fig. 2C). Immunofluorescence staining revealed that the number of Bcl-xL-positive hepatocytes in mutant livers was significantly lower compared with controls (55.4±8.0 cells/field in control vs. 36.9±12.7 in mutant livers at 36hr post injury [n=5; field=0.14mm2, P=0.017]), although liver is composed of several different cell types, including hepatocytes, hepatic stellate cells, sinusoidal endothelial cells, Kupffer cells, and cholangiocytes (Fig. 2D,E). These findings suggest that fibronectin plays an important role in supporting hepatocyte survival following acute liver injury.
Lack of fibronectin leads to significantly increased hepatocyte proliferation with increased levels of HGF following acute liver injury
CCl4 is primarily cytotoxic to hepatocytes and causes centrilobular hepatocellular necrosis. The responses to carbon tetrachloride CCl4-induced acute liver injury are the hepatocyte necrosis that is followed by the subsequent hepatocyte regeneration, and there is an overlapping in these two processes (26–27). Since fibronectin deficiency resulted in significantly increased apoptosis in hepatocytes post injury (Fig. 2A,B), we hypothesized that the absence of fibronectin may result in elevated liver damage. Serum alanine aminotransferase (ALT) levels (a marker for hepatocyte injury) did not show significant differences between mutant and control livers and their peak was ~36hrs. However, a slight elevation in mutant livers was observed at 24 and 36hrs post injury (~12% [6,495±3,205 in control vs. 7,798±4,633 in mutant] and ~8% [9,933±5,297 in control vs. 10,686±3,855 in mutant], respectively; Fig. 3A). Those findings were mirrored in histopathological analysis until 72hrs post injury when fibronectin-null livers displayed significantly smaller damaged areas (Fig. 3B). Therefore, these results strongly suggest that, although fibronectin plays an important role in supporting hepatocyte survival, the lack of fibronectin induces an earlier onset of regenerative response and accelerated hepatocyte proliferation after injury.
Fig. 3. Analysis of liver damage following acute injury.
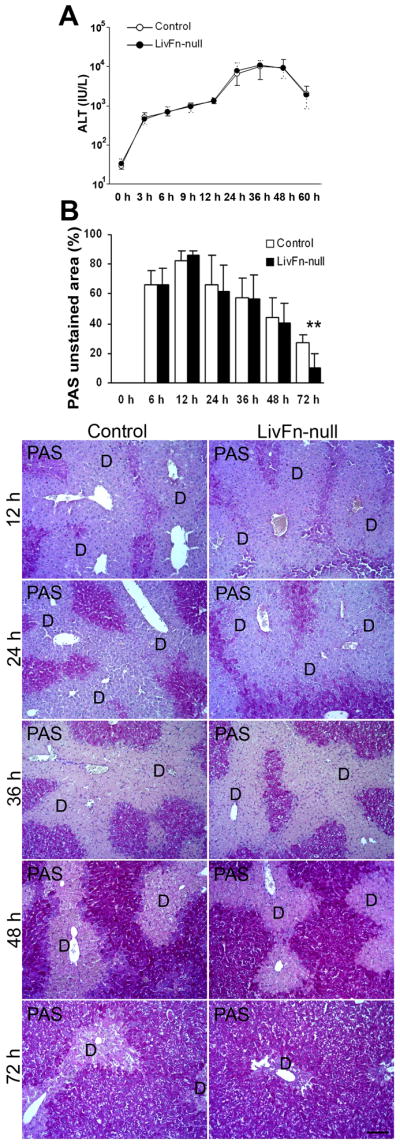
(A) Serum ALT levels in control and fibronectin-null mice post injury. Data are means±S.D. (n=10 for each group).
(B) Upper panel: Time course of the quantification of damaged areas using Periodic Acid-Schiff (PAS)-stained tissue sections. Data are the means±S.D. (n=5 for each group). **, P<0.01. Lower panels: PAS staining of livers at 12, 24, 36, 48, and 72hrs after injury. D, damaged areas. Bar=100μm.
The proliferative response post injury was then examined. Fibronectin-null livers indeed showed an earlier onset of regenerative response, as evidenced by a markedly increased number of Ki67-positive cells post injury (Fig. 4A). Double immunofluorescence staining for Ki67 and albumin revealed a significantly increased number of double-positive hepatocytes in mutant livers from 36 to 72hrs compared with controls (~13.6-fold increase in mutant livers at 36hrs post injury; Fig. 4B, P<0.01). The significantly increased hepatocyte proliferation in fibronectin-null liver was further confirmed by double immunofluorescence staining for phosphorylated Histone H3 (to detect chromosome condensation during mitosis) and albumin at 36hrs post injury (3.1±1.2 cells/field in control vs. 32.5±3.1 cells/field in mutant [[field=0.14 mm2; n=5 for each group; P<0.01]) (Fig. 4C).
Fig. 4. Fibronectin deficiency significantly increases hepatocyte proliferation post injury.

(A) Immunostaining for Ki67 (in brown) at 36 and 72hrs post injury. Sections were counterstained with hematoxylin. Arrowheads indicate positive cells. CV, central vein. Bar=50μm.
(B) Upper panels: Triple immunofluorescence staining for Ki67 (in red), albumin (in green) and DAPI (in blue) at 36hrs post injury. Arrowheads indicate Ki67 and albumin double-positive hepatocytes. Bar=25μm. Lower panel: Analysis of Ki67-positive hepatocytes. Data are the means±S.D. (cells/field; field=0.14mm2 [n=5 for each group]). **, P<0.01.
(C) Triple immunofluorescence staining for phosphorylated Histone H3 (pHistoneH3, in red), albumin (in green) and DAPI (in blue) at 36hrs post injury. Arrowheads indicate pHistoneH3 and albumin double-positive hepatocytes. Bar=25μm.
Activation and transdifferentiation of hepatic stellate cells (Ito cells or fat-storing cells) into myofibroblasts is the central event following liver injury (3). Using cytoglobin as a marker for hepatic stellate cells (28), and α-SMA as a marker for the activated form, immunofluorescence analyses revealed that the number of both un-activated and activated cells increased in fibronectin-null livers at 36 and 72hrs post injury (Fig. 5A (14)). Since cross-talk between hepatic stellate cells and hepatocytes drives hepatocyte proliferation and liver regeneration (29–30), an increased amount of diffusible factor secreted from activated hepatic stellate cells in fibronectin-null livers was proposed as a paracrine mediator for enhanced hepatocyte proliferation following injury. One of the key mediators in promoting hepatocyte proliferation is HGF and activated hepatic stellate cells are the major cellular source of HGF, including in CCl4-induced liver damage (31–32). HGF binds to cell surface receptor Met and such ligand-binding triggers trans-phosphorylation of Met tyrosine residues. Significantly elevated HGF levels in fibronectin-null livers at 36hrs post injury (~1.7-fold increase, P<0.01) were found (Fig. 5B). Indeed, double immunofluorescence staining revealed a specific colocalization of HGF with α-SMA in mutant livers at 36hrs post injury (Fig. 5C). Furthermore, a specific colocalization of phosphorylated-Met with the hepatocyte marker albumin was confirmed in mutant livers (Fig. 5D).
Fig. 5. Fibronectin deficiency significantly upregulates liver HGF levels post injury.

(A) Double immunofluorescence staining for cytoglobin (in red)/DAPI (in blue) and α-SMA (in red)/DAPI (in blue) at 36 and 72hrs post injury. CV, central vein. Bar=50μm.
(B) Hepatic HGF levels at 36hrs post injury. Data are means±S.D. (n=4 for each group). **, P<0.01.
(C) Triple immunofluorescence staining for α-SMA (in red), HGF (in green), and DAPI (in blue) in mutant livers at 36hrs post injury. Arrowheads indicate α-SMA and HGF double-positive activated hepatic stellate cells (orange to yellow color). Bar=25μm.
(D) Double immunofluorescence staining for phospho-Met (pMet, in red) and albumin (in green) in the mutant liver at 36hrs post injury. Arrowheads indicate double-positive hepatocytes (orange to yellow color). Bar=25μm.
In response to liver injury, HGF-Met binding leads to the activation of intracellular signaling pathways that involve Ras/extracellular signal-regulated kinase (Erk) and PI3K/Akt pathways, which thereby provide clues to the initiating signals for hepatocyte proliferation (29, 33–34). Considerable phosphorylation of Erk1/2 was observed in fibronectin-null livers (~9.3- and ~7.7-fold induction at 36 and 48hrs post injury, respectively, compared with 0hr [untreated]) with the induction peak around 36hrs. In contrast the pErk1/2 induction levels in control livers were remarkably lower (~3.5- and ~1.9-fold induction at 36 and 48hrs post injury, respectively, compared with 0hr [untreated]). The Erk1/2 phosphorylation levels between control and fibronectin-null livers at each time point did not show significant differences (Fig. 6A). However, importantly, immunohistochemical observation revealed that the number of pERK-positive hepatocytes in fibronectin-null livers was significantly higher compared with controls at both 36 and 48hrs post injury (Fig. 6B,C). PI3-kinase/Akt and focal adhesion kinase (FAK)/p53 pathways are involved in transduction of survival signals (22, 35). There were no obvious differences in the levels of phosphorylated Akt except that mutant livers showed decreased phosphorylation (~1.7-fold reduction) at 24hrs post injury (Fig. 6A). The expression levels of p53 post injury were similar between control and mutant livers (Fig. 6A). These findings indicate that fibronectin is not essential for hepatocyte proliferation and suggest that the PI3-kinase/Akt pathway might play a role in hepatocyte apoptosis following acute liver injury.
Fig. 6. Analysis of intracellular signalings following liver injury.

(A) Western blot analysis of phospho-ERK (pERK), total ERK, phospho-Akt (pAkt), total Akt, p53, and HSC70 (loading control) in control and mutant livers. Pooled samples from 3 mouse livers from each strain were used for the analysis. Band intensity was measured by densitometry, and each pERK and pAkt intensity was normalized to total ERK and total Akt, respectively; each p53 intensity was normalized to HSC70. Then the intensity at 0hr in the control liver was set to 1. Each intensity is shown relative to the control value.
(B) Immunostaining for pERK (in brown) at 36 and 48hrs post injury Sections were counterstained with hematoxylin. CV, central vein. Bar=50μm. Inset: Double immunofluorescence staining for pERK (in red) and albumin (in green) in control and fibronectin-null livers at 36hrs post injury. Arrowheads indicate double-positive hepatocytes (orange to yellow color). Bar=50μm.
(C) Analysis of pERK-positive hepatocytes. Data are means±S.D. (cells/field; field=0.14mm2 [n=5 for each group]). **, P<0.01.
Fibronectin-mediated matrix-survival signal is transduced through the PI3-kinase-Bcl-xL signaling axis in primary hepatocytes in vitro
There is evidence that reactive oxygen species (ROS) are produced in hepatocytes following CCl4-induced liver injury and such ROS have cytotoxic effects on hepatocytes (36). To further determine whether fibronectin is directly involved in the Bcl-xL-mediated anti-apoptotic effects on hepatocytes, in vitro studies were performed with the potent ROS inducer H2O2. Fibronectin-null primary hepatocytes were used to exclude an endogenous production of cellular fibronectin. When these cells were plated on type I collagen and on fibronectin-coated substrata, the number of Bcl-xL-positive hepatocytes and Bcl-xL expression levels were similar in both conditions (Fig. 7A,B). However, when plated following 1 mM H2O2 treatment, the number of Bcl-xL-positive hepatocytes and Bcl-xL expression levels were significantly higher on fibronectin (Fig. 7A,B). Since the decreased Akt phosphorylation was noted in mutant livers at 24hrs post injury (Fig. 6A), the transduction pathway for survival signals from fibronectin was investigated. Indeed, application of PI3-kinase inhibitor LY294002 (5μM; IC50=1.4μM) following H2O2 treatment significantly inhibited (~60.0%) Bcl-xL expression on fibronectin (Fig. 7C). MEK1/2 inhibitor PD98059 (up to 20μM; IC50=2μM), JNK inhibitor 420119 (up to 1μM; IC50=40nM), and p38 MAP kinase inhibitor SB203580 (up to 5μM; IC50=34nM) had no obvious effect on Bcl-xL expression on fibronectin (Fig. 7C and data not shown). Taken together, these results indicate that a deficiency in fibronectin leads to lack of fibronectin-mediated matrix-survival signals transduced through the PI3-kinase-Bcl-xL signaling axis and induces hepatocyte apoptosis in response to acute liver damage.
Fig. 7. Fibronectin-mediated matrix-survival signals are transduced through PI3-kinase/Bcl-xL in primary hepatocytes in vitro.

(A) Double immunofluorescence staining for Bcl-xL (in red) and DAPI (in blue) in fibronectin-null primary hepatocytes on collagen type I (Col1) and on plasma fibronectin (pFn) substrata with or without the treatment of H2O2. Bar=50μm.
(B) Analysis of the number of Bcl-xL-positive hepatocytes (/field: field=0.07mm2, left panels) and Bcl-xL intensity in hepatocytes (right panels) on Col1 and pFn substrata with or without the treatment of H2O2. Data are means±S.D. n.s., non-significant. **, P<0.01.
(C) The effect of MEK1/2 inhibitor PD98059, JNK inhibitor 420119, and PI3-kinase inhibitor LY294002 on Bcl-xL expression. Primary hepatocytes with or without pre-application of PD98059 (20μM), 420119 (1μM), and LY294002 (5μM) were treated with H2O2, subsequently plated on pFn, and incubated for 15hrs. Data are means±S.D. **, P<0.01.
Discussion
In the current study, evidence is provided that shows 1) a deficiency in fibronectin induces significantly increased hepatocyte apoptosis, which is accompanied by a significant downregulation of Bcl-xL protein expression; 2) a lack of fibronectin leads to an earlier onset of regeneration and an accelerating hepatocyte proliferation with increased levels of HGF; and 3) fibronectin-mediated matrix-survival signals are transduced through the PI3-kinase-Bcl-xL signaling axis in primary hepatocytes in vitro. We clearly demonstrate the functional links between fibronectin and hepatocyte survival both in vivo and in vitro.
Since skin wounds heal normally in mice lacking plasma-type fibronectin (10), an absolute requirement for fibronectin in response to adult tissue damage has been speculative. Although there is a study showing that exogenously administered plasma-type fibronectin can prevent D-galactosamine/lipopolysaccharide-induced lethal hepatic failure in mice (37), the high level of fibronectin used for the analysis (~4mg/ml) is far beyond physiological levels (~200μg/ml in normal plasma (38)). This present genetic study with a fibronectin-deficient model provides compelling evidence that fibronectin acts as an anti-apoptotic factor for hepatocytes in response to acute liver injury. In agreement, several in vitro studies have shown that fibronectin can be a survival factor inhibiting apoptosis in a variety of cell types (20–22). There is an established notion that cells sense their location through specific interactions with the ECM and that inappropriate cell-ECM interactions and environments induce apoptosis (23). A lack of plasma coagulation factor XIII (one of the transglutaminase family members (39–40)) causes a defect in plasma fibronectin and fibrin(ogen) covalent cross-linking complexes and results in significant hepatocyte apoptosis and a delay in hepatocyte proliferation following acute liver injury. Furthermore, those phenotypes are accompanied by a significantly high induction of p53-p21 expression, suggesting that FXIII-mediated covalently cross-linked matrix survival signals could be transduced through p53-p21 signaling axis (17). Although fibronectin-mediated survival signals are transduced through FAK-p53 signaling axis in vitro in specific conditions such as in serum free culture (22, 41), the present findings have demonstrated that hepatocyte survival signals are transduced from fibronectin via PI3-kinase/Akt to Bcl-xL in response to acute injury. This implies that fibronectin regulates Bcl-xL expression with PI3-kinase/Akt as one of the upstream mediators. Bcl-xL is expressed in hepatocytes and the disruption of the bcl-xL gene leads to continuous hepatocyte apoptosis even in intact (un-injured) livers (42). Furthermore, Bcl-xL is up-modulated during liver regeneration following partial hepatectomy (25), suggesting enhanced hepatocyte survival.
Accumulating evidence proposes that apoptosis and cell proliferation are closely linked. Indeed, Bcl-2 family members play dual roles, serving as mediators of both processes, with the expression of anti-apoptotic Bcl-xL retarding cell cycle progression (43–44). However, in contrast to the anti-apoptotic function mediated by the fibronectin/Bcl-xL signaling axis following liver injury, the lack of fibronectin resulted in earlier onset of regeneration and the acceleration of hepatocyte proliferation. This is contrary to in vitro evidence that fibronectin can promote EGF-initiated hepatocyte proliferation in culture (16). We have previously demonstrated that the lack of fibronectin in the liver shows elevated active TGF-β levels in response to CCl4-induced acute liver injury, which results in more activation and proliferation of hepatic stellate cells (14). In the present study, significantly elevated levels of HGF produced by hepatic stellate cells were found in fibronectin-null livers following injury, and this is a known key mediator for promoting hepatocyte proliferation (29, 34). Therefore, these findings support the following hypothesis: Although fibronectin plays a pivotal role in protecting against hepatocyte apoptosis following acute liver damage, a deficiency in fibronectin results in elevated activation and proliferation of hepatic stellate cells and thereby increased HGF production, which in turn surpasses the fibronectin-mediated hepatocyte proliferation activity. Ultimately, greater activity of the Erk pathway, as suggested by higher phosphorylation levels at 36 and 48hrs in fibronectin-null livers, enhances hepatocyte proliferation and results in significantly smaller damaged areas at 72hrs post injury. Thus, fibronectin is essential for survival but is dispensable for proliferation of hepatocytes in response to acute liver injury.
Acknowledgments
Grant Support: This work was supported by National Institutes of Health research grant DK074538 (to T.S.).
We thank Dr. Thorsten Burmester for cytoglobin antibody, and Dr. Emma Stephenson for editorial assistance.
Abbreviations
- ALT
alanine aminotransferase
- α-SMA
α-smooth muscle actin
- Bcl-2
B-cell lymphoma-2
- CCl4
carbon tetrachloride
- DAPI
4′6-diamidino-2-phenylindole
- ECM
extracellular matrix
- EGF
epidermal growth factor
- Erk
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- FITC
fluorescein isothiocyanate
- HGF
hepatocyte growth factor
- JNK
c-Jun N-terminal kinase
- mAb
monoclonal antibody
- MEK
mitogen-activated protein kinase kinase
- pAb
polyclonal antibody
- PI3-kinase
phosphatidylinositol 3-kinase
- pI-pC
polyinosinic-polycytidic acid
- PVDF
polyvinylidene fluoride
Footnotes
Conflict of interest: The authors disclose no conflict of interest.
Author contributions: Takao Sakai conceived ideas, designed experiments, and supervised the project. Kei Moriya, Keiko Sakai, and Michel Yan performed experiments. Kei Moriya, Keiko Sakai, and Takao Sakai analyzed the data. Takao Sakai wrote and edited the manuscript.
References
- 1.Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet. 2010;376:190–201. doi: 10.1016/S0140-6736(10)60274-7. [DOI] [PubMed] [Google Scholar]
- 2.Pathikonda M, Munoz SJ. Acute liver failure. Ann Hepatol. 2010;9:7–14. [PubMed] [Google Scholar]
- 3.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark RAF. The molecular and cellular biology of wound repair. 2. xxiii. New York: Plenum Press; 1996. p. 611. [611] leaf of plates. [Google Scholar]
- 5.Mosher DF. Fibronectin. xviii. San Diego: Academic Press; 1989. p. 474. [Google Scholar]
- 6.Hynes RO. Fibronectins. xv. New York: Springer-Verlag; 1990. p. 546. [Google Scholar]
- 7.Schwarzbauer JE. Fibronectin: from gene to protein. Curr Opin Cell Biol. 1991;3:786–791. doi: 10.1016/0955-0674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- 8.Gressner OA, Weiskirchen R, Gressner AM. Biomarkers of hepatic fibrosis, fibrogenesis and genetic pre-disposition pending between fiction and reality. J Cell Mol Med. 2007;11:1031–1051. doi: 10.1111/j.1582-4934.2007.00092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakai T, Johnson KJ, Murozono M, Sakai K, Magnuson MA, Wieloch T, Cronberg T, et al. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- 11.Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J Biol Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- 12.Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13:3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sottile J, Shi F, Rublyevska I, Chiang HY, Lust J, Chandler J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol Cell Physiol. 2007;293:C1934–1946. doi: 10.1152/ajpcell.00130.2007. [DOI] [PubMed] [Google Scholar]
- 14.Moriya K, Bae E, Honda K, Sakai K, Sakaguchi T, Tsujimoto I, Kamisoyama H, et al. A fibronectin-independent mechanism of collagen fibrillogenesis in adult liver remodeling. Gastroenterology. 2011;140:1653–1663. doi: 10.1053/j.gastro.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caputi M, Melo CA, Baralle FE. Regulation of fibronectin expression in rat regenerating liver. Nucleic Acids Res. 1995;23:238–243. doi: 10.1093/nar/23.2.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawada N, Tomomura A, Sattler CA, Sattler GL, Kleinman HK, Pitot HC. Extracellular matrix components influence DNA synthesis of rat hepatocytes in primary culture. Exp Cell Res. 1986;167:458–470. doi: 10.1016/0014-4827(86)90186-2. [DOI] [PubMed] [Google Scholar]
- 17.Tsujimoto I, Moriya K, Sakai K, Dickneite G, Sakai T. Critical role of factor XIII in the initial stages of carbon tetrachloride-induced adult liver remodeling. Am J Pathol. 2011;179:3011–3019. doi: 10.1016/j.ajpath.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashi H, Sakai K, Baba H, Sakai T. Thrombospondin-1 is a novel negative regulator of liver regeneration after partial hepatectomy via TGF-βετα1 activation in mice. Hepatology. 2011 doi: 10.1002/hep.24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda T, Sakabe T, Sunaga A, Sakai K, Rivera AL, Keene DR, Sasaki T, et al. Conversion of mechanical force into TGF-beta-mediated biochemical signals. Curr Biol. 2011;21:933–941. doi: 10.1016/j.cub.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruoslahti E. Fibronectin and its integrin receptors in cancer. Adv Cancer Res. 1999;76:1–20. doi: 10.1016/s0065-230x(08)60772-1. [DOI] [PubMed] [Google Scholar]
- 21.Globus RK, Doty SB, Lull JC, Holmuhamedov E, Humphries MJ, Damsky CH. Fibronectin is a survival factor for differentiated osteoblasts. J Cell Sci. 1998;111 ( Pt 10):1385–1393. doi: 10.1242/jcs.111.10.1385. [DOI] [PubMed] [Google Scholar]
- 22.Almeida EA, Ilic D, Han Q, Hauck CR, Jin F, Kawakatsu H, Schlaepfer DD, et al. Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH(2)-terminal kinase. J Cell Biol. 2000;149:741–754. doi: 10.1083/jcb.149.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilmore AP. Anoikis. Cell Death Differ. 2005;12 (Suppl 2):1473–1477. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- 24.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzung SP, Fausto N, Hockenbery DM. Expression of Bcl-2 family during liver regeneration and identification of Bcl-x as a delayed early response gene. Am J Pathol. 1997;150:1985–1995. [PMC free article] [PubMed] [Google Scholar]
- 26.Wu J, Norton PA. Animal models of liver fibrosis. Scand J Gastroenterol. 1996;31:1137–1143. doi: 10.3109/00365529609036901. [DOI] [PubMed] [Google Scholar]
- 27.Rojkind M, Greenwel P. Animal models of liver fibrosis. Adv Vet Sci Comp Med. 1993;37:333–355. [PubMed] [Google Scholar]
- 28.Schmidt M, Gerlach F, Avivi A, Laufs T, Wystub S, Simpson JC, Nevo E, et al. Cytoglobin is a respiratory protein in connective tissue and neurons, which is up-regulated by hypoxia. J Biol Chem. 2004;279:8063–8069. doi: 10.1074/jbc.M310540200. [DOI] [PubMed] [Google Scholar]
- 29.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 30.Balabaud C, Bioulac-Sage P, Desmouliere A. The role of hepatic stellate cells in liver regeneration. J Hepatol. 2004;40:1023–1026. doi: 10.1016/j.jhep.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Maher JJ. Cell-specific expression of hepatocyte growth factor in liver. Upregulation in sinusoidal endothelial cells after carbon tetrachloride. J Clin Invest. 1993;91:2244–2252. doi: 10.1172/JCI116451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Passino MA, Adams RA, Sikorski SL, Akassoglou K. Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science. 2007;315:1853–1856. doi: 10.1126/science.1137603. [DOI] [PubMed] [Google Scholar]
- 33.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura T, Mizuno S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:588–610. doi: 10.2183/pjab.86.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005;24:425–439. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- 36.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- 37.Qiu Z, Kwon AH, Tsuji K, Kamiyama Y, Okumura T, Hirao Y. Fibronectin prevents D-galactosamine/lipopolysaccharide-induced lethal hepatic failure in mice. Shock. 2006;25:80–87. doi: 10.1097/01.shk.0000185797.04589.5c. [DOI] [PubMed] [Google Scholar]
- 38.Tomasini-Johansson B, Mosher DF. Plasma fibronectin concentration in inbred mouse strains. Thromb Haemost. 2009;102:1278–1280. doi: 10.1160/TH09-03-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorand L. Factor XIII: structure, activation, and interactions with fibrinogen and fibrin. Ann N Y Acad Sci. 2001;936:291–311. doi: 10.1111/j.1749-6632.2001.tb03516.x. [DOI] [PubMed] [Google Scholar]
- 40.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 41.Ilic D, Almeida EA, Schlaepfer DD, Dazin P, Aizawa S, Damsky CH. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takehara T, Tatsumi T, Suzuki T, Rucker EB, 3rd, Hennighausen L, Jinushi M, Miyagi T, et al. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology. 2004;127:1189–1197. doi: 10.1053/j.gastro.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 43.Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006;13:1351–1359. doi: 10.1038/sj.cdd.4401987. [DOI] [PubMed] [Google Scholar]
- 44.Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat. 2007;10:13–29. doi: 10.1016/j.drup.2007.01.003. [DOI] [PubMed] [Google Scholar]