

Abstract
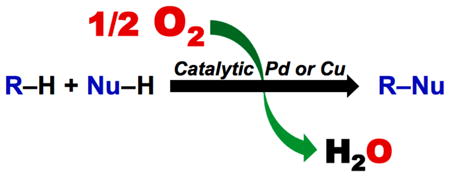
Oxidation reactions are key transformations in organic chemistry because they can increase chemical complexity and incorporate heteroatom substituents into carbon-based molecules. This principle is manifested in the conversion of petrochemical feedstocks into commodity chemicals and in the synthesis of fine chemicals, pharmaceuticals, and other complex organic molecules. The utility and function of such molecules correlate directly with the presence and specific placement of oxygen and nitrogen heteroatoms and other functional groups within the molecules.
Methods for selective oxidation of C–H bonds have expanded significantly over the past decade, and their role in the synthesis of organic chemicals will continue to increase. Our group’s contributions to this field are linked to our broader interest in the development and mechanistic understanding of aerobic oxidation reactions. Molecular oxygen (O2) is the ideal oxidant. Its low cost and lack of toxic by-products make it a highly appealing reagent that can address key ‘green chemistry’ priorities in industry. Economic and environmental incentives provide strong motivation to use O2, and the commodity chemicals industry often uses aerobic oxidation reactions. In contrast, O2 is seldom used to prepare smaller-volume, more-complex chemicals, a limitation that reflects, in part, the limited synthetic scope and utility of existing aerobic reactions.
Pd-catalyzed reactions represent some of the most versatile methods for selective C–H oxidation, but they often require stoichiometric transition-metal or organic oxidants, such as CuII, AgI or benzoquinone. This Account describes recent strategies that we have identified to use O2 in these reactions. In Pd-catalyzed C–H oxidation reactions that form carbon-heteroatom bonds, the stoichiometric oxidant is often needed to promote difficult reductive elimination steps in the catalytic mechanism. To address this issue, we have identified new ancillary ligands for Pd that promote reductive elimination, or replaced Pd with a Cu catalyst that undergoes facile reductive elimination from a CuIII intermediate. Both strategies have enabled O2 to be used as the sole stoichiometric oxidant in the catalytic reactions. C–H oxidation reactions that form the product via β-hydride or C–C reductive elimination steps tend to be more amenable to the use of O2. The use of new ancillary ligands has also overcome some of the limitations in these methods. Mechanistic studies are providing insights into some (but not yet all) of these advances in catalytic reactivity.
1. Introduction
Over the past decade, catalytic C–H functionalization reactions have begun to emerge as viable alternatives to traditional synthetic methods in organic chemistry. These methods exhibit substantial diversity, with mechanisms ranging from hydrogen-atom abstraction and C–H insertion (e.g., by carbenes, carbenoids and related species) to organometallic C–H activation.1 The mechanistic variations lead to unique selectivity patterns, with important implications for organic synthesis.2
Organometallic C–H oxidation and oxidative-coupling reactions offer important advantages over widely used catalytic cross-coupling methods and traditional C–C and C–X (X = heteroatom) bond-forming reactions. By eliminating the requirement for prefunctionalized substrate(s), such as alkyl/aryl halides or organometallic reagents, these methods offer the prospect of broader substrate scope and improved atom-economy. In order for these benefits to be fully realized, however, at least two conditions must be met: (1) the reaction must achieve site-selective functionalization of a C–H bond in an organic substrate and (2) the cost and/or waste associated with the stoichiometric oxidant should not offset the benefits of using a less-functionalized substrate. Aerobic oxidative-coupling reactions that proceed with high chemo-, regio- and stereoselectivity represent near-ideal embodiments of these criteria.
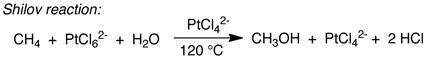
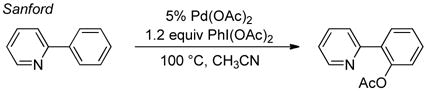
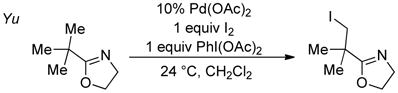
Platinum-catalyzed oxidation of methane, reported by Shilov nearly four decades ago, provides historical context for the field of organometallic C–H oxidation.3 This reaction achieves selective oxidation of methane to methanol (and methyl chloride) in aqueous solution, with PtIICl42− as the catalyst and PtIVCl62− as the stoichiometric oxidant (eq 1), and the mechanism involves three key steps (Scheme 1):4 (1) electrophilic C–H activation of methane by PtII, (2) two-electron oxidation of a PtII–CH3 intermediate by PtIV, and (3) nucleophilic attack of water on the resulting PtIV–CH3 species, formally an SN2-type reductive elimination, to afford CH3OH. This so-called “Shilov reaction” is one of the earliest catalytic demonstrations of organometallic C–H oxidation, and it continues to serve as a paradigm for new developments in the field. For example, PdII/PdIV-catalyzed C–H oxidation reactions (e.g., eqs 2–3)5 proceed by a mechanism that closely resembles Scheme 1, involving sequential C–H activation, oxidation, and C–X reductive elimination steps.
Scheme 1.

Proposed Catalytic Mechanism of the Shilov Reaction.
 |
(1) |
 |
(2) |
 |
(3) |
The use of PtIV as a stoichiometric oxidant in the Shilov reaction represents a significant drawback that has been difficult to address.6 Oxidants that are commonly used in PdII/PdIV-catalyzed reactions, such as PhI(OAc)2, diaryliodonium salts, N-bromosuccinimide, and electrophilic fluorine reagents,7 are more user friendly (e.g., soluble in organic solvents) and less expensive than PtIV; however, their use still contributes to lower atom economy, thereby reducing their appeal vis-à-vis traditional cross-coupling methods and hindering large-scale applications. Separately, many recently reported PdII-catalyzed C–H oxidation reactions employ stoichiometric benzoquinone, CuII or AgI as stoichiometric oxidants.7 As this field expands and the opportunities afforded by these reactions impact mainstream organic synthesis, the importance of addressing this “oxidant problem” will become increasingly apparent.
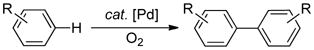
Our research group has had a long-standing interest in Pd-catalyzed aerobic oxidation reactions.8 These reactions typically proceed via a PdII/Pd0 pathway and differ from Shilov and PdII/PdIV-catalyzed C–H oxidations in that the stoichiometric oxidant (O2) does not participate directly in the substrate oxidation step(s) (Scheme 2). Important precedents exist for C–H oxidation reactions of this type. For example, PdII-catalyzed homocoupling of arenes was first reported in the mid-1960s (eq 4),9 but these reactions received little attention relative to the more-selective, non-oxidative cross-coupling reactions of aryl halides. The growing importance of C–H functionalization reactions and environmentally benign methods for chemical synthesis provides strong motivation to reinvestigate reactions of this type. Such efforts will benefit from four decades of advances in mechanistic characterization of organometallic reactions (including C–H activation), insights into ancillary ligand effects in catalysis, and principles of O2 activation and reduction by transition metals.
Scheme 2.
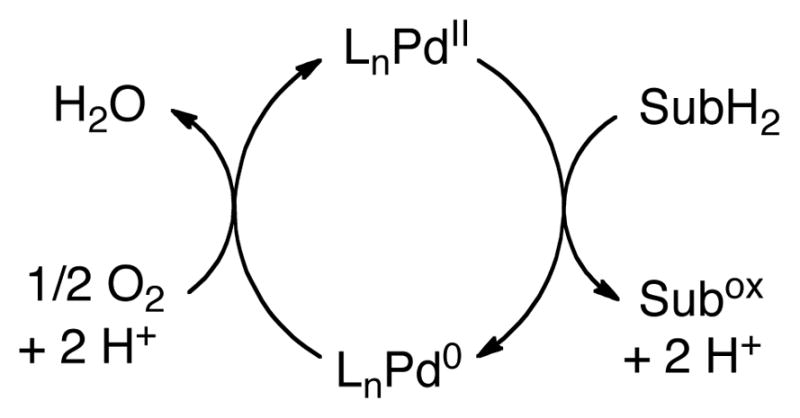
Simplified Catalytic Cycle for PdII/Pd0-catalyzed Aerobic Oxidation Reactions.
 |
(4) |
2. Pd-Catalyzed Aerobic Oxidation Reactions: β-Hydride vs. Reductive Elimination Reactions
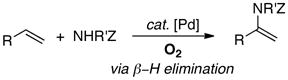
Pd-catalyzed oxidation reactions that proceed via a PdII/Pd0 cycle can be divided into two general classes, distiguished by the product-forming step in the catalytic mechanism: β-hydride elimination versus reductive elimination. Examples of the former class include alcohol oxidation and oxidative-coupling reactions of alkenes (Table 1, Reactions A–D). The latter class includes oxidative-coupling reactions of arenes and allylic oxidation reactions (Table 1, Reactions E–G). A survey of the literature7,8,10 reveals that PdII-catalyzed oxidation reactions that proceed via β-hydride elimination are often compatible with O2 as the oxidant, whereas reactions that proceed via reductive elimination typically employ alternative oxidants, such as benzoquinone, CuII and AgI, among others. Such trends are evident in our studies of Pd-catalyzed oxidative amination of alkenes:8c aza-Wacker reactions that form enamide products via β-hydride elimination from an alkyl-PdII intermediate are compatible with O2 (eq 5), but the aminoacetoxylation of alkenes requires PhI(OAc)2 (eq 6), reflecting the need to oxidize a PdII(alkyl)(OAc) intermediate to a higher oxidation state to promote C–O reductive elimination.
Table 1.
Representative Pd-Catalyzed Oxidation Reaction and the Identity of the Product-Forming Step in the Catalytic Mechanism.
| Oxidative Transformation | Product-Forming Step |
|---|---|
| A. Alcohol Oxidation |
|
|
| |
| B. Wacker-Type Oxidative Cyclization | |
|
| |
| C. Aza-Wacker | |
|
| |
| D. Oxidative Heck Coupling | |
|
| |
|
| |
| E. Oxidative Biaryl Coupling |
|
|
| |
| F. Allylic C–H Oxidation | |
|
| |
| G. Oxidative Heterofunctionalization of Arenes | |
|
| |
Nu = heteroatom nucleophile; Z = electron-withdrawing group
 |
(5) |
 |
(6) |
β-Hydride and reductive elimination steps are quite different with respect to the redox changes that take place at Pd. β-Hydride eliminations from PdII–alkoxide or –alkyl intermediates generate PdII–hydrides, which involves no formal change in the PdII oxidation state. In contrast, reductive eliminations from PdII(Ar)(Ar′) or PdII(Ar)(Nu) intermediates afford Pd0, reflecting two-electron reduction of the Pd center. If the PdII center in these catalytic intermediates is not sufficiently oxidizing, the reductive elimination step may be slow or even thermodynamically unfavorable. In many cases, oxidation of the organopalladium(II) intermediate (e.g., to a PdIII or PdIV species) is needed to promote reductive elimination.11 Our studies of aerobic C–H oxidation reactions presented below address both classes of reactions (β-hydride and reductive elimination).
3. Allylic C–H Acetoxylation: A Ligand-Based Strategy to Replace Benzoquinone with O2
Pd-catalyzed oxidative heterofunctionalization of allylic and aromatic C–H bonds (cf. Table 1, F and G) are rarely compatible with O2.12 Allylic C–H oxidation reactions commonly employ benzoquinone (BQ) as the stoichiometric oxidant.13 BQ has been shown to promote the product-forming reductive elimination step,14 which involves inner-sphere or outer-sphere nucleophilic attack on an allyl-PdII intermediate, and it also serves as the stoichiometric oxidant in the conversion of Pd0 to PdII (Scheme 3).15 We speculated that an ancillary ligand could be used to destabilize the allyl-PdII intermediate and enable reductive elimination to occur in the absence of BQ. If the same ligand could promote oxidation of Pd0 to PdII with O2,16 it should be possible to achieve aerobic allylic C–H oxidation reactions.17
Scheme 3.

Proposed Mechanism for Pd-Catalyzed Allylic Acetoxylation Using BQ as the Oxidant
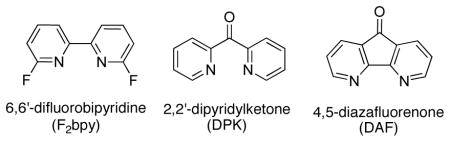
Nitrogen-based ligands are common in Pd-catalyzed aerobic oxidation reactions, and we reasoned that electron-deficient ligands would increase the PdII/Pd0 reduction potential and thereby facilitate reductive elimination. Three of our initial ideas included 6,6′-difluorobipyridine (6,6′-F2bpy), 2,2′-dipyridylketone (DPK) and 4,5-diazafluorenone (DAF). The fluorine substituents of 6,6′-F2-bpy significantly reduce the basicity of the nitrogen lone pair via their inductive effect, while the carbonyl groups in DPK and DAF could remove electron density on PdII center via π-backbonding, formally leading to a PdIV resonance contribution (eq 7).
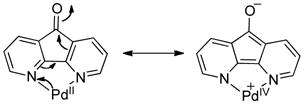 |
(7) |
These ligands were screened together with other nitrogen-based ligands in an effort to achieve allylic C–H actetoxylation of allyl benzene under 1 atm O2 (Chart 1). Traditional ligands, including pyridine, bipyridine, and phenanthroline, were almost completely ineffective. Bipyrimidine, which is effective in BQ-promoted allylic acetoxylation,13b 6,6′-F2-bpy and DPK were also unsuccessful. Diazafluorenone (DAF) proved to be uniquely successful, enabling an 81% yield of cinnamyl acetate. We also tested 9,9-dimethyl-4,5-diazafluorene (Me2DAF), and obtained a 50% yield. The latter result is not better than that observed with DAF, but the improved results relative to all other ligands suggests that the beneficial effect of DAF reflects the unique geometry of the diazafluorene structure. The DAF-Pd(OAc)2 catalyst system was successfully applied to aerobic acetoxylation of a number of terminal alkenes, including those bearing synthetically useful functional groups (Chart 2). Linear allylic acetates were obtained exclusively, with a strong preference for the E stereoisomer. This reactivity was implemented to achieve net anti-Markovnikov hydration of terminal alkenes via sequential aerobic acetoxylation, alcoholysis of the acetate and hydrogenation of the allylic alcohol (Scheme 4). In this one-pot sequence, the heterogeneous Pd catalyst for the hydrogenation step was formed in situ by adding activated carbon to the crude reaction mixture and placing the vessel under an atmosphere of hydrogen gas.
Chart 1.

Ligand Effects in Pd-Catalyzed Aerobic Allylic Acetoxylation.
Chart 2.

Aerobic Allylic Acetoxylation of Terminal Alkenes
Scheme 4.

Net Anti-Markovnikov Hydration of Terminal Alkenes Involving Aerobic Allylic Acetoxylation, Acetate Cleavage, Alkene Hydrogenation Sequence.
Mechanistic studies have begun to provide insights into the ability of the diazafluorenone ligand to support aerobic, BQ-free catalytic turnover. Initial studies confirmed that DAF promotes reductive elimination from PdII. The reactivity of two [(L)PdII(η3–allyl)]OAc complexes, 1 (L = DAF) and 2 (L = tBu2bpy), toward reductive elimination was investigated under three different conditions: in the absence of oxidant and in the presence of O2 or BQ (Figure 1). The DAF-ligated complex 1 formed allyl acetate under all three conditions, and the rate was faster in the presence of an oxidant. The tBu2bpy-ligated complex 2 was completely unreactive, except in the presence of BQ, demonstrating that BQ can induce acetoxylation of an otherwise unreactive π–allyl–PdII complex and that O2 cannot serve as a direct surrogate for BQ. The C–O reductive elimination step was shown to be reversible with 1, as revealed by the incorporation of deuterium-labeled acetate into cinammyl acetate in the presence of 25 mol % [(DAF)PdII(allyl)]OAc (Figure 2A). Analogous exchange was not observed in the presence of the tBu2bpy complex 2. A mechanism that accounts for these observations is shown in Figure 2B. Reductive elimination of allyl acetate initially affords an Pd0–alkene adduct, and this species can revert to the allyl-PdII species (krev), undergo alkene exchange (kexch), or react with an oxidant (kox). Evidence for competition between the kox and kexch steps was evident from reduced isotopic exchange at elevated O2 pressures. The ability of an oxidant to compete with the krev step explains the faster rate of allyl acetate reductive elimination from 1 in the presence of an oxidant.
Figure 1.

Ligand effects on stoichiometric acetoxylation of π–allyl–Pd complexes.
Figure 2.


Evidence for reversible C–O reductive elimination from (DAF)Pd(η3–allyl) complexes (A), and mechanistic rationale for (L)Pd(η3–allyl)-catalyzed acetate exchange in cinnamyl acetate and the influence of an oxidant on the extent of exchange (B).
Additional studies reveal that ligand-promoted reductive elimination is not sufficient to achieve aerobic catalytic acetoxylation. [(6,6′-F2bpy)PdII(allyl)]OAc undergoes stoichiometric reductive elimination of allyl acetate under aerobic conditions (eq 8), but the 6,6′-F2bpy ligand does not support aerobic catalytic turnover (Chart 1). These observations suggest the ancillary ligand influences other steps in the catalytic mechanism (e.g., C–H activation). Additional mechanistic studies will be needed to fully elucidate the unique influence of DAF on catalytic reactivity.
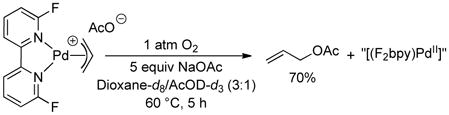 |
(8) |
4. Regioselective Aerobic Oxidative Coupling of Arenes without Directing Groups
Pd-catalyzed methods for oxidative homocoupling of arenes were among the first examples of aerobic organometallic C–H oxidation (eq 4).9 The synthetic utility of these methods were limited by low regioselectivity, however. Here, we summarize two recent studies that address issues of regioselectivity while incorporating the use O2 as the oxidant.
4.1. Selective Homocoupling of o-Xylene
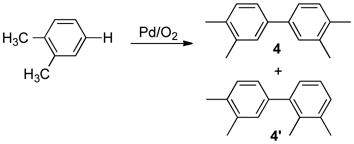
A rare, commercially important application of Pd-catalyzed homocoupling of arenes is the reaction of dimethyl-ortho-phthalate (eq 9). This reaction is employed in the commercial synthesis of 4,4′-biphthalic anhydride (3),18 a monomer for the high-performance polyimide resin, Upilex® (eq 10).19 The production of 3 originates with o-xylene and requires five overall steps including esterification and alcoholysis/hydrolysis reactions. This route could be streamlined by the development of a selective method for direct oxidative coupling of o-xylene (eq 11).20 The best reported conditions for this reaction employ a Pd(OAc)2/1,3-cyclohexanedione catalyst system at 140 °C.20b The reaction exhibits modest regioselectivity (4/4′ = 3.3:1), but low overall chemoselectivity (40%), with significant formation of higher-molecular-weight oligomers. Prompted by recent industrial interest in improved arene homocoupling methods,21 we decided to explore this reaction further, with the hypothesis that higher regio- and chemoselectivity could be achieved by developing a more active catalyst that operates at lower temperatures.22
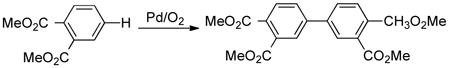 |
(9) |
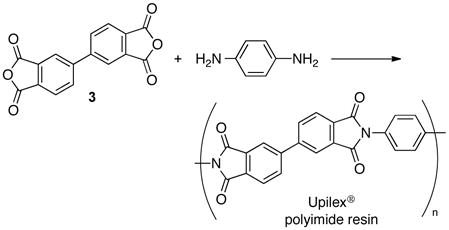 |
(10) |
 |
(11) |
Our initial screening studies focused on the oxidative coupling of o-xylene (eq 10) at 80 °C, 60 degrees lower than the previously optimized conditions, and a variety of oxidatively stable nitrogen ligands were tested in combination with Pd(OAc)2 or Pd(TFA)2 (TFA = trifluoroacetate). Chelating ligands such as bipyridine and phenanthroline, which is used in the oxidative coupling of dimethyl o-phthalate, inhibited the oxidative coupling of o-xylene. Monodentate pyridine derivatives, especially electron-deficient derivatives were promising, and the best results were obtained with 2-fluoropyridine (2Fpy). The identity of the anionic ligand also influenced the reaction, with a 1:1 ratio of acetate and trifluoroacetate (TFA) anions being optimal, and cocatalytic Cu(OTf)2 (Pd/Cu = 1:1) improved the results at low catalyst loading. Unpublished mechanistic data suggest CuII influences multiple steps in the catalytic mechanism, including C–H activation, transmetalation of aryl ligands between PdII centers, and, potentially, biaryl reductive elimination and reoxidation of Pd0. Further investigation of these fundamental steps is ongoing. With the optimized reaction conditions [4.4 M o-xylene in AcOH, 0.1% Pd(OAc)(TFA)/0.2% 2Fpy, 0.1% Cu(OTf)2, 1 atm O2, 80 °C], the oxidative coupling of o-xylene proceeded with improved regioselectivity (7.3:1) and much higher chemoselectivity (94%) relative to the previous results.
Systematic analysis of ancillary ligand effects on this reaction provided valuable insights. Initial rates of the oxidative coupling reactions were obtained with a wide range of substituted-pyridine ligands, and the log of the rate constant was plotted vs. the pyridinium pKa. This analysis revealed a “volcano plot” in which 2Fpy is at the peak (Figure 3). Pyridines more basic than 2Fpy are expected attenuate the electrophilicity of the Pd catalyst and reduce catalyst activity. In contrast, 1H NMR spectroscopic studies showed that pyridines less basic than 2Fpy are poor ligands and appear to be unable to activate the catalyst, for example, by breaking up binuclear and trinuclear aggregates of Pd-carboxylates.
Figure 3.

Dependence of the rate of o-xylene homocoupling on the identity of the ancillary pyridine ligands. The pKa values correspond to those for the pyridinium ions in DMSO.
A large deuterium kinetic isotope effect, kH/kD = 10.7(2.0)], observed in a competition experiment with o-xylene/o-xylene-d10 (1:1), is consistent with C–H activation of o-xylene as the turnover-limiting step of the catalytic mechanism. Electron-deficient ligands, such as 2Fpy and TFA, should enhance the electrophilicity of the PdII catalyst and facilitate C–H activation. The beneficial effect of acetate (1 equiv relative to Pd) on the reaction can be rationalized within the framework of a “concerted metalation-deprotonation” (CMD) pathway for PdII-mediated C–H activation,23 wherein a basic ligand, such as acetate, participates in deprotonation of a C–H bond as the Pd–Caryl bond is forming.
Overall, this work led to a significantly improved method for aerobic oxidative coupling of o-xylene, and it highlights the influence of electron-deficient ligands in promoting Pd-catalyzed oxidation reactions. The ability of N-donor ligands to stabilize Pd catalysts in aerobic oxidation reactions is widely recognized, but they also typically diminish catalytic activity.24 In the present case, however, the electron-deficient ancillary donor ligand enhances the catalyst activity.25 The identification of oxidatively stable ligands that support aerobic catalytic turnover and also enhance Pd-catalyst activity has important implications for the development of improved C–H oxidation reactions.
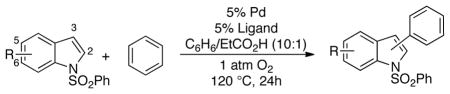
4.2. Regioselective Oxidative Cross-Coupling of Indoles and Benzene
The synthesis of biaryls via direct oxidative cross-coupling of two arenes represents a near-ideal transformation; however, such reactions face major challenges associated with regioselectivity, homo- vs. cross-coupling and the identity of the oxidant. Fagnou and coworkers reported an important breakthrough in this area in the Pd-catalyzed coupling of indoles and arenes (Scheme 5).26 Good control over regioselectivity was achieved by the choice of oxidant: Cu(OAc)2 favored C3 arylation and AgOAc favored C2 arylation of the indole. One-electron oxidants such as CuII and AgI can promote C–C reductive elimination from PdII,27 but the oxidative cross-coupling of indole and benzene was also possible with stoichiometric PdII (Scheme 5A). These observations prompted us to consider whether similar oxidative coupling reactions could be achieved with O2 as the oxidant, ideally while retaining control over the reaction regioselectivity.28
Scheme 5.

Oxidative Cross-Coupling of Indoles and Benzene with Stoichiometric Transition-Metal Oxidants.26
A number of ligands were evaluated in the Pd-catalyzed oxidative cross-coupling of N-pivaloyl indole with benzene under 1 atm O2 (Chart 3). Most ligands performed poorly; however, respectable product yields were obtained with DAF and Me2DAF (45% and 66%, respectively). Further optimization studies revealed that the identities of the ancillary ligand (DAF vs. Me2DAF) and anionic ligands affected the product regioselectivity. Selectivities were observed as high as 5:1 favoring the C2 isomer with a Pd(TFA)2/Me2DAF catalyst system, and 5.8:1 favoring the C3 isomer with a Pd(OPiv)2/DAF catalyst system (Table 2). In all cases, O2 served as the sole stoichiometric oxidant with no additional transition metal addtive or cocatalyst (Table 2).28
Chart 3.

Identification of a Ligand for Aerobic Pd-Catalyzed Indole-Arene Cross-Coupling
Table 2.
Regioselective Oxidative Cross-Coupling of Indole with Simple Arenes
| |||
|---|---|---|---|
| R | Catalyst | Yield | C2:C3 Selectivity |
| H | Pd(TFA)2/Me2DAF | 66% | 1:5.8 |
| Pd(OPiv)2/DAF | 76% | 2:1 | |
| 5-Cl | Pd(TFA)2/Me2DAF | 71% | 1:1.3 |
| Pd(OPiv)2/DAF | 68% | 5:1 | |
| 6-Cl | Pd(TFA)2/Me2DAF | 71% | 1.4:1 |
| Pd(OPiv)2/DAF | 66% | 3.7:1 | |
| 5-OMe | Pd(TFA)2/Me2DAF | 54% | 1:3.9 |
| Pd(OPiv)2/DAF | 71% | 2.3:1 | |
| 6-OMe | Pd(TFA)2/Me2DAF | 70% | 1:1.3 |
| Pd(OPiv)2/DAF | 65% | 2.6:1 | |
Modest primary deuterium kinetic isotope effects (kH/kD = 2.8–3.8) were observed in competition experiments with 1:1 C6H6/C6D6. These values are smaller than observed in the homocoupling of o-xylene, but they are consistent with rate-limiting C–H activation. Additional studies will be needed to gain further insights into the catalytic mechanism. For example, these reactions could involve C–H activation of the two substrates at different PdII centers, followed by transmetalation, to afford a PdII(Ph)(indolyl) intermediate. Or, the same intermediate could form via sequential C–H activation at a single PdII center. The biaryl product can be generated via C–C reductive elimination from this intermediate. More work is also required to understand the origin of the reaction regioselectivity. Experimental and computational studies to probe these issues are ongoing. Despite such uncertainties, the catalytic results highlight the potential ability of ancillary ligands to support regioselective aerobic oxidative cross-coupling of arenes.
5. Aerobic Dehydrogenation of Cyclohexanones to Phenols
Conversion of saturated carbon-carbon bonds to alkenes is an important class of C–H functionalization.29 The recognition that Pd-catalyzed reactions involving β-hydride elimination are often compatible with O2 (see Section 2) prompted us to begin exploring this class of reaction. Such reactions could proceed via PdII-mediated C–H activation and β-hydride elimination (Scheme 6, steps A and B), followed by aerobic oxidation of the resulting PdII–H (Scheme 6, steps C–E).30 If successful, such methods could provide environmentally benign alternatives to commonly used stoichiometric dehydrogenation reagents, such as DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) and IBX (2-iodoxybenzoic acid) Our initial studies focused on aerobic dehydrogenation of cyclohexanones to phenols (Scheme 7).31
Scheme 6.

Proposed Mechanism for PdII-Catalyzed Dehydrogenation of Saturated C–C Bonds.
Scheme 7.

Stepwise Sequence of PdII-Mediated Dehydrogenation of Cyclohexanones.
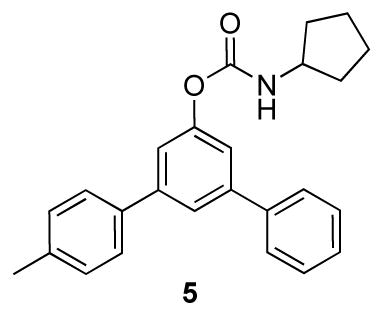
3-Substituted cyclohexenones and cyclohexanones are readily accessible, and their dehydrogenation leads to meta-substituted phenols that can be difficult to access by traditional aromatic substitution reactions. 3-Methylcyclohexanone was one of the substrates used in our catalyst screening efforts, which evaluated a number of PdII sources and ancillary ligands under 1 atm O2 (Chart 4). We examined a number of pyridine-derived ligands, including electron-deficient derivatives similar to those described above. Moderate success was observed with 2-fluoropyridine (44%), but even better results were obtained when 2-dimethylaminopyridine (2NMe2py) was used in combination with p-toluenesulfonic acid (TsOH). The mechanistic basis for this success is still under investigation, but our working hypothesis is that the pyridine nitrogen coordinates to Pd and the tertiary amine is protonated to afford a more electron-deficient ligand. A ligand with a quaternary ammonium substituent, [2-Me3Npy]+, is also effective (63%, unpublished), but not as good as the 2NMe2py/TsOH combination. The optimized catalyst system was effective in the dehydrogenation of a number of substituted cyclohexanone and cyclohexenone substrates (Chart 5). Dehydrogenation reactions of this type represent efficient alternatives to traditional cross-coupling reactions in the preparation of substituted aromatic molecules. This prospect was illustrated in an efficient route to the phenolic fragment of an allosteric inhibitor of the luteinizing hormone receptor (5), which had been prepared previously by a sequence of low-yielding cross-coupling reactions.31
Chart 4.

Ligand Effects for Aerobic Dehydrogenation of 3-Methyl Cyclohexanone.
Chart 5.

Phenols Prepared via Dehydrogenation of Cyclohexanone or Cyclohexenone Derivatives.
a3% Pd(TFA)2/6% NMe2py/12% TsOH
b 5% Pd(TFA)2/10% NMe2py/20% TsOH
The time course of cyclohexanone dehydrogenation reveals the intermediate formation of cyclohexenone, which rapidly converts to phenol under the reaction conditions. Kinetic analysis reveals the second dehydrogenation step is approximately three-fold faster than the first (k2 ≈ 3·k1, Scheme 8). Recent studies have led to a new catalyst system [Pd(DMSO)2(TFA)2/AcOH] that achieves “interrupted” dehydrogenation of cyclohexanone, in which the first dehydrogenation step proceeds much faster than the second on the basis of kinetic simulation of the reaction time course (k1:k2 > 30:1; Scheme 8). This method has been applied to a number of synthetically useful targets (Chart 6).32 Further development of these and other aerobic dehydrogenation reactions, together with mechanistic characterization of these new reactions, represent important goals of future studies. Overall, these results support the analysis in Section 2 that C–H functionalization reactions that proceed via β-hydride elimination are particularly amenable to the use of O2 as the terminal oxidant.
Scheme 8.

Aerobic Dehydrogenation Provides access to Phenols or Cyclohexenones.
Chart 6.

Enones Prepared via Dehydrogenation of Cycloketone Derivatives.
6. Copper(II)-Catalyzed Aerobic C–H Oxidation
Recent advances in copper catalysis have introduced new opportunities to achieve aerobic C–H oxidation.8d,33 Our work in this area originated with a mechanistic study of Cu-catalyzed methods for aerobic oxidative coupling of arylboronic acids and heteroatom nucleophiles (“Chan-Lam reactions”; eq 11), in which aryl-copper(III) species have been proposed as intermediates.34,35 We also investigated C–N reductive elimination reactions involving a well-defined macrocyclic aryl-copper(III) species 6 (eq 12).36,37 Reductive elimination from CuIII is extremely facile, rivaling (or surpassing) the ease of reductive elimination from PdIV, and our recent studies suggest that such reactions may play a key role in aerobic C–H oxidations.38
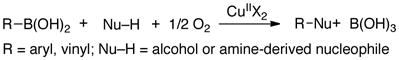 |
(11) |
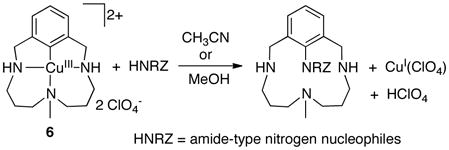 |
(12) |
The mechanism by which organocopper(III) intermediates could form in CuII-catalyzed C–H oxidation reactions is not necessarily obvious, but recent studies reveal that CuII-mediated C–H activation can proceed via disproportionation of CuII into CuI and an organocopper(III) species,39 illustrated in the synthesis of the aryl-CuIII complex 6, reported by Ribas et al. (eq 13).40 Reactions of this type have been shown to proceed via one of two possible pathways: (1) stepwise C–H activation by CuII followed by oxidation to the aryl-CuIII species, or (2) concerted, proton-coupled electron transfer (Scheme 8).39,41
Scheme 8.

Possible Mechanisms for CuII-Mediated C–H Activation and Formation of Aryl-CuIII Species.
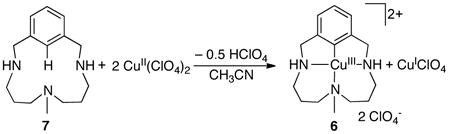 |
(13) |
In the course of our work, we recognized that Cu-mediated C–H activation and reductive elimination steps (e.g., eqs 12 and 13) could be combined with aerobic oxidation of CuI to provide a catalytic pathway for aerobic C–H oxidation (Scheme 9). This concept was demonstrated in the Cu-catalyzed oxidative functionalization of arene 7 under 1 atm O2, using methanol and pyridone as the heteroatom nucleophiles (Scheme 10).38 Kinetic and spectroscopic studies of the catalytic reaction provided direct evidence for the intermediacy of the aryl-CuIII complex 6, supporting the mechanism in Scheme 9.
Scheme 9.

Stepwise Mechanism for Cu-Catalyzed Aerobic C–H Oxidation.
Scheme 10.

Oxidative Functionalization of 6 Catalyzed by CuII
In parallel with these fundamental studies, we developed a CuII-catalyzed method for aerobic oxidative amidation of terminal alkynes. The combination of 20 mol % CuCl2 with 2 equiv pyridine and 2 equiv Na2CO3 in toluene under 1 atm O2 enables oxidative coupling of terminal alkynes with a variety of nitrogen nucleophiles (Scheme 11).42 Numerous related Cu-catalyzed aerobic C–H oxidation methods of this type have been published recently by others.8d The mechanisms of these reactions, including those in Scheme 11, are poorly understood and generally remain unexplored. The results described above, however, suggest that a catalytic mechanism involving an organocopper(III) intermediate may be a plausible pathway.
Scheme 11.

Aerobic Copper-Catalyzed Synthesis of Ynamides from Terminal Alkynes
The mechanism in Scheme 9 exhibits an intriguing relationship to the Shilov mechanism presented in the Introduction (Scheme 1). Both reactions feature C–H activation steps intiated by a divalent metal ion (CuII or PtII) and the resulting organometallic intermediate must be oxidized to a higher-valent species (CuIII–Ar and PtIV–CH3) before C–X reductive elimination takes place. Both reactions utilize a metal-based oxidant, CuII or PtIV, to oxidize the organometallic intermediate. A key difference between the two reactions, however, is the nature of the reduced metal byproduct. CuI, which forms in the C–H activation process and upon reductive elimination from CuIII, can react readily with O2 to regenerate active CuII catalyst. In contrast, PtII is kinetically inert toward reaction with O2. This difference enables copper catalysts to overcome the “oxidant problem” experienced by many Pt and Pd catalysts. Future work will undoubtedly clarify the scope and limitations of this reactivity.
6. Conclusion
The results presented here highlight a number of different strategies to address the “oxidant problem” in Pd-catalyzed C–H oxidation reactions. A common theme throughout these studies is the important role of the ancillary ligands in promoting aerobic catalytic turnover. Such ligands can promote reductive elimination, thereby avoiding the need for BQ or other undesirable stoichiometric oxidants to promote the reductive elimination step. In other cases, ligands facilitate key C–H activation and/or β-hydride elimination steps and/or contribute to regioselective C–H functionalization. The mechanistic basis for these ligand effects is not yet fully understood, but the empirical results described here point to the abundant opportunities to develop new aerobic C–H oxidation reactions. Finally, Cu-catalyzed aerobic oxidation reactions appear to provide a mechanistically novel strategy to address some of the most challenging C–H oxidation reactions that proceed via C–X reductive elimination.
Acknowledgments
We are grateful for generous support from the NIH (R01-GM67163/SSS and F32-GM087890/ANC) and the DOE (DE-FG02-05ER15690) throughout the course of this work. Additional valuable support was provided by the Dreyfus Foundation and Bristol-Myers Squibb, and we thank Mitsui Chemicals, Inc. and Mitsubishi Chemical Corporation for supporting visiting scientists (T. Hamada and Y. Izawa), who played a key role in the work described here.
Biographies
Alison Campbell received a B.S. in biochemistry from Lafayette College in 2004 and a Ph.D. in 2009 from the University of North Carolina under the supervision of Professor Michel R. Gagné. She was then an NIH Postdoctoral Fellow at the University of Wisconsin with Professor Shannon S. Stahl studying new methods for palladium-catalyzed aerobic C–H oxidations, and she recently joined the process research group at Eli Lilly (Indianapolis, IN).
Shannon Stahl is a Professor of Chemistry at the University of Wisconsin-Madison. He was an undergraduate at the University of Illinois at Urbana-Champaign, and subsequently attended Caltech (Ph.D., 1997), where he was an NSF predoctoral fellow with Prof. John E. Bercaw. From 1997–1999, he was an NSF postdoctoral fellow with Prof. Stephen J. Lippard at MIT.
References
- 1.For leading references, see the February 2010 and March 2011 issues of Chem. Rev. and the April 2011 issue of Chem. Soc. Rev.
- 2.Newhouse T, Baran PS. If C–H Bonds Could Talk: Selective C-H Bond Oxidation. Angew Chem Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gol’dshleger NF, Es’kova VV, Shteinman AA, Shilov AE. Reactions of Alkanes in Solutions of Chloride Complexes of Platinum. Zh Fiz Khim (Engl Transl) 1972;46:785–786. [Google Scholar]
- 4.Stahl SS, Labinger JA, Bercaw JE. Homogeneous Oxidation of Alkanes by Electrophilic Late Transition Metals. Angew Chem Int Ed. 1998;37:2181–2192. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2180::AID-ANIE2180>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 5.(a) Dick AR, Hull KL, Sanford MS. A Highly Selective Catalytic Method for the Oxidative Functionalization of C-H Bonds. J Am Chem Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; (b) Giri R, Chen X, Yu JQ. Palladium-Catalyzed Asymmetric Iodination of Unactivated C-H Bonds under Mild Conditions. Angew Chem Int Ed. 2005;44:2112–2115. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]
- 6.For efforts to replace PtIV, see the following and references cited therein: Lin M, Shen C, Garcia-Zayas EA, Sen A. Catalytic Shilov Chemistry: Platinum Chloride-Catalyzed Oxidation of Terminal Methyl Groups by Dioxygen. J Am Chem Soc. 2001;123:1000–1001. doi: 10.1021/ja001926+.Bar-Nahum I, Khenkin AM, Neumann R. Mild, Aqueous, Aerobic, Catalytic Oxidation of Methane to Methanol and Acetaldehyde Catalyzed by a Supported Bipyrimidinylplatinum-Polyoxometalate Hybrid Compound. J Am Chem Soc. 2004;126:10236–10237. doi: 10.1021/ja0493547.Weinberg DR, Labinger JA, Bercaw JE. Competitive Oxidation and Protonation of Aqueous Monomethylplatinum(II) Complexes: A Comparison of Oxidants. Organometallics. 2007;26:167–172.
- 7.For additional information, see recent reviews of Pd-catalyzed C–H oxidation: Chen X, Engle KM, Wang DH, Yu JQ. Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew Chem Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273.Lyons TW, Sanford MS. Palladium-Catalyzed Ligand-Directed C-H Functionalization Reactions. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e.Yeung CS, Dong VM. Catalytic Dehydrogenative Cross-Coupling: Forming Carbon-Carbon Bonds by Oxidizing Two Carbon-Hydrogen Bonds. Chem Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d.Liu C, Zhang H, Shi W, Lei A. Bond Formation between Two Nucleophiles: Transition Metal Catalyzed Oxidative Cross-Coupling Reactions. Chem Rev. 2011;111:1780–1824. doi: 10.1021/cr100379j.
- 8.For reviews, see: Stahl SS. Palladium Oxidase Catalysis: Selective Oxidation of Organic Chemicals by Direct Dioxygen-Coupled Turnover. Angew Chem Int Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630.Stahl SS. Palladium-Catalyzed Oxidation of Organic Chemicals with O2. Science. 2005;309:1824–1826. doi: 10.1126/science.1114666.Kotov V, Scarborough CC, Stahl SS. Palladium-Catalyzed Aerobic Oxidative Amination of Alkenes: Development of Intra- and Intermolecular Aza-Wacker Reaction. Inorg Chem. 2007;46:1910–1923. doi: 10.1021/ic061997v.Wendlandt AE, Suess AM, Stahl SS. Copper-Catalyzed Aerobic Oxidative C–H Functionalizations: Recent Trends and Mechanistic Insights. Angew Chem, Int Ed. doi: 10.1002/anie.201103945.
- 9.(a) Davidson JM, Triggs C. Reactions of Palladium Complexes with Benzene and Toluene. Chem Ind. 1966:457. [Google Scholar]; (b) Davidson JM, Triggs C. Reaction of Metal Ion Complexes with Hydrocarbons. I. Palladation and Some Other New Electrophilic Substitution Reactions Preparation of Palladium(I) J Chem Soc (A) 1968:1324–1330. [Google Scholar]
- 10.Gligorich KM, Sigman MS. Recent Advancements and Challenges of Palladium(II)-Catalyzed Oxidation Reactions with Molecular Oxygen as the Sole Oxidant. Chem Commun. 2009:3854–3867. doi: 10.1039/b902868d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.See the following and references therein: Backvall JE, Bystrom SE, Nordberg RE. Stereoselective and Regioselective Palladium-Catalyzed 1,4-Diacetoxylation of 1,3-Dienes. J Org Chem. 1984;49:4619–4631.Chen X, Goodhue CE, Yu JQ. Palladium-Catalyzed Alkylation of sp2 and sp3 C–H Bonds with Methylboroxine and Alkylboronic Acids: Two Distinct C–H Activation Pathways. J Am Chem Soc. 2006;128:12634–12635. doi: 10.1021/ja0646747.Lanci MP, Remy MS, Kaminsky W, Mayer JM, Sanford MS. Oxidatively Induced Reductive Elimination from (tBu2bpy)Pd(Me)2: Palladium(IV) Intermediates in a One-Electron Oxidation Reaction. J Am Chem Soc. 2009;131:15618–15620. doi: 10.1021/ja905816q.Khusnutdinova JR, Rath NP, Mirica LM. Stable Mononuclear Organometallic Pd(III) Complexes and Their C-C Bond Formation Reactivity. J Am Chem Soc. 2010;132:7303–7306. doi: 10.1021/ja103001g.
- 12.Exceptions exist; see, for example: Mitsudome T, Umetani T, Nosaka N, Mori K, Mizugaki T, Ebitani K, Kaneda K. Convenient and Efficient Pd-Catalyzed Regioselective Oxyfunctionalization of Terminal Olefins by Using Molecular Oxygen as Sole Reoxidant. Angew Chem Int Ed. 2006;45:481–485. doi: 10.1002/anie.200502886.Zhang J, Khaskin E, Anderson NP, Zavalij PY, Vedernikov AN. Catalytic Aerobic Oxidation of Substituted 8-Methylquinolines in Pd(II)-2,6-Pyridinedicarboxylic Acid Systems. Chem Commun. 2008:3625–3627. doi: 10.1039/b803156h.Tsang WCP, Munday RH, Brasche G, Zheng N, Buchwald SL. Palladium-Catalyzed Method for the Synthesis of Carbazoles Via Tandem C-H Functionalization and C-N Bond Formation. J Org Chem. 2008;73:7603–7610. doi: 10.1021/jo801273q.Liu GS, Yin GY, Wu L. Palladium-Catalyzed Intermolecular Aerobic Oxidative Amination of Terminal Alkenes: Efficient Synthesis of Linear Allylamine Derivatives. Angew Chem Int Ed. 2008;47:4733–4736. doi: 10.1002/anie.200801009.Zhang YH, Yu JQ. Pd(II)-Catalysed Hydroxylation of Arenes with 1 atm O2 or Air. J Am Chem Soc. 2009;131:14654–14655. doi: 10.1021/ja907198n.
- 13.See the following and references therein: Chen MS, Prabagaran N, Labenz NA, White MC. Serial Ligand Catalysis: A Highly Selective Allylic C-H Oxidation. J Am Chem Soc. 2005;127:6970–6971. doi: 10.1021/ja0500198.Lin BL, Labinger JA, Bercaw JE. Mechanistic Investigations of Bipyrimidine-Promoted Palladium-Catalyzed Allylic Acetoxylation of Olefins. Can J Chem. 2009;87:264–271.
- 14.For a leading reference, see: Bäckvall JE, Gogoll A. Evidence for (π,-Allyl)Palladium(II)(Quinone) Complexes in the Palladium-Catalyzed 1,4-Diacetoxylation of Conjugated Dienes. Tet Lett. 1988;29:2243–2246.
- 15.Grennberg H, Gogoll A, Backvall JE. Acid-Induced Transformation of Palladium(0) Benzoquinone Complexes to Palladium(II) and Hydroquinone. Organometallics. 1993;12:1790–1793. [Google Scholar]
- 16.Stahl SS, Thorman JL, Nelson RC, Kozee MA. Oxygenation of Nitrogen-Coordinated Palladium(0): Synthetic, Structural, and Mechanistic Studies and Implications for Aerobic Oxidation Catalysis. J Am Chem Soc. 2001;123:7188–7189. doi: 10.1021/ja015683c. [DOI] [PubMed] [Google Scholar]
- 17.Campbell AN, White PB, Guzei IA, Stahl SS. Allylic C-H Acetoxylation with a 4,5-Diazafluorenone-Ligated Palladium Catalyst: A Ligand-Based Strategy To Achieve Aerobic Catalytic Turnover. J Am Chem Soc. 2010;132:15116–15119. doi: 10.1021/ja105829t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.For leading references, see: Shiotani A, Itatni H, Inagaki T. Selective Coupling of Dimethyl Phthalate with Palladium Catalysts at Atmospheric Pressure. J Mol Catal. 1986;34:57–66.Itatani H, Yoshimoto H, Shiotani A, Yokota A, Yoshikiyo M. Process for Producing Biphenyl Compounds. 4,294,976. US Patent. 1981
- 19.Kreuz JA, Edman JR. Polyimide Films. Adv Mater. 1998;10:1229–1232. [Google Scholar]
- 20.For leading references, see ref. 9a and the following: Yoshimoto H, Itatani H. Palladium-Catalyzed Competitive Reaction of Aromatic Compounds. J Catal. 1973;31:8–12.Itatani H, Shiotani A, Yoshimoto H, Yoshikiyo M, Yokota A. Manufacturing Method of Biphenyl Derivatives. 79324. JP Patent. 1980
- 21.A NineSigma™ proposal request (#11222-1) was issued in Oct. 2008 by a “a multi-billion dollar chemical manufacturer” focused on this topic.
- 22.Izawa Y, Stahl SS. Aerobic Oxidative Coupling of o-Xylene: Discovery of 2-Fluoropyridine as a Ligand to Support Selective Pd-Catalyzed C-H Functionalization. Adv Synth Catal. 2010;352:3223–3229. doi: 10.1002/adsc.201000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorelsky SI, Lapointe D, Fagnou K. Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates. J Am Chem Soc. 2008;130:10848–10849. doi: 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]
- 24.See, for example: Steinhoff BA, Guzei IA, Stahl SS. Mechanistic Characterization of Aerobic Alcohol Oxidation Catalyzed by Pd(Oac)2/Pyridine Including Identification of the Catalyst Resting State and the Origin of Nonlinear [Catalyst] Dependence. J Am Chem Soc. 2004;126:11268–11278. doi: 10.1021/ja049962m.
- 25.See also: Engle KM, Wang D-H, Yu J-Q. Ligand-Accelerated C-H Activation Reactions: Evidence for a Switch of Mechanism. J Am Chem Soc. 2010;132:14137–14151. doi: 10.1021/ja105044s.
- 26.(a) Stuart DR, Fagnou K. The Catalytic Cross-Coupling of Unactivated Arenes. Science. 2007;316:1172–1175. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]; (b) Stuart DR, Villemure E, Fagnou K. Elements of Regiocontrol in Palladium-Catalyzed Oxidative Arene Cross-Coupling. J Am Chem Soc. 2007;129:12072–12703. doi: 10.1021/ja0745862. [DOI] [PubMed] [Google Scholar]
- 27.See refs. 11c, 11d, and references cited therein.
- 28.Campbell AN, Meyer EB, Stahl SS. Regiocontolled Aerobic Oxidative Coupling of Indoles and Benzene Using Pd Catalysts with 4,5-Diazafluorene Ligands. Chem Commun. 2011;47:10257–10259. doi: 10.1039/c1cc13632a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi J, MacArthur AHR, Brookhart M, Goldman AS. Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes. Chem Rev. 2011;111:1761–1779. doi: 10.1021/cr1003503. [DOI] [PubMed] [Google Scholar]
- 30.Konnick MM, Stahl SS. Reaction of Molecular Oxygen with a PdII-Hydride To Produce a PdII-Hydroperoxide: Experimental Evidence for an HX-Reductive-Elimination Pathway. J Am Chem Soc. 2008;130:5753–5762. doi: 10.1021/ja7112504. [DOI] [PubMed] [Google Scholar]
- 31.Izawa Y, Pun D, Stahl SS. Palladium-Catalyzed Aerobic Dehydrogenation of Substituted Cyclohexanones to Phenols. Science. 2011;333:209–213. doi: 10.1126/science.1204183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diao T, Stahl SS. Synthesis of Cyclic Enones via Direct Palladium-Catalyzed Aerobic Dehydrogenation of Ketones. J Am Chem Soc. 2011;133:14566–14569. doi: 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.For an important early example, see: Chen X, Hao X-S, Goodhue CE, Yu J-Q. Cu(II)-Catalyzed Functionalizations of Aryl C-H Bonds Using O2 as an Oxidant. J Am Chem Soc. 2006;128:6790–6791. doi: 10.1021/ja061715q.
- 34.Qiao JX, Lam PYS. Copper-Promoted Carbon-Heteroatom Bond Cross-Coupling with Boronic Acids and Derivatives. Synthesis. 2011;2011:829–856. [Google Scholar]
- 35.King AE, Brunold TC, Stahl SS. Mechanistic Study of Copper-Catalyzed Aerobic Oxidative Coupling of Arylboronic Esters and Methanol: Insights into an Organometallic Oxidase Reaction. J Am Chem Soc. 2009;131:5044–5045. doi: 10.1021/ja9006657. [DOI] [PubMed] [Google Scholar]
- 36.Huffman LM, Stahl SS. Carbon-Nitrogen Bond Formation Involving Well-Defined Aryl-Copper(III) Complexes. J Am Chem Soc. 2008;130:9196–9197. doi: 10.1021/ja802123p. [DOI] [PubMed] [Google Scholar]
- 37.(a) Casitas A, King AE, Parella T, Costas M, Stahl SS, Ribas X. Direct Observation of CuI/CuIII Redox Steps Relevant to Ullmann-Type Coupling Reactions. Chem Sci. 2010:326–330. [Google Scholar]; (b) Huffman LM, Casitas A, Font M, Canta M, Costas M, Ribas X, Stahl SS. Observation and Mechanistic Study of Facile C–O Bond Formation between a Well-Defined Aryl-Copper(III) Complex and Oxygen Nucleophiles. Chem, Eur J. 2011;17:10643–10650. doi: 10.1002/chem.201100608. [DOI] [PubMed] [Google Scholar]
- 38.King AE, Huffman LM, Casitas A, Costas M, Ribas X, Stahl SS. Copper-Catalyzed Aerobic Oxidative Functionalization of an Arene C-H Bond: Evidence for an Aryl-Copper(III) Intermediate. J Am Chem Soc. 2010;132:12068–12073. doi: 10.1021/ja1045378. [DOI] [PubMed] [Google Scholar]
- 39.See Section 4 of ref. 8d for a full discussion of this class of reactivity.
- 40.Ribas X, Jackson DA, Donnadieu B, Mahia J, Parella T, Xifra R, Hedman B, Hodgson KO, Llobet A, Stack TDP. Aryl C–H Activation by CuII to Form an Organometallic Aryl–CuIII Species: A Novel Twist on Copper Disproportionation. Angew Chem Int Ed. 2002;41:2991–2994. doi: 10.1002/1521-3773(20020816)41:16<2991::AID-ANIE2991>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 41.Ribas X, Calle C, Poater A, Casitas A, Gómez L, Xifra R, Parella T, Benet-Buchholz J, Schweiger A, Mitrikas G, Solà M, Llobet A, Stack TDP. Facile C–H Bond Cleavage Via a Proton-Coupled Electron Transfer Involving a C-H···CuII Interaction. J Am Chem Soc. 2010;132:12299–12306. doi: 10.1021/ja101599e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamada T, Ye X, Stahl SS. Copper-Catalyzed Aerobic Oxidative Amidation of Terminal Alkynes: Efficient Synthesis of Ynamides. J Am Chem Soc. 2008;130:833–835. doi: 10.1021/ja077406x. [DOI] [PubMed] [Google Scholar]