

Abstract
GnRH binds to its receptor on gonadotropes and activates multiple members of the MAPK signaling family that in turn regulates the expression of several immediate early genes (IEGs) including Jun, Fos, Atf3, and Egr1. These IEGs confer hormonal responsiveness to gonadotrope-specific genes including Gnrhr, Cga, Fshb, and Lhb. In this study we tested the hypothesis that GnRH specifically regulates the accumulation of Jun and Atf3 mRNA through a pathway that includes intracellular Ca2+, calcineurin, and nuclear factor of activated T cells (NFAT). Our results indicate that pretreatment of murine LβT2 cells with 1, 2-bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl)-ester, a Ca2+ chelator, reduced the expression of all the IEGs to varying degrees, whereas treatment with thapsigargin, an intracellular Ca2+ protein pump inhibitor, increased the expression of the IEG. Furthermore, cyclosporin A, a calcineurin-specific inhibitor, reduced the ability of GnRH to regulate accumulation of Jun and Atf3 mRNA and to a lesser extent Fos. In contrast, Egr1 mRNA was unaffected. NFATs are transcription factors regulated by calcineurin and were detected in LβT2 cells. GnRH increased luciferase activity of an NFAT-dependent promoter reporter that was dependent on intracellular Ca2+ and calcineurin activity. Additionally, although small interfering RNA specific for Nfat4 only marginally reduced GnRH regulation of Jun, Fos, and Atf3 mRNA accumulation, activity of an activator protein-1-responsive reporter construct was reduced by 48%. Together these data suggest that calcineurin and NFAT are new members of the gonadotrope transcriptional network that confer hormonal responsiveness to several key genes required for gonadotropin synthesis and secretion.
Normal reproductive function requires LH and FSH, gonadotropins that are synthesized and secreted from gonadotropes of the anterior pituitary. LH and FSH are heterodimeric glycoproteins composed of a common α-subunit (encoded by Cga) and distinct β-subunits (encoded by Lhb and Fshb, respectively) that confer receptor specificity (reviewed in Refs. 1–3). GnRH secreted from the hypothalamus binds to its receptor (encoded by Gnrhr) and activates multiple signaling pathways ultimately leading to increased expression of the gonadotrope specific genes Cga, Lhb, Fshb, and Gnrhr (2–4). Hormonal responsiveness of the gonadotrope-specific genes also requires several immediate early genes (IEGs) including Jun, Fos, Atf3, and Egr1, which act to communicate the GnRH signal by binding to their response elements on Fshb (JUN/FOS) and Gnrhr (JUN/FOS), Cga [JUN/activating transcription factor (ATF3)], and Lhb (early growth response protein 1) (reviewed in Refs. 1, 3, and 5–8).
GnRH signals through Gαq proteins ultimately leading to increased activity of several members of the MAPK family (9). These include the ERK (10–12), c-JUN N-terminal kinase (JNK) (10, 11) and p38 MAPK (10, 13–15). GnRH also increases Ca2+ mobilization and influx through voltage-gated calcium channels (VGCC) (15–19), suggesting extensive cross talk that involves multiple signaling pathways.
Calcium is an important second messenger that has a plethora of intracellular targets. GnRH signaling leads to increased Ca2+ that in turn interacts with calmodulin, leading to activation of calmodulin-dependent protein kinases, including type I and type II (CaMKII) (20–23). Calmodulin kinase type I has been shown to repress transcription of Fshb via phosphorylation of histone deacetylase (HDAC) within gonadotropes (22), whereas GnRH-regulated CaMKII phosphorylation leads to increases in Cga mRNA (20, 21) and activity of a rat Lhb-Luciferase reporter (18, 19) in LβT2 cells. CAMKII has also been shown to regulate a c-Fos-Luciferase reporter downstream of GnRH and ultimately impacts activity of an Fshb-Luciferase reporter (23).
Another Ca2+/calmodulin target in gonadotrope cells is calcineurin, a calcium-dependent protein phosphatase (22, 24, 25). Calcineurin is regulated in αT3 cells, an immortalized gonadotrope-specific cell line (22), and its activity is required for accumulation of ATF3 that depends on activation of both JNK and ERK (24). GnRH has also been shown to increase the nuclear localization of the Ca2+/calcineurin-regulated nuclear factor of activated T-cells 2 (NFAT2) 2 using a fluorescently tagged expression construct transfected into both HeLa cells and LβT2 cells, another immortalized gonadotrope-specific cell line (26, 27).
Significant cross talk occurs between the MAPK family members and Ca2+-dependent signaling targets (2–5, 7, 8, 16, 28, 29). Calcium influx through VGCC affects ERK activation, whereas mobilization of intracellular Ca2+ affects JNK activity in gonadotrope derived cell lines (16, 30). The role of JNK in the regulation of JUN posttranslational phosphorylation is well documented in gonadotrope cell lines (3, 16, 24, 28, 30, 31); however, the transcriptional regulation of Jun downstream of GnRH is only partially defined. This regulation involves T cell factor (TCF), a transcription factor originally identified in immune cells, and the coactivator β-catenin (32).
The contribution of Ca2+ mobilization to JNK activation suggests that the two signaling pathways may be a point of convergence for regulation of Jun transcription. This would allow the second messenger to activate several pathways, which in turn can activate specific downstream targets. For example, Ca2+/CaMKII regulates transcription of Fos, whereas transcriptional effects on Jun and other basic leucine zipper domain (bZIP) encoding mRNA remain unclear (23). Nevertheless, the ability of GnRH to selectively regulate expression of bZIP proteins would provide further refined regulation of gene transcription in gonadotropes.
Calcium signals occur quickly in LβT2 cells, and the ability of GnRH to regulate calcineurin-dependent cellular localization of NFAT proteins rapidly, within 15 min (26, 27), suggests that the IEGs, including Jun and Atf3, could be endogenous transcriptional targets of NFATs. In this regard, NFAT proteins were originally identified as transcriptional activators necessary for T cell and B cell function (33–35) in the immune system. NFAT are also functionally active in nonimmune cell types and can regulate transcription directly by binding to regulatory elements in DNA (33, 34, 36, 37) and cooperate with bZIP proteins to regulate target genes including cytokines, IL-2 (38), and TNFα (39) in addition to others (33, 35, 36). Interestingly, regulation of TNFα occurs downstream of MAPK in pancreatic islet cells (39), providing precedence for a link between MAPK activation and transcriptional targets of NFAT. Because JUN autoregulates its own promoter through distally located activator protein-1 (AP1) sites (40), it is plausible that this IEG is a target of NFAT/AP1 in gonadotropes.
Herein we present data demonstrating that JNK/Ca2+/calcineurin activity selectively regulates TCF-dependent luciferase activity and expression of Jun, Atf3, and potentially Fos downstream of a GnRH signal. Furthermore, we demonstrate that LβT2 cells express Nfat1–4 and show that GnRH can regulate the transcriptional activity of an NFAT-responsive promoter reporter construct dependent on mobilization of Ca2+ and activation of calcineurin. Finally, we show that reduction of Nfat4 dampens GnRH-mediated accumulation of Jun, Fos, and Atf3 mRNA as well as activity of the AP1-responsive CGA-Luciferase reporter in LβT2 cells. Together these findings demonstrate a previously unappreciated role for a Ca2+/calcineurin/NFAT pathway in GnRH-regulated expression of Jun, Atf3, and potentially Fos. These are the bZIP proteins that confer GnRH responsiveness to several downstream genes including Cga.
Results
JNK selectively activates Jun and Atf3 mRNA accumulation downstream of GnRH
Members of the MAPK signaling family are key components of GnRH signaling and the regulation of gonadotrope specific genes (16, 30, 31, 41); therefore, we first investigated the role of several MAPK family members. JNK phosphorylates JUN leading to increased transcriptional activity of the oncogene and is also necessary for ATF3 accumulation (42). Work in LβT2 cells has examined the posttranscriptional accumulation of ATF3 and phosphorylated JUN (13, 24, 42), whereas the transcriptional regulation of the IEGs is not completely understood. To investigate the role of JNK in IEG transcription, LβT2 cells were serum starved and then pretreated with the JNK-specific inhibitor SP600125 (25 μm) for 1 h before the subsequent treatment with GnRH (10 nm) for 1 h. mRNAs were reverse transcribed, and quantitative real-time PCR (qRT-PCR) was performed using primers specific for IEG Jun, Fos, Atf3, and Egr1.
Inhibition of JNK significantly reduced Jun transcription (61% reduction) in response to GnRH (Fig. 1A), and Atf3 transcription (Fig. 1C) (51% reduction). GnRH induction of Fos was enhanced (68% increase) in the presence of the JNK inhibitor (Fig. 1B), whereas Egr1 mRNA was not significantly affected (Fig. 1D). Inhibitors for other MAPK family members including ERK did not affect Jun mRNA accumulation in the presence of GnRH, whereas Fos (21% reduction), Atf3 (37% reduction), and Egr1 (58% reduction) transcripts are regulated downstream of ERK (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Additionally, an inhibitor specific for p38 MAPK reduced Atf3 transcript (22% reduction), whereas the other IEGs examined were not altered (Supplemental Fig. 2). These results demonstrate that JNK activation by GnRH contributes to maximum accumulation of both Jun and Atf3 transcripts. Although inhibition of JNK activity reduced GnRH-dependent accumulation of both transcripts, residual activity remained, suggesting that additional signaling pathways are also required.
Fig. 1.

GnRH selectively regulates Jun and Atf3 via JNK activation. LβT2 cells were serum starved overnight and then pretreated with DMSO or the JNK-specific inhibitor SP600125 (25 μm) for 1 h. After SP600125 treatment, cells were treated with PBS (vehicle) or GnRH (10 nm) for 1 h. qRT-PCR was performed using primers specific for Jun (A), Fos (B), Atf3 (C), and Egr1 (D). Cyclophilin B was used to normalize the samples, and the expression of each gene is shown relative to DMSO- and vehicle-treated cells. Data shown are the mean ± sem from three independent experiments performed on separate days. qRT-PCR samples were run in at least duplicate. Data were analyzed via one-way ANOVA. **, P < 0.01; ***, P < 0.001.
GnRH regulation of IEG mRNA accumulation requires calcium
JNK activation downstream of GnRH requires mobilization of intracellular Ca2+, whereas ERK activation requires Ca2+ influx from VGCC (16). To investigate the role of Ca2+ mobilization in GnRH regulation of Jun and Atf3, we used 1, 2-bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl)-ester (BAPTA-AM), a Ca2+ chelator. In these experiments, LβT2 cells were pretreated with BAPTA-AM (50 μm) for 1 h followed by a subsequent treatment with vehicle or GnRH (10 nm) for 1 h. The changes in IEG mRNA were then examined by qRT-PCR using primers specific for the Jun, Fos, Atf3, and Egr1.
GnRH-regulated accumulation of Jun (Fig. 2A), Fos (Fig. 2B), Atf3 (Fig. 2C), and Egr1 (Fig. 2D) was inhibited by BAPTA to varying degrees. The bZIP proteins, Jun (64% reduction), Fos (51% reduction), and Atf3 (87% reduction) as well as the zinc finger protein Egr1 (58% reduction) showed a requirement of Ca2+ for maximal induction by GnRH. These results are consistent with previous studies that reported BAPTA-AM blocked activity of a Jun-Luciferase reporter construct and accumulation of ATF3 in αT3 cells (17, 24). Collectively these results demonstrate that GnRH-regulated transcription of endogenous Jun, Fos, Atf3, and Egr1 requires Ca2+ at the transcriptional level.
Fig. 2.

GnRH regulation of IEG accumulation requires calcium. LβT2 cells were serum starved overnight and pretreated with DMSO, BAPTA-AM (50 μm), or thapsigargin (1 μm) for 1 h. After treatment, cells were then treated with PBS (vehicle) or GnRH (10 nm) for 1 h. qRT-PCR was performed using primers specific for Jun (A and E), Fos (B and F), Atf3 (C and G), and Egr1 (D and H). Cyclophilin B was used to normalize samples, and each gene is expressed relative to DMSO- and vehicle-treated cells. Data shown are the mean ± sem from three independent experiments performed on different days. qRT-PCR samples were run in at least duplicate. Data were analyzed via one-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further explore the role of cytosolic Ca2+ in the GnRH signaling pathway, we used thapsigargin, a noncompetitive inhibitor of the sarco/endoplasmic reticulum Ca2+/ATPase pump that increases the concentration of cytosolic Ca2+. Accordingly, we treated serum-starved LβT2 cells with vehicle [dimethyl sulfoxide (DMSO)] or thapsigargin (1 μm) for 1 h. The cells were then treated with vehicle (PBS) or GnRH (10 nm) for an additional hour in the continued presence of thapsigargin. Changes in IEG were then quantitated by qRT-PCR as described previously.
Although thapsigargin appeared to increase the basal expression of Jun (Fig. 2E), and to a lesser extent Fos (Fig. 2F), Atf3 (Fig. 2G), and Egr1 (Fig. 2H), these changes were not statistically significant (P > 0.05). In contrast, thapsigargin significantly enhanced GnRH regulated expression of all the IEGs: Jun (89% increase), Fos (86% increase), Atf3 (50% increase), and Egr1 (79% increase). Together the results with BAPTA-AM and thapsigargin indicate that cytosolic Ca2+ mediates GnRH-regulated expression of Jun, Fos, Atf3, and Egr1.
We also noted that the GnRH-dependent fold induction of Atf3 and Egr1 were reproducibly higher in the thapsigargin vs. BAPTA experiments. We attribute this difference to an effect of DMSO because BAPTA and thapsigargin were dissolved with different dilutions of the solvent (1:200 and 1:1000, respectively). Apparently the DMSO effect is specific for Atf3 and Egr1 because the GnRH-dependent fold inductions were more consistent for Jun and Fos.
The involvement of Ca2+ in regulating changes in accumulation of IEG mRNA suggests that Ca2+/calmodulin-dependent kinases may be required. To explore this possibility, LβT2 cells were treated with the CaMKII inhibitor KN93 and the calmodulin kinase kinase (CaMKK) inhibitor STO609. Inhibition of CaMKII did not affect the transcriptional accumulation of endogenous IEG mRNA accumulation (Supplemental Fig. 3). This is contradictory to a report by Ely et al. (23), who demonstrate that Fos-Luciferase activity is down-regulated in the presence of CaMKII inhibitor, suggesting that the chromatin status of the promoter may contribute to CaMKII regulation. Similar results were observed in cells pretreated with CaMKK inhibitor (Supplemental Fig. 4). Neither inhibitor had an effect on GnRH-regulated expression of Jun, Fos, or Atf3, suggesting that other Ca2+/calmodulin-dependent targets, such as calcineurin, may be necessary.
Calcineurin activity is required for Jun and Atf3 transcription downstream of GnRH
Increased calcium can activate several calmodulin-dependent downstream effectors including calcineurin, a Ca2+-dependent protein phosphatase (22, 24, 43). To examine whether GnRH regulated expression of IEGs required calcineurin, LβT2 cells were serum starved, pretreated with a calcineurin inhibitor, cyclosporin A (20 μm), for 1 h followed by treatment with vehicle (PBS) or GnRH (10 nm) for an additional hour. qRT-PCR was performed using primers specific for the IEGs as described previously.
Cyclosporin A reduced the amount of Jun transcript regulated by GnRH (46% reduction) (Fig. 3A), suggesting a requirement for the phosphatase. A previous study reported that inhibition of calcineurin reduced levels of ATF3 protein in αT3 cells (24), and herein we demonstrate that the Atf3 transcript is also reduced in LβT2 cells by 45% (Fig. 3C). Although cyclosporin A dampened GnRH-regulated expression of Fos, the change was not statistically significant, but the trend suggests that Fos is a potential target of calcineurin. GnRH-induced accumulation of Egr1 mRNA was unaffected by treatment with cyclosporin A. These findings demonstrate that GnRH activates calcineurin, presumably downstream of Ca2+, and that this signaling cascade is necessary for the transcriptional regulation of Jun, Atf3, and possibly Fos.
Fig. 3.

Calcineurin activity is required for Jun and Atf3 transcription downstream of GnRH. LβT2 cells were serum starved overnight and pretreated with DMSO or cyclosporin A (20 μm) for 1 h. After treatment, cells were then treated with PBS (vehicle) or GnRH (10 nm) for 1 h. qRT-PCR was performed using primers specific for Jun (A), Fos (B), Atf3 (C), and Egr1 (D). Cyclophilin B was used to normalize samples, and the expression of each gene is shown relative to the amount of expression in DMSO- and vehicle-treated cells. Data shown are the mean ± sem from three independent experiments performed on separate days. qRT-PCR samples were run in at least duplicate. Data were analyzed via one-way ANOVA. *, P < 0.05.
LβT2 cells express Nfat family members
Calcineurin is a calcium-dependent serine/threonine protein phosphatase that regulates the transcriptional activity of NFAT proteins (34, 37, 44–46). GnRH regulates an NFAT-dependent luciferase promoter reporter in HeLa and LβT2 cells (27), suggesting that Nfats are endogenously expressed in the gonadotrope cell line. To confirm expression of Nfat family members in LβT2 cells, semiquantitative PCR was performed using primers specific for each of the four classical members of the NFAT family (Nfat1–4) (47). The expression of another Nfat family member, Nfat5 (also known as Nfatz), was not examined because it is not calcineurin dependent (34, 48). In these experiments, LβT2 cells were serum starved overnight and then treated with vehicle (PBS) or GnRH (10 nm) for 1 h.
As shown in Fig. 4A, LβT2 cells express four NFAT family members including Nfat1/Nfatp (officially Nfatc2), Nfat2/Nfatc (officially Nfatc1), Nfat3 (officially Nfatc4), and Nfat4/Nfatx (officially Nfatc3). Of these mRNA, Nfat4 appears to be the most highly expressed. Immunoblots were also performed to confirm protein expression using an antibody specific for NFAT4 in cells treated with vehicle or GnRH. NFAT proteins are traditionally not transcriptionally regulated, and as expected, GnRH did not regulate levels of NFAT4 (Fig. 4B). This result is consistent with previous reports indicating that transcriptional activity of NFAT proteins is regulated by posttranslational phosphorylation (35, 46). Thus, in the absence of signal, NFAT proteins are phosphorylated leading to accumulation in the cytoplasm and exclusion from the nucleus. These phosphate groups are removed by calcineurin, a Ca2+-dependent protein phosphatase, allowing nuclear translocation of the NFAT proteins and increased transcriptional activity (34, 37, 44–46).
Fig. 4.
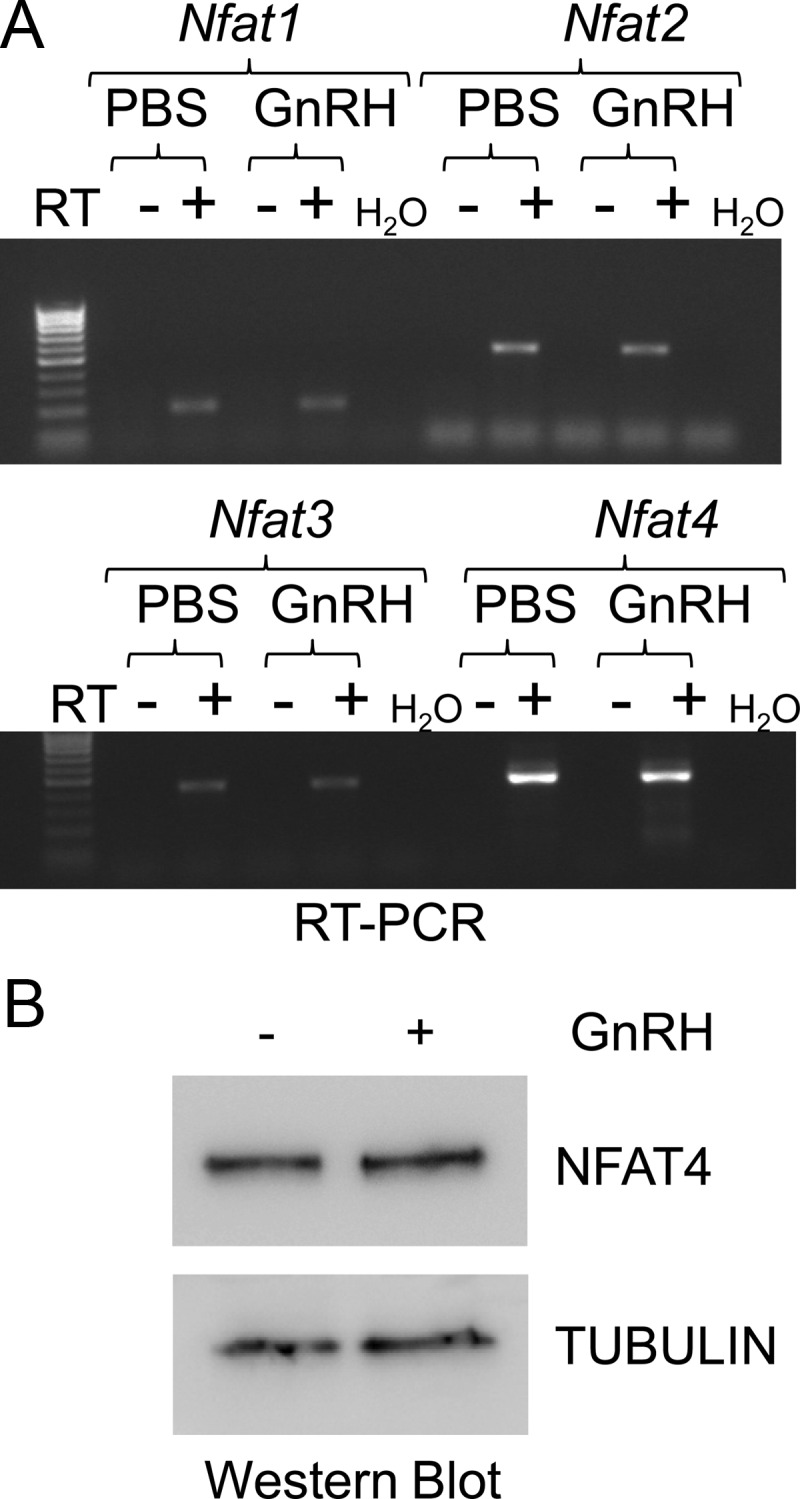
LβT2 cells express several Nfat family members. LβT2 cells were serum starved and treated with vehicle (PBS) or GnRH (10 nm) for 1 h. After RNA isolation and reverse transcription of cDNA, semiquantitative PCR was performed using primers specific for Nfat1, Nfat2, Nfat3, and Nfat4 (A). Whole-cell lysates from LβT2 cells were run on a SDS polyacrylamide gel and then immunoblotted for NFAT4 and tubulin, used as a loading control (B). Data shown are representative of three independent experiments. RT, Reverse transcriptase.
GnRH regulation of NFAT-dependent transcription requires Ca2+ mobilization and calcineurin activity
GnRH has been reported to regulate activity of an Nfat-Luciferase promoter reporter (27). Because LβT2 cells express Nfat1–4 (Fig. 4), we next examined the signaling pathways involved in NFAT-dependent transcriptional regulation. Here we used an NFAT-responsive, artificial reporter (49). In the absence of hormone, the activity of the Nfat-Luciferase construct was low, whereas GnRH significantly increased luciferase activity [Fig. 5 and Refs. 26 and 27)].
Fig. 5.

GnRH regulates NFAT-dependent transcription through Ca2+ and calcineurin. An artificial NFAT-Luc reporter construct was transiently transfected into LβT2 cells as described in Materials and Methods. After transfection, cells were pretreated for 1 h with DMSO or ethanol (as vehicle) or one of the following inhibitors: the intracellular Ca2+ chelator BAPTA-AM (50 nm) (A), the calcineurin inhibitor cyclosporin A (200 μm) (B), STO609 (25 μm), an inhibitor of CaMKK (C), or the JNK inhibitor SP600125 (25 μm) (D). After treatment with either PBS or GnRH (100 nm), cells were lysed and luciferase activity was measured. Data were normalized as described by Valcu and Valcu (65). Data shown are the mean ± sem for three independent experiments, each performed in triplicate, with the exception of D, which is mean ± sd from two independent experiments performed in triplicate. Data were analyzed via one-way ANOVA. ***, P < 0.001.
To confirm that GnRH regulation of NFAT transcriptional activity requires release of intracellular Ca2+, LβT2 cells were transfected with the Nfat-Luciferase reporter and then pretreated with BAPTA-AM for 1 h before treatment with vehicle or GnRH (100 nm) for an additional 8 h. GnRH-induced transcriptional activation of the Nfat-Luciferase promoter reporter and pretreatment with BAPTA-AM abolished luciferase activity (Fig. 5A). This demonstrates that GnRH regulation of NFAT-dependent transcriptional activity requires intracellular Ca2+.
To examine whether the activation of the Nfat-Luciferase reporter is dependent on calcineurin activity, cells were transfected and pretreated with cyclosporin A (200 μm) for 1 h followed by treatment with vehicle or GnRH (100 nm) for 8 h. GnRH-mediated activation of NFAT transcriptional activity required calcineurin (Fig. 5B), whereas inhibitors specific for other Ca2+ targets such as CaMKK (Fig. 5C) did not affect luciferase activity. Together these data suggest specific Ca2+/calcineurin signals regulate NFAT-dependent transcription in response to GnRH.
We also examined the role of the MAPK member JNK on NFAT-dependent transcriptional activation by GnRH. Cells were transfected with NFAT-luciferase, pretreated with SP600125 (25 μm) for 1 h followed by vehicle or GnRH (100 nm) for 8 h. Inhibition of JNK did not reduce luciferase activity (Fig. 5D), suggesting that GnRH-regulated expression of an NFAT-dependent artificial promoter occurs independently of JNK. This was in contrast to TCF/lymphoid enhancer factor (LEF)-dependent transcription, which was found to require JNK/Ca2+/calcineurin (Supplemental Fig. 5), suggesting partial cross talk between the pathways that regulate the two transcription factors (TCF and NFAT) could ultimately impact Jun transcription.
Maximal GnRH induction of Jun, Fos, and Atf3 mRNA requires NFAT
Members of the NFAT family can bind JUN and regulate AP1-dependent genes (34–36, 38, 39), suggesting that GnRH regulated expression of Jun also requires NFAT. To explore this possibility, LβT2 cells were transiently transfected with small interfering RNA (siRNA) specific for Nfat4, the most highly expressed Nfat in LβT2 cells (Fig. 4A and unpublished data) or with a nonspecific scrambled siRNA (NS siRNA) as a control.
Seventy-two hours after transfection of the siRNA, serum starved LβT2 cells were treated with vehicle (PBS) or GnRH (100 nm) for 1 h. Analysis by qRT-PCR indicated that Nfat4 siRNA reduced Nfat4 mRNA levels by approximately 90% (Fig. 6A), whereas Nfat3 mRNA levels remained unchanged (data not shown). NFAT4 protein levels were reduced 36% in cells transfected with the Nfat4 siRNA (Fig. 6B). Reduction of Nfat4 transcript coincided with a decrease in GnRH induction of several IEGs, including bZIP proteins Jun (11% reduction), Fos (13% reduction), and Atf3 (18% reduction) (Fig. 6, C–E). Although Egr1 transcript trended toward a reduction, a statistically significant reduction was not observed (Fig. 6F). Although these reductions in Jun, Fos, and Atf3 are modest, they probably reflect the fact that the NFAT protein was reduced to a lesser extent than Nfat mRNA (36 vs. 90%). This differential suggests that NFAT protein has a longer half-life relative to its mRNA. It is also important to recognize that AP1 proteins function as heterodimers to regulate specific gene targets. JUN, FOS, and ATF3 are marginally detectable in the absence of GnRH (50), suggesting that their concentrations are well below the dissociation constant (Kd) of their respective heterodimers. If this is true, then the concentration of the heterodimer should increase exponentially until the concentration of monomers approach their Kd concentration. Conversely, a small decrease in these bZIP proteins should translate into a larger decrease in the concentration of AP1 heterodimers. Accordingly, siRNA-mediated reduction of Nfat that causes modest changes in Jun, Fos, and Atf3, and presumably their encoded proteins, would be expected to have a greater impact on a promoter that binds AP1 heterodimers such as Cga (50), a notion that will be tested below.
Fig. 6.

Nfat4 is required for maximal Jun, Fos, and Atf3 mRNA accumulation downstream of GnRH. LβT2 cells were transfected with a scrambled sequence NS siRNA or siRNA specific for Nfat4. Seventy-two hours after transfection, serum-starved LβT2 cells were treated with vehicle (PBS) or GnRH (100 nm) for 1 h. After treatment, RNA was isolated and reverse transcribed, and real-time PCR was performed using primers specific for Nfat4 mRNA and the IEG Jun, Fos, Atf3, and Egr1 (A and C–F). Fold changes for cells treated with GnRH and NS siRNA were compared with fold changes of cells treated with GnRH and Nfat4 siRNA using the comparative (CT) method described by Schmittgen and Livak (66). For example, the equation below was used to determine the GnRH induced fold change (2-ΔΔCt) in Jun mRNA in cultures transfected with NS siRNA: 2-ΔΔCt (Jun/NS siRNA) where ΔΔCT = [(CT Jun − CT Gapdh internal control) [GnRH treated] − (CT Jun − CT Gapdh internal control) [vehicle treated]]. 2-ΔΔCt values for each IEG mRNA in cultures transfected with either NS siRNA or Nfat siRNA are the mean ± sem from four independent experiments performed consecutively on different days. qRT-PCR samples were run in duplicate. Data were analyzed via a two-tailed unpaired t test. *, P < 0.05; **, P < 0.01. B, Whole-cell lysates from LβT2 cells transfected with siRNA were run on a SDS polyacrylamide gel and then immunoblotted for NFAT4 and tubulin, used as a loading control. These transfections were done together, with a subset collected for RNA and another for protein as described in Materials and Methods.
Reduced Nfat4 expression alters GnRH induction of an AP1-responsive promoter
The human CGA promoter responds to GnRH via binding of JUN and ATF3 (50, 51). We have previously demonstrated that TCF is necessary for both Jun accumulation and maximal activity of the JUN target CGA downstream of GnRH (32). This provides an AP1-responsive promoter to examine the role of NFAT in GnRH signaling to target genes. Transient expression of siRNA specific for Nfat4 reduced the induction of Jun, Fos, and Atf3 downstream of GnRH (Fig. 6), suggesting AP1 targets such as CGA may also be reduced. To investigate this, LβT2 cells were transiently transfected with siRNA specific for Nfat4 (or NS siRNA as control) and a CGA-Luciferase reporter construct. After 72 h, cells were treated with vehicle (PBS) or GnRH (10 nm) for 8 h and then assayed for luciferase activity.
GnRH increases the activity of CGA-Luciferase in the presence of NS siRNA; however, expression of Nfat4-specific siRNA reduces GnRH induction by 48% (Fig. 7). This is consistent with the notion that small siRNA-mediated reductions in bZIP monomers leads to a much greater change in activity of AP1 heterodimers. Residual activity of the reporter construct after treatment with the Nfat siRNA could reflect either incomplete loss of NFAT or contributions made by other compensatory pathways. Similar observations were made when a dominant-negative TCF was coexpressed with the CGA-Luciferase (32), suggesting that AP1 targets are partially responsible for GnRH mediated activation of Cga/CGA transcription. Collectively these data place Nfat4 in the gonadotrope transcriptional network and suggests other gene targets of AP1 may also require NFAT.
Fig. 7.

CGA-Luciferase activation downstream of GnRH requires Nfat4. LβT2 cells were transiently transfected with a scrambled sequence NS siRNA or siRNA specific for Nfat4 and a −1500/+45 CGA-Luciferase reporter construct. Seventy-two hours after transfection, LβT2 cells were treated with vehicle (PBS) or GnRH (10 nm) for 8 h. The cells were lysed, and the luciferase activity was measured. Data were normalized as described by Valcu and Valcu (65). Data shown are the mean ± sem for three independent experiments each performed in triplicate. Data were analyzed via a one-way ANOVA. ***, P < 0.001.
Discussion
GnRH pulses from the hypothalamus initiate several signaling pathways, including members of the MAPK signaling family, that culminate in the regulated expression of at least 76 genes in gonadotropes (4, 6, 18, 20, 21, 32, 43, 50). Previous work from our laboratory indicated that maximal levels of Jun expression in response to GnRH required a functional interaction between TCF7L2 and β-catenin. Alterations in the activity of TCF7L2 or levels of β-catenin also had a secondary impact on activity of the CGA promoter, a JUN/ATF3-dependent downstream target. In addition, the TCF7L2/β-catenin dependency of GnRH-regulated expression of Jun appeared to be unique to this IEG because hormone-regulated expression of Egr1 or Atf3 occurred independently of these two coactivators (32, 50). The work reported in this manuscript extends these findings by demonstrating that GnRH-regulated expression of Jun also requires JNK- and Ca2+-dependent regulation of calcineurin and its downstream target NFAT. Similar signaling pathways are also necessary for the regulation of TCF-dependent transcription (Supplemental Fig. 5), suggesting a point of convergence for Jun mRNA accumulation, and downstream targets including Cga/CGA. We posit that this complex signaling network allows gonadotropes to respond efficiently and exquisitely to the rise and fall of GnRH that occurs with each pulsatile burst of the neurohormone.
JNK has been shown to regulate NFAT-dependent transcription in a cell-specific manner, providing both positive and negative influences on gene transcription (39, 52, 53). GnRH activation of JNK takes approximately 30 min to reach its peak (12, 15, 54). Interestingly, JNK has also been shown to export NFAT4 from the nucleus in BHK cells (55), suggesting that JNK may also antagonize the function of NFAT4, depending on cellular context. In this regard, calcineurin activation downstream of GnRH regulates NFAT-dependent transcription and facilitates NFAT2 nuclear import and increased transcriptional activity (26, 27). Therefore, it is possible that GnRH stimulates nuclear import as demonstrated by Armstrong et al. (27), which is followed soon thereafter by export. This is intriguing because Jun/JUN responds to GnRH pulses, such that it needs to be rapidly up regulated and then down-regulated to be primed to respond to the next GnRH pulse. Consequently, JNK may be important for both import and export of NFAT, providing both positive and negative controls for Jun transcription. Further studies using immunocytochemistry to examine cellular localization of endogenous NFAT and the cellular mechanisms that control these events could provide more insight regarding their functional role in gonadotropes.
NFAT proteins have been shown to cooperate with JUN bound to AP1 sites on the promoters of several genes important for the immune response, including IL-2, IL-3, IL-5, and FasL as reviewed elsewhere (36, 44, 46). For example, maximal transcription of FasL involves calcineurin and activation of NFAT and AP1 (56, 57). Furthermore, cooperative interactions between protein kinase C (PKC)-θ and calcineurin have also been reported to induce JNK and subsequent activation of IL-2 in T cells (58). In addition, regulation of TNFα in pancreatic islet cells requires MAPK activity in addition to calcineurin (39). Thus, the ability of JUN and NFAT proteins to cooperate suggests that NFAT may also contribute to the transcriptional regulation of JUN-dependent genes in the gonadotrope including Cga, Gnrhr, and Fshb. This possibility is supported by our current studies indicating that NFAT4 is necessary for maximal accumulation of Jun, Fos, and Atf3 mRNA and the activity of an AP1-responsive Cga promoter-reporter in response to GnRH in gonadotropes (Figs. 6 and 7). Finally, NFAT proteins have also been shown to cooperate with GATA transcription factors (34), which are important in pituitary development (59, 60) and regulation of gonadotrope gene expression (61). Collectively these results point to a role for NFAT in the gonadotrope transcriptional network through the regulation of Jun, Fos, and Atf3 and suggest downstream targets including Cga/CGA or Fshb are also affected.
Our report that calcineurin is required for GnRH regulation of Jun and Atf3, and potentially Fos, is consistent with an early report indicating that the neurohormone regulates transcription of calcineurin in αT3 cells (22). Additional support for the role of calcineurin in regulation of AP1-dependent genes in gonadotropes comes from a report by Armstrong et al. (27), indicating that cyclosporin A reduced GnRH activation of reporter luciferase constructs for αGSU (human), rat LHβ, and FSHβ in αT3 cells. Calcineurin is also necessary for Fshb regulation through an association with HDAC (22). This interaction between calcineurin and HDAC represses Fshb, leading to differences in Lhb and Fshb transcriptional regulation (22), which is important, particularly during the LH surge. NFAT proteins have been shown to also interact with HDAC and repress gene transcription (35, 62), suggesting another potential role for NFAT proteins in gonadotropes.
Although our studies indicate that GnRH signals through Ca2+ mobilization and activation of calcineurin to regulate NFAT target genes including Jun and Atf3, and potentially Fos, it is also possible that GnRH regulates expression of AP1-dependent genes in gonadotropes through other Ca2+ targets. In this regard, Ely et al. (23) recently demonstrated that GnRH signals through CaMKII to regulate Fos-Luciferase, suggesting that the Fshb gene, an endogenous FOS target, is also affected. Surprisingly, however, we did not observe a reduction of endogenous Fos mRNA in the presence of KN93, a CaMKII specific inhibitor, suggesting that chromatin status may play a role in the ability of GnRH to activate IEG encoding genes.
Nfat4 reduction leads to a modest reduction in maximal accumulation of Jun, Fos, and Atf3 induced by GnRH presumably due to only partial reduction of NFAT4 protein (Fig. 6). In contrast, activity of the AP1-dependent Cga promoter was much more dramatically affected (48% reduction, Fig. 7). As discussed earlier, this is consistent with the notion that small changes in the concentration of bZIP proteins would be expected to have a much greater effect on the concentration of AP1 heterodimers if concentration of the monomers is below the Kd of the heterodimer. Nevertheless, although incomplete reduction of NFAT4 could explain the remaining activity of the CGA promoter reporter after siRNA treatment, it is also important to note that mice with isoform-specific NFAT knockout do not have overt phenotypes; rather, compound knockouts are required to see an immune phenotype (33, 35, 48). Indeed, the redundancy of the NFAT in immune cell function underscores their physiological significance. Similarly, the presence of multiple Nfat family members in LβT2 cells (Fig. 4A) suggests that compensatory mechanisms may also underlie their function in gonadotropes to ensure that AP1-dependent genes like Cga remain responsive to GnRH.
While this manuscript was in submission, another study reported that calcineurin mediates GnRH-regulated expression of Cga and Fshb in αT3 and LβT2 cells via two downstream targets, NFAT3 and transducer of regulated cAMP response element-binding protein (CREB) activity (TORC1, officially CREB regulated transcription coactivator) (63). Interestingly, these authors postulate that NFAT3 exerts its affect by binding directly to the Cga promoter in a region known to be required for GnRH responsiveness (63). They further suggest that TORC enhances the response to GnRH through a functional interaction with NFAT3. In contrast to Cga, calcineurin activated NFAT3 appears to mediate GnRH-regulated transcription of Fshb through an indirect mechanism that includes another NFAT responsive IEG, Nur77 (63). Although NUR77 interacts synergistically with TORC and CREB to increase activity of the Fshb promoter, the authors suggest that these interactions are more important for basal rather than GnRH-stimulated activity of the promoter (63). Thus, this study, along with our current findings, underscores the importance of calcineurin and NFAT in mediating GnRH regulation of gonadotropin gene expression. These combined results also raise the intriguing possibility that NFAT mediates and amplifies the GnRH signal by binding to the promoters of Jun, Atf3, and potentially Fos as well as to the promoter of their distal target, namely Cga.
Our current study, along with several previous studies (1, 4, 6, 23, 32, 63), provide a growing body of data indicating that the four signature genes of gonadotropes, Cga, Fshb, Gnrhr, and Lhb, respond to GnRH indirectly via regulated expression of distinct IEGs. Regulated expression of Lhb is dependent on Egr1 (5, 64) and shown here to be independent of GnRH induced changes in Ca2+-dependent activation of calcineurin. In contrast, GnRH regulated expression of Cga, Fshb, and Gnrhr requires bZIP family members that include JUN, FOS, and ATF3 (reviewed in Refs. 3 and 6). Previously we reported that GnRH-regulated expression of Jun requires the binding of a TCF/β-catenin complex (32) (modeled in Fig. 8). Herein we demonstrate that TCF/β-catenin transcriptional activity mediated by GnRH is also dependent on calcineurin and JNK. It remains to be determined whether GnRH-regulated expression of Atf3 also requires TCF/β-catenin. Calcineurin and JNK also mediate GnRH regulated expression of Jun and Atf3. Likely targets of JNK on the Jun promoter include JUN and the TCF/β-catenin complex. Targets of calcineurin on the Jun promoter include NFAT and the TCF/β-catenin complex.
Fig. 8.

Ca2+, calcineurin, and NFAT mediate GnRH-regulated expression of Jun, Atf3, and the downstream target Cga. Calcineurin and JNK mediate GnRH-regulated expression of Jun, Atf3, and the downstream target Cga through several transcription factors including NFAT, JUN, and TCF/β-catenin. β-CAT, β-Catenin; CaM, calmodulin; CaMK, calmodulin kinase; CaMKK, calmodulin kinase kinase; DAG, diacylglycerol; EGR1, early growth response protein 1; IP3, inositol trisphospate; PKC, protein kinase C; PLC, phospholipase C.
Based on previously published reports (36, 38), we suspect that NFAT binds to the Jun promoter and interacts cooperatively with JUN though AP1 sites. Furthermore, we hypothesize that the TCF/β-catenin complex may also interact cooperatively with NFAT. With respect to the Atf3 promoter, it remains to be determined whether NFAT interacts with EGR1, either directly by binding DNA or indirectly, to augment activity of the promoter. The target of JNK on the Atf3 promoter also remains to be determined. The proximal promoter of CGA is the ultimate target of GnRH induced increases in JUN, FOS, and ATF3. As mentioned above, the recent report of Pnueli et al. (63) suggests that NFAT may also bind directly to the Cga promoter, establishing a second site of action for this transcription factor. Although complex, these GnRH-dependent pathways should allow for exquisite control of gonadotropin gene expression, which is necessary for maintaining reproductive fitness.
Materials and Methods
Chemicals
GnRH, DMSO, BAPTA-AM, thapsigargin, and PBS were purchased from Sigma Chemical Company (St. Louis, MO); TRIzol, Lipofectamine, Lipofectamine 2000, and OPTI-MEM were purchased from Invitrogen Life Technologies (Carlsbad, CA); the p38 MAPK inhibitor SB203580, CamKII inhibitor KN93/KN92, CAMKK inhibitor STO609, and cyclosporin A were purchased from Calbiochem (EMD Biosciences Inc., La Jolla, CA); the ERK inhibitor PD98059 was purchased from Alexis Biochemicals (San Diego, CA), and the JNK inhibitor SP600125 was purchased from Biosource International Inc. (Camarillo, CA).
DNA constructs, cell culture, and transient transfections
The NFATΔNEO-Luc construct was kindly provided by the laboratory of Dr. Neil A. Clipstone (Loyola University Medical School, Chicago, IL). The construct was modified by removal of the neocassette from the NFAT-luciferase described previously (49). The TCF/LEF luciferase reporter construct (TOPflash) was purchased from Upstate Biotechnology (Lake Placid, NY).
LβT2 cells (provided by P. Mellon, University of California, San Diego, San Diego, CA) were maintained at 37 C with 5% CO2 in high-glucose DMEM supplemented with 10% fetal-bovine serum (FBS) and 1% penicillin-streptomycin (Invitrogen Life Technologies) for the duration of experiments. For transient transfection of TOPflash (Upstate Biotechnology) and NFAT-luciferase reporters, LβT2 cells were plated (250,000 cells/well in a 24 well plate) in complete media for 24 h. A transfection mixture containing DNA, Lipofectamine (Invitrogen Life Technologies; 1.6 μl/well), and DMEM was incubated at room temperature for 30 min to allow liposomes to form. The mixture was then added to cells, which had been washed once with PBS. After overnight incubation (12–16 h), vehicle or inhibitor was added to the cells (in DMEM/1% penicillin-streptomycin) for 60 min followed by treatment with PBS or GnRH (100 nm) for 8 h. Cells were maintained in the presence of the inhibitor listed for the duration of the experiment. After 8 h, cells were washed with PBS and lysed, and the activity of the reporter was assayed (single luciferase reporter assay kit; Promega Corp., Madison WI).
The human CGA-Luciferase promoter (−1500/+45 bp) plasmid has been described previously (51). For these experiments, LβT2 cells (500,000/well) were plated in DMEM with 10% FBS the day before transfection. A transfection cocktail containing 1 μl Lipofectamine 2000 (Invitrogen Life Technologies), 5 pmol siRNA, and 200 ng reporter vector in OPTI-MEM (Invitrogen Life Technologies; 100 μl final volume per reaction). The transfection cocktail was added to each well containing 500 μl media. The media were changed 24 h later. Cells were serum starved for 12–15 h and then treated with vehicle (PBS) or GnRH (10 nm) for 8 h at 72 h after transfection. Cells were then lysed and activity of the reporter was assayed (single luciferase reporter assay kit; Promega).
For examination of RNA, LβT2 cells were plated (500,000 cells/well, 35 mm plates) and incubated in complete media overnight. Twenty-four hours after plating, the cells were serum starved overnight (12–16 h), followed by pretreatment with the inhibitor for 60 min. Inhibitor concentrations were determined following a dose curve (data not shown) or from published reports in the same cell types. Cells were then treated with PBS or 10 nm GnRH for 1 h in the presence of vehicle or inhibitor before washing with PBS and adding 1 ml TRIzol (Invitrogen Life Technologies).
RNA isolation and cDNA synthesis
Total RNA was isolated from the cells using TRIzol (Invitrogen Life Technologies) following the manufacturer's protocol and resuspended with diethylpyrocarbonate-treated water. Half a microgram of total RNA was used to synthesize cDNA using 4 μl Quanta cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD) in a total volume of 20-μl reactions (total volume) following the manufacturer's protocol. cDNA samples were diluted 1:10 in diethylpyrocarbonate-treated water and stored at −20 C.
Semiquantitative PCR
To investigate the expression of the Nfat family members, standard PCR was performed using 5 μl of diluted cDNA and primers specific for Nfat1 (forward, 5′-GTGGCCACTCTCCAATCAGT-3′ and reverse, 5′-TCATCTGCTGTCCCAATGAA-3′) or Nfat2, Nfat3, and Nfat4 as described by (62). Standard PCR methods were followed and run for 35 cycles. For each PCR reaction, a master mix containing AmpliTaq DNA polymerase with Buffer II, MgCl2, and 10 mm deoxynucleotide triphosphates (Applied Biosystems, Foster City, CA) was mixed with primers and added to 5 μl of diluted cDNA. The final volume of a RT-PCR reaction was 20 μl. RT-PCR reaction conditions for Nfat1, Nfat2, and Nfat3 were: 94 C for 2 min, followed by 35 cycles of 95 C for 15 sec, 63 C for 30 sec, and 72 C for 45 sec followed by a final extension of 72 C for 5 min. The cycling was similar for Nfat4, with the exception that the annealing temperature was 70 C rather than 63 C. The samples were then run on a 1.5% agarose gel and bands were visualized by UV light.
Western blot
Whole-cell lysates were collected from LβT2 cells and protein concentrations were determined as previously described (32). Proteins were then separated on a 12% polyacrylamide sodium dodecyl sulfate (SDS) gel and transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories, Hercules, CA) in Towbin buffer. The membranes were blocked in 5% nonfat milk in Tris-buffered saline (pH 8.0) and then incubated overnight at 4 C in antibodies specific for NFAT4 (Santa Cruz Biotechnologies, Santa Cruz, CA) or tubulin (Abcam, Cambridge, MA). Membranes were rinsed three times with Tris-buffered saline and incubated in horseradish peroxidase-conjugated secondary antibody (Pierce, Rockford, IL) at room temperature for 1 h. The membrane was rinsed three times followed by detection of antigen-antibody complexes by chemiluminesence (Immobilon chemiluminescence horseradish peroxidase substrate; Millipore, Billerica, MA). Chemiluminescence was visualized on a LAS4000 (FUJIFILM Medical Systems, Stamford, CT) and quantitated.
Quantitative real-time PCR
Relative mRNA levels were determined using qRT-PCR and the ABI Prism 7500 FAST real-time PCR system using version 2.0.3 software (Applied Biosystems). For each qRT-PCR reaction, a master mix containing 10 μl of Fast SYBR Green master mix (Applied Biosystems), and primers were added to 5 μl of cDNA. The final volume of a qRT-PCR reaction was 20 μl and samples were performed in duplicate. qRT-PCR reaction conditions were: 50 C for 2 min and 95 C for 10 min; then 40 cycles of 95 C for 3 sec and 60 C for 30 sec. Primer sequences were as follows: Jun (forward, 5′-AATGGGCACATCACCACTACAC-3′, and reverse, 5′-TGCTCGTCGGTCACGTTCT-3′); Fos (forward, 5′-GAGGAGGGAGCTGACAGATACACT-3′, and reverse, 5′-TGCAACGCAGACTTCTCATCTT-3′); Egr1 (forward, 5′-GGGAGCCGAGCGAACAA-3′, and reverse, 5′-TCAGAGCGATGTCAGAAAAGGA-3′). Primers specific for Atf3 and cyclophilin B (to normalize cDNA samples) were used as previously described (32).
siRNA transfection of LβT2 cells
LβT2 cells (4 × 106) were suspended in 100 μl SG cell line solution (Lonza, Inc., Walkersville, MD) containing 30 pmol of nonspecific siRNA or Nfat4 siRNA (Ambion, Austin, TX) and transfected on a Nucleofector 4D (Lonza) using CA137 pulse program. DMEM with 10% FBS (400 μl) was added to each cuvette and then split between two wells of a six-well plate containing 2 ml of DMEM with 10% FBS. Cells were cultured for 72 h, with the final 12–16 h in the absence of serum and then treated for 1 h with either vehicle (PBS) or GnRH (100 nm). Cells were washed with PBS, and 1 ml TRIzol (Invitrogen Life Technologies) was added and RNA isolated. qRT-PCR was performed using primers specific for Jun, Fos, Egr1, and Atf3 (32) as described above. The following primers were used for detection of Nfat4 (forward, 5′-CCATTGGTCTACAGGATATCACCTTA-3′, and reverse, 5′-CCGTGGCTTGGGAAACAG-3′); Nfat3 (forward, 5′-CAACTTCCTGCCAGACTCTAAAGTG-3′, and reverse, 5′-ACACCCGCTTGTTGCTGTACT-3′); and Gapdh (forward, 5′-TGTGTCCGTCGTGGATCTGA-3′, and reverse, 5′-CCTGCTTCACCACCTTCTTGA-3′) (to normalize cDNA samples).
Statistics
Individual experiments assessing IEG mRNA levels were repeated on at least 3 separate days. Collected samples were assayed in a single qRT-PCR assay using technical duplicates. Reporter assays were performed in triplicate and then repeated two (SP600125) or three times on separate days. Data were normalized as described by Valcu and Valcu (65) and assessed using Prism GraphPad software (GraphPad Inc., San Diego, CA). For the experiments analyzed by one-way ANOVA with Tukey's post hoc analysis (Figs. 1–5 and 7), all 2-ΔΔCt values (66) of control and treated samples were divided by the mean of the control sample. The GnRH-dependent fold differences in Fig. 6 were determined by normalization to their respective PBS controls for either NS siRNA or Nfat siRNA within each experiment. The means of these fold differences were then analyzed by a two-tailed unpaired t test. P values for each experiment are listed in the figure legends.
Supplementary Material
Acknowledgments
We thank Drs. Amelia Karlsson and Neil Clipstone for the NFAT-Luc construct used in this study and Dr. Mary Hunzicker-Dunn for thoughtful discussion. Additionally, we thank Veronica Windell for her help during a laboratory rotation. Finally, we acknowledge Derek Pouchnik and the facilities at the Washington State Core Laboratory for Bioanalysis and Biotechnology for assistance with qRT-PCR.
This work was supported by the National Institutes of Health Grant R01 HD055776 (to J.H.N).
Current Address for A.K.B.: National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina 27709.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AP1
- Activator protein-1
- BAPTA-AM
- 1, 2-bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl)-ester
- bZIP
- basic leucine zipper domain
- CaMKII
- calmodulin kinase type II
- CaMKK
- calmodulin kinase kinase
- CREB
- cAMP response element-binding protein
- DMSO
- dimethyl sulfoxide
- FBS
- fetal bovine serum
- HDAC
- histone deacetylase
- IEG
- immediate early gene
- JNK
- c-jun N-terminal kinase
- Kd
- dissociation constant
- LEF
- lymphoid enhancer factor
- NFAT2
- nuclear factor of T cells 2
- NS siRNA
- nonspecific scrambled siRNA
- PKC
- protein kinase C
- qRT-PCR
- quantitative RT-PCR
- SDS
- sodium dodecyl sulfate
- siRNA
- silent interfering RNA
- TCF
- T cell factor
- VGCC
- voltage-gated calcium channel.
References
- 1. Jorgensen JS, Quirk CC, Nilson JH. 2004. Multiple and overlapping combinatorial codes orchestrate hormonal responsiveness and dictate cell-specific expression of the genes encoding luteinizing hormone. Endocr Rev 25:521–542 [DOI] [PubMed] [Google Scholar]
- 2. Gharib SD, Wierman ME, Shupnik MA, Chin WW. 1990. Molecular biology of the pituitary gonadotropins. Endocr Rev 11:177–199 [DOI] [PubMed] [Google Scholar]
- 3. Bliss SP, Navratil AM, Xie J, Roberson MS. 2010. GnRH signaling, the gonadotrope and endocrine control of fertility. Front Neuroendocrinol 31:322–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thackray VG, Mellon PL, Coss D. 2010. Hormones in synergy: regulation of the pituitary gonadotropin genes. Mol Cell Endocrinol 314:192–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salisbury TB, Binder AK, Grammer JC, Nilson JH. 2007. Maximal activity of the luteinizing hormone β-subunit gene requires β-catenin. Mol Endocrinol 21:963–971 [DOI] [PubMed] [Google Scholar]
- 6. Salisbury TB, Binder AK, Nilson JH. 2008. Welcoming beta-catenin to the gonadotropin-releasing hormone transcriptional network in gonadotropes. Mol Endocrinol 22:1295–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gardner S, Pawson AJ. 2009. Emerging targets of the GnRH receptor: novel interactions with Wnt signalling mediators. Neuroendocrinology 89:241–251 [DOI] [PubMed] [Google Scholar]
- 8. Naor Z. 2009. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol 30:10–29 [DOI] [PubMed] [Google Scholar]
- 9. Ferris HA, Shupnik MA. 2006. Mechanisms for pulsatile regulation of the gonadotropin subunit genes by GNRH1. Biol Reprod 74:993–998 [DOI] [PubMed] [Google Scholar]
- 10. Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, Naor Z. 2004. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone β-subunit promoter. Endocrinology 145:2228–2244 [DOI] [PubMed] [Google Scholar]
- 11. Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z. 2002. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHβ-subunit promoter. Endocrinology 143:1018–1025 [DOI] [PubMed] [Google Scholar]
- 12. Harris D, Chuderland D, Bonfil D, Kraus S, Seger R, Naor Z. 2003. Extracellular signal-regulated kinase and c-Src, but not Jun N-terminal kinase, are involved in basal and gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone α-subunit promoter. Endocrinology 144:612–622 [DOI] [PubMed] [Google Scholar]
- 13. Haisenleder DJ, Burger LL, Walsh HE, Stevens J, Aylor KW, Shupnik MA, Marshall JC. 2008. Pulsatile gonadotropin-releasing hormone stimulation of gonadotropin subunit transcription in rat pituitaries: evidence for the involvement of Jun N-terminal kinase but not p38. Endocrinology 149:139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kanasaki H, Bedecarrats GY, Kam KY, Xu S, Kaiser UB. 2005. Gonadotropin-releasing hormone pulse frequency-dependent activation of extracellular signal-regulated kinase pathways in perifused LβT2 cells. Endocrinology 146:5503–5513 [DOI] [PubMed] [Google Scholar]
- 15. Roberson MS, Zhang T, Li HL, Mulvaney JM. 1999. Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology 140:1310–1318 [DOI] [PubMed] [Google Scholar]
- 16. Mulvaney JM, Roberson MS. 2000. Divergent signaling pathways requiring discrete calcium signals mediate concurrent activation of two mitogen-activated protein kinases by gonadotropin-releasing hormone. J Biol Chem 275:14182–14189 [DOI] [PubMed] [Google Scholar]
- 17. Mulvaney JM, Zhang T, Fewtrell C, Roberson MS. 1999. Calcium influx through L-type channels is required for selective activation of extracellular signal-regulated kinase by gonadotropin-releasing hormone. J Biol Chem 274:29796–29804 [DOI] [PubMed] [Google Scholar]
- 18. Weck J, Anderson AC, Jenkins S, Fallest PC, Shupnik MA. 2000. Divergent and composite gonadotropin-releasing hormone-responsive elements in the rat luteinizing hormone subunit genes. Mol Endocrinol 14:472–485 [DOI] [PubMed] [Google Scholar]
- 19. Weck J, Fallest PC, Pitt LK, Shupnik MA. 1998. Differential gonadotropin-releasing hormone stimulation of rat luteinizing hormone subunit gene transcription by calcium influx and mitogen-activated protein kinase-signaling pathways. Mol Endocrinol 12:451–457 [DOI] [PubMed] [Google Scholar]
- 20. Haisenleder DJ, Ferris HA, Shupnik MA. 2003. The calcium component of gonadotropin-releasing hormone-stimulated luteinizing hormone subunit gene transcription is mediated by calcium/calmodulin-dependent protein kinase type II. Endocrinology 144:2409–2416 [DOI] [PubMed] [Google Scholar]
- 21. Haisenleder DJ, Burger LL, Aylor KW, Dalkin AC, Marshall JC. 2003. Gonadotropin-releasing hormone stimulation of gonadotropin subunit transcription: evidence for the involvement of calcium/calmodulin-dependent kinase II (Ca/CAMK II) activation in rat pituitaries. Endocrinology 144:2768–2774 [DOI] [PubMed] [Google Scholar]
- 22. Lim S, Luo M, Koh M, Yang M, bin Abdul Kadir MN, Tan JH, Ye Z, Wang W, Melamed P. 2007. Distinct mechanisms involving diverse histone deacetylases repress expression of the two gonadotropin β-subunit genes in immature gonadotropes, and their actions are overcome by gonadotropin-releasing hormone. Mol Cell Biol 27:4105–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ely HA, Mellon PL, Coss D. 2011. GnRH induces the c-Fos gene via phosphorylation of SRF by the calcium/calmodulin kinase II pathway. Mol Endocrinol 25:669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mayer SI, Dexheimer V, Nishida E, Kitajima S, Thiel G. 2008. Expression of the transcriptional repressor ATF3 in gonadotrophs is regulated by Egr-1, CREB, and ATF2 after gonadotropin-releasing hormone receptor stimulation. Endocrinology 149:6311–6325 [DOI] [PubMed] [Google Scholar]
- 25. Natarajan K, Ness J, Wooge CH, Janovick JA, Conn PM. 1991. Specific identification and subcellular localization of three calmodulin-binding proteins in the rat gonadotrope: spectrin, caldesmon, and calcineurin. Biol Reprod 44:43–52 [DOI] [PubMed] [Google Scholar]
- 26. Armstrong SP, Caunt CJ, Finch AR, McArdle CA. 2011. Using automated imaging to interrogate gonadotrophin-releasing hormone receptor trafficking and function. Mol Cell Endocrinol 331:194–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Armstrong SP, Caunt CJ, Fowkes RC, Tsaneva-Atanasova K, McArdle CA. 2009. Pulsatile and sustained gonadotropin-releasing hormone (GnRH) receptor signaling: does the Ca2+/NFAT signaling pathway decode GnRH pulse frequency? J Biol Chem 284:35746–35757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu F, Austin DA, Mellon PL, Olefsky JM, Webster NJ. 2002. GnRH activates ERK1/2 leading to the induction of c-fos and LHβ protein expression in LβT2 cells. Mol Endocrinol 16:419–434 [DOI] [PubMed] [Google Scholar]
- 29. Liu F, Austin DA, Webster NJ. 2003. Gonadotropin-releasing hormone-desensitized LβT2 gonadotrope cells are refractory to acute protein kinase C, cyclic AMP, and calcium-dependent signaling. Endocrinology 144:4354–4365 [DOI] [PubMed] [Google Scholar]
- 30. Roberson MS, Bliss SP, Xie J, Navratil AM, Farmerie TA, Wolfe MW, Clay CM. 2005. Gonadotropin-releasing hormone induction of extracellular-signal regulated kinase is blocked by inhibition of calmodulin. Mol Endocrinol 19:2412–2423 [DOI] [PubMed] [Google Scholar]
- 31. Burger LL, Haisenleder DJ, Aylor KW, Marshall JC. 2009. Regulation of Lhb and Egr1 gene expression by GNRH pulses in rat pituitaries is both c-Jun N-terminal kinase (JNK)- and extracellular signal-regulated kinase (ERK)-dependent. Biol Reprod 81:1206–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Salisbury TB, Binder AK, Grammer JC, Nilson JH. 2009. GnRH-regulated expression of Jun and JUN target genes in gonadotropes requires a functional interaction between TCF/LEF family members and β-catenin. Mol Endocrinol 23:402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Macian F. 2005. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5:472–484 [DOI] [PubMed] [Google Scholar]
- 34. Hogan PG, Chen L, Nardone J, Rao A. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17:2205–2232 [DOI] [PubMed] [Google Scholar]
- 35. Im SH, Rao A. 2004. Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol Cells 18:1–9 [PubMed] [Google Scholar]
- 36. Macían F, López-Rodríguez C, Rao A. 2001. Partners in transcription: NFAT and AP-1. Oncogene 20:2476–2489 [DOI] [PubMed] [Google Scholar]
- 37. Soto-Nieves N, Puga I, Abe BT, Bandyopadhyay S, Baine I, Rao A, Macian F. 2009. Transcriptional complexes formed by NFAT dimers regulate the induction of T cell tolerance. J Exp Med 206:867–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nguyen TN, Kim LJ, Walters RD, Drullinger LF, Lively TN, Kugel JF, Goodrich JA. 2010. The C-terminal region of human NFATc2 binds cJun to synergistically activate interleukin-2 transcription. Mol Immunol 47:2314–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lawrence MC, Naziruddin B, Levy MF, Jackson A, McGlynn K. 2011. Calcineurin/nuclear factor of activated T cells and MAPK signaling induce TNF-α gene expression in pancreatic islet endocrine cells. J Biol Chem 286:1025–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Angel P, Hattori K, Smeal T, Karin M. 1988. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 55:875–885 [DOI] [PubMed] [Google Scholar]
- 41. Dobkin-Bekman M, Naidich M, Pawson AJ, Millar RP, Seger R, Naor Z. 2006. Activation of mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol Cell Endocrinol 252:184–190 [DOI] [PubMed] [Google Scholar]
- 42. Maudsley S, Naor Z, Bonfil D, Davidson L, Karali D, Pawson AJ, Larder R, Pope C, Nelson N, Millar RP, Brown P. 2007. Proline-rich tyrosine kinase 2 mediates gonadotropin-releasing hormone signaling to a specific extracellularly regulated kinase-sensitive transcriptional locus in the luteinizing hormone β-subunit gene. Mol Endocrinol 21:1216–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Melamed P. 2008. Histone deacetylases and repression of the gonadotropin genes. Trends Endocrinol Metab 19:25–31 [DOI] [PubMed] [Google Scholar]
- 44. Oh-hora M, Rao A. 2009. The calcium/NFAT pathway: role in development and function of regulatory T cells. Microbes Infect 11:612–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rao A. 2009. Signaling to gene expression: calcium, calcineurin and NFAT. Nat Immunol 10:3–5 [DOI] [PubMed] [Google Scholar]
- 46. Rao A, Luo C, Hogan PG. 1997. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15:707–747 [DOI] [PubMed] [Google Scholar]
- 47. Vihma H, Pruunsild P, Timmusk T. 2008. Alternative splicing and expression of human and mouse NFAT genes. Genomics 92:279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mancini M, Toker A. 2009. NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer 9:810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Helms WS, Jeffrey JL, Holmes DA, Townsend MB, Clipstone NA, Su L. 2007. Modulation of NFAT-dependent gene expression by the RhoA signaling pathway in T cells. J Leukoc Biol 82:361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xie J, Bliss SP, Nett TM, Ebersole BJ, Sealfon SC, Roberson MS. 2005. Transcript profiling of immediate early genes reveals a unique role for activating transcription factor 3 in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol Endocrinol 19:2624–2638 [DOI] [PubMed] [Google Scholar]
- 51. Heckert LL, Schultz K, Nilson JH. 1996. The cAMP response elements of the α subunit gene bind similar proteins in trophoblasts and gonadotropes but have distinct functional sequence requirements. J Biol Chem 271:31650–31656 [DOI] [PubMed] [Google Scholar]
- 52. Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD. 2003. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J 22:5079–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Molkentin JD. 2004. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res 63:467–475 [DOI] [PubMed] [Google Scholar]
- 54. Yokoi T, Ohmichi M, Tasaka K, Kimura A, Kanda Y, Hayakawa J, Tahara M, Hisamoto K, Kurachi H, Murata Y. 2000. Activation of the luteinizing hormone β promoter by gonadotropin-releasing hormone requires c-Jun NH2-terminal protein kinase. J Biol Chem 275:21639–21647 [DOI] [PubMed] [Google Scholar]
- 55. Chow CW, Rincón M, Cavanagh J, Dickens M, Davis RJ. 1997. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science 278:1638–1641 [DOI] [PubMed] [Google Scholar]
- 56. Jayanthi S, Deng X, Ladenheim B, McCoy MT, Cluster A, Cai NS, Cadet JL. 2005. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc Natl Acad Sci USA 102:868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holtz-Heppelmann CJ, Algeciras A, Badley AD, Paya CV. 1998. Transcriptional regulation of the human FasL promoter-enhancer region. J Biol Chem 273:4416–4423 [DOI] [PubMed] [Google Scholar]
- 58. Werlen G, Jacinto E, Xia Y, Karin M. 1998. Calcineurin preferentially synergizes with PKC-θ to activate JNK and IL-2 promoter in T lymphocytes. EMBO J 17:3101–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kerr J, Wood W, Ridgway EC. 2008. Basic science and clinical research advances in the pituitary transcription factors: Pit-1 and Prop-1. Curr Opin Endocrinol Diabetes Obes 15:359–363 [DOI] [PubMed] [Google Scholar]
- 60. Rosenfeld MG, Briata P, Dasen J, Gleiberman AS, Kioussi C, Lin C, O'Connell SM, Ryan A, Szeto DP, Treier M. 2000. Multistep signaling and transcriptional requirements for pituitary organogenesis in vivo. Recent Prog Horm Res 55:1–13; discussion 13–14 (Review) [PubMed] [Google Scholar]
- 61. Fowkes RC, King P, Burrin JM. 2002. Regulation of human glycoprotein hormone alpha-subunit gene transcription in LβT2 gonadotropes by protein kinase C and extracellular signal-regulated kinase 1/2. Biol Reprod 67:725–734 [DOI] [PubMed] [Google Scholar]
- 62. Ranger AM, Gerstenfeld LC, Wang J, Kon T, Bae H, Gravallese EM, Glimcher MJ, Glimcher LH. 2000. The nuclear factor of activated T cells (NFAT) transcription factor NFATp (NFATc2) is a repressor of chondrogenesis. J Exp Med 191:9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pnueli L, Luo M, Wang S, Naor Z, Melamed P. 2011. Calcineurin Mediates the Gonadotropin-Releasing Hormone Effect on Expression of Both Subunits of the Follicle-Stimulating Hormone through Distinct mechanisms. Mol Cell Biol 31:5023–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wolfe MW, Call GB. 1999. Early growth response protein 1 binds to the luteinizing hormone-β promoter and mediates gonadotropin-releasing hormone-stimulated gene expression. Mol Endocrinol 13:752–763 [DOI] [PubMed] [Google Scholar]
- 65. Valcu M, Valcu CM. 2011. Data transformation practices in biomedical sciences. Nat Methods 8:104–105 [DOI] [PubMed] [Google Scholar]
- 66. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.