

Abstract
Recently, we have identified serum response factor (SRF) as a mediator of clinically relevant androgen receptor (AR) action in prostate cancer (PCa). Genes that rely on SRF for androgen responsiveness represent a small fraction of androgen-regulated genes, but distinguish benign from malignant prostate, correlate with aggressive disease, and are associated with biochemical recurrence. Thus, understanding the mechanism(s) by which SRF conveys androgen regulation to its target genes may provide novel opportunities to target clinically relevant androgen signaling. Here, we show that the small GTPase ras homolog family member A (RhoA) mediates androgen-responsiveness of more than half of SRF target genes. Interference with expression of RhoA, activity of the RhoA effector Rho-associated coiled-coil containing protein kinase 1 (ROCK), and actin polymerization necessary for nuclear translocation of the SRF cofactor megakaryocytic acute leukemia (MAL) prevented full androgen regulation of SRF target genes. Androgen treatment induced RhoA activation, increased the nuclear content of MAL, and led to MAL recruitment to the promoter of the SRF target gene FHL2. In clinical specimens RhoA expression was higher in PCa cells than benign prostate cells, and elevated RhoA expression levels were associated with aggressive disease features and decreased disease-free survival after radical prostatectomy. Overexpression of RhoA markedly increased the androgen-responsiveness of select SRF target genes, in a manner that depends on its GTPase activity. The use of isogenic cell lines and a xenograft model that mimics the transition from androgen-stimulated to castration-recurrent PCa indicated that RhoA levels are not altered during disease progression, suggesting that RhoA expression levels in the primary tumor determine disease aggressiveness. Androgen-responsiveness of SRF target genes in castration-recurrent PCa cells continued to rely on AR, RhoA, SRF, and MAL and the presence of intact SRF binding sites. Silencing of RhoA, use of Rho-associated coiled-coil containing protein kinase 1 inhibitors, or an inhibitor of SRF-MAL interaction attenuated (androgen-regulated) cell viability and blunted PCa cell migration. Taken together, these studies demonstrate that the RhoA signaling axis mediates clinically relevant AR action in PCa.
Prostate cancer (PCa) remains the most frequently diagnosed cancer of an internal organ and the second leading cause of cancer-related death in Western men. In 2012, estimates indicate that 241,740 men will be diagnosed with PCa and 28,170 men will succumb to this disease in the United States alone (1). Standard treatment for localized disease consists of surgery or radiation therapy (2). Patients who present with locally advanced or metastatic PCa or whose cancer recurs after initial therapy have limited treatment options and are routinely treated with androgen deprivation therapies (ADTs) (3). This therapeutic approach exploits the well-known reliance of PCa cells on androgens for proliferation and survival and leads to an overall decrease in tumor burden and a favorable clinical response. Androgen deprivation, however, does not cure PCa and, in the vast majority of patients, disease eventually reappears as castration-recurrent PCa (CRPC), which is invariably fatal. Inappropriate activation of the androgen receptor (AR) is essential for proliferation of CRPC cells (4–7). The latter insight has led to the development of novel androgen deprivation approaches that demonstrate antitumor activity in a substantial subset of CRPC patients (8–10). However, effects are partial and temporary, and AR remains activated in CRPC which fails second-line ADT (11, 12). Thus, AR activity is important throughout PCa progression but is not inhibited fully by available ADT.
The AR is a ligand-dependent transcription factor that binds to regulatory regions known as androgen response elements (AREs) in or near target genes to control their transcription (13). Despite the continued reliance of PCa cells on AR signaling, the AR-dependent events that drive and support disease progression remain elusive. By preventing the biosynthesis of androgens or the binding of androgens to AR, traditional and novel ADTs target all androgen-dependent gene expression in PCa. However, a significant number of AR target genes may not have an impact on, or exert mitigating effects on disease progression. Therapies directed against androgen-dependent events that are responsible for clinical progression are likely to be more effective.
Systems biology approaches indicate that the manner in which AR regulates effector genes varies considerably and in many cases involves action of secondary transcription factors (TFs) (14–19). Recently, we have isolated a novel mechanism of androgen action in which androgen regulation of effector genes is independent of AR-ARE interaction but mediated by action of the TF serum response factor (SRF) on its consensus binding motif, a CArG box (20, 21). This mode of androgen action governs the expression of less than 6% of androgen-responsive genes and is associated with functions in cell cycle, cell movement, and cell morphology, all of which are relevant to cancer (21). Moreover, in patient specimens, the SRF- and androgen-dependent gene signature is sufficient to distinguish benign from malignant prostate specimens and correlates with aggressive disease. Compared with similarly sized sets of AR target genes or random gene sets, the AR- and SRF-dependent gene expression profile is associated robustly with disease progression and biochemical failure. In addition, expression of AR- and SRF-dependent genes is enriched in PCa cells, whereas ARE-driven genes are expressed more strongly in benign prostate (21).
In view of the clinical significance of this distinct mode of AR action, the molecular mechanism(s) by which AR imparts androgen regulation to SRF target genes warrants further exploration. In the classical model of SRF action, SRF is bound constitutively to CArG boxes in target genes, where its activity is regulated by signaling cascades and/or by interaction with one or more of its cofactors (22). Because SRF conveys androgen regulation to a small fraction of androgen-responsive genes, and the number of androgen- and SRF-responsive genes (159) represents only a subset (∼12%) of SRF target genes (21), we hypothesized that androgen regulation of SRF target genes may reflect androgen control over select SRF-activating pathways and/or SRF cofactors.
The RhoA/actin/megakaryocytic acute leukemia (MAL) signaling axis is a well-known activating pathway upstream of SRF (22, 23). Extracellular stimuli induce RhoA GTPase activity, which engages RhoA effectors. Activated RhoA effectors then facilitate actin polymerization, leading to the release of the SRF coactivator (MAL) from monomeric actin. Liberated MAL translocates from the cytoplasm to the nucleus, where it regulates SRF-mediated transcription. Here, we show that androgen stimulation activates RhoA, which conveys androgen responsiveness to a subset of SRF target genes, and present evidence that RhoA is overexpressed in clinical PCa specimens, where it is associated with aggressive disease features and poor outcome after surgical intervention.
Taken together, our work identifies the RhoA signaling axis, which harbors drugable targets, as a mediator of clinically relevant androgen action in PCa.
Materials and Methods
Cell culture
LNCaP, LN-Rf, and C4–2 cells were obtained and cultured as described previously (21, 24). Behavior of cells was monitored throughout the study by assessing overall androgen-responsiveness, morphology, and transcriptional regulation, which were consistent with previous observations for these cell lines.
Reagents
Methyltrienolone (R1881) was purchased from DuPont (Boston, MA). Antibodies were purchased as follows: RhoA and SRF from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); β-actin from Cell Signaling (Beverly, MA); MAL from Novus Biologicals (Littleton, CO); and FLAG tag from Sigma-Aldrich (St.Louis, MO). The antibody directed against FHL2 was a kind gift from Dr. Roland Schule. Y-27632, CCG-1423, hydroxyfasudil, latrunculin B, and phorbol 12-myristate 13-acetate were obtained from Enzo Life Sciences (Plymouth Meeting, PA). siGenome SmartPools targeting RhoA, MAL, SRF, DGAT2, CYR61, WFS1, SLITRK6, RAB3IP, and AR and nontargeting control SmartPools were purchased from Thermo-Scientific (Lafayette, CO). Geneticin was obtained from Invitrogen (Carlsbad, CA).
Generation of stably transfected LNCaP cell lines
LNCaP cells were seeded at a density of 1 × 106 cells in 10-cm dishes in medium without antibiotics. The next day, cells were transfected with empty pcDNA3.1 vector of pcDNA3.1 vector encoding wild-type RhoA using TransFast (Promega Corp., Madison, WI) according to the manufacturer's instructions. Geneticin was added to the culture medium (250 μl Geneticin/ml) 2 d after transfection. Selection medium was replaced every 2 d. After formation of Geneticin-resistant colonies, dishes were trypsinized to generate polyclonal stably transfected LNCaP-pcDNA3.1 and LNCaP-pcDNA3.1-RhoA sublines.
RhoA activity assay
LNCaP cells were seeded at a density of 3 × 106 cells in 10-cm dishes in medium supplemented with charcoal-stripped fetal bovine serum (FBS). Medium was replaced 3 d later, and cells were treated with 5 nm of the synthetic androgen R1881 for the indicated periods of time. The entire RhoA activity assay was performed in a cold room. Cells were washed twice on ice with ice-cold Tris-buffered saline supplemented with Complete Mini protease inhibitors (Roche, Mannheim, Germany). Dishes were drained of Tris-buffered saline and 700 μl ice-cold lysis buffer [50 mm Tris-HCl (pH 7.2), 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 500 mm sodium chloride, 10 mm magnesium chloride, Complete mini] was pipetted onto the cells. Cells were scraped into prechilled microcentrifuge tubes and spun for 5 min at 13,000 rpm at 4 C. RhoA Assay Reagent (30 μg) (Millipore, Billerica, MA) was added to 550 μl of lysate, and the mixture was subjected to rotation for 45 min at 4 C with gentle agitation. The agarose pellet was obtained after a 10-sec centrifugation at 14,000 × g at 4 C, and washed three times using 500 μl ice-cold wash buffer [50 mm Tris-HCl (pH 7.2), 1% Triton X-100, 150 mm sodium chloride, 10 mm magnesium chloride, Complete mini] at 4 C. The final agarose bead pellet was resuspended in 30 μl of 2× sample buffer [200 mm Tris-HCl (pH 6.7), 20% glycerol, 10% β-mercaptoethanol, 5.0% sodium dodecyl sulfate] and boiled at 95 C for 5 min. Dithiothreitol (1 m, 2 μl) was added before boiling. The resulting supernatant was used for Western blot analysis. After cell lysis, 20 μl of the cell lysate were set aside, the same volume of 2× sample buffer was added, and the resulting sample was boiled for 5 min at 95 C. This sample served to assess total RhoA levels in Western blot analysis.
RNA isolation
RNA isolation from cell cultures and xenograft tissues was done as described previously (21, 24, 25).
Real-time RT-PCR
Real time RT-PCR was performed as described elsewhere (21, 24). Primers targeting RhoA (forward (F) primer; 5′-ccggaagaaactggtgattg-3′ and reverse (R) primer; 5′-gctttccatccacctcgata-3′) and MAL (F primer; 5′-gccaggtgaactatcccaaa-3′ and R primer; 5′-cacagaaccctgggactcat-3′) were obtained from the Mayo Clinic DNA synthesis core facility. Primers targeting CYR61, SDK1, FMO5, DHRS2, WWTR1, CALD1, THG1L, COL6A, WFS1, and RAB3IP were obtained from Integrated DNA Technologies (IDT, San Diego,CA) and are listed in Supplemental Table 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org. Glyceraldehyde-3-phosphate dehydrogenase primers were purchased from Applied Biosystems (Foster City, CA).
Transient transfections
In experiments using promoter-reporter constructs, cells were seeded in six-well plates at a density of 200,000 cells per well in 2 ml regular growth medium without antibiotics. The next day, transfection mixtures were prepared. For each well, 0.75 μg of promoter-reporter construct and 0.25 μg of expression construct or empty vector was added to 250 μl of Optimem-I medium (Invitrogen) (mixture A). In a second reaction mixture, 1 μl of Lipofectamin 2000 transfection reagent (Invitrogen) was added to 250 μl of Optimem-I medium (mixture B). In the case of cotransfection of plasmid with small interfering RNA (siRNA), 1 μl of a 20 mm siRNA SmartPool was added to 1 μg of total DNA in mixture A. Both reaction mixtures were incubated for 5 min at room temperature after which mixture A was added to mixture B and the combined mixture was incubated for 20 min at room temperature before it was added to the cells. The next day, medium was replaced. For experiments that assessed androgen effects, cells were washed once with medium supplemented with 9% charcoal-stripped serum (CSS), fresh CSS-supplemented medium was added, and cells were treated with 5 nm R1881 or ethanol vehicle. After 2 d, cells were washed with PBS (Invitrogen) and lysed in 250 μl passive lysis buffer (Promega). Aliquots (10 μl) of cleared lysates were analyzed for luciferase activity as before (20). Bio-Rad Protein Assays (Bio-Rad Laboratories, Inc., Hercules, CA) were performed on cell lysates to control for potential differences in cell numbers.
Site-directed mutagenesis
V14-RhoA and N19-RhoA constructs were generated using a QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and mutagenic primers 5′-ctggtgattgttggtgatgtagcctgtggaaagacatgc-3′(V14-RhoA forward primer), 5′-gcatgtctttccacaggctacatcaccaacaatcaccag-3′ (V14-RhoA reverse primer), 5′-gatggagcctgtggaaagaactgcttgctcatagtcttc-3′ (N19-RhoA forward primer), and 5′-gaagactatgagcaagcagttctttccacaggctccatc-3′(N19-RhoA reverse primer). Constructs were sequenced at Roswell Park Cancer Institute Biomolecular Resource Core Facility to verify sequence integrity.
Western blot analysis
Whole-cell extracts were prepared and analyzed by Western blotting as described elsewhere (21, 24).
Cell fractionation
LNCaP cells were seeded in medium supplemented with CSS. Medium was replaced 2 d later and cells were treated with 5 nm R1881 or vehicle for 4, 16, or 48 h. Nuclear and cytoplasmic extracts were prepared using a Nuclear Extract Kit (Active Motif, Carlsbad, CA) following the manufacturer's instructions and stored in a −80 C freezer until analysis.
Cell viability assay
Cell viability was analyzed as described previously (24).
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as described in the Supplemental data.
Immunocytofluoresence
LNCaP cells were electroporated with an expression construct for FLAG-tagged MAL (FLAG-MAL) or empty vector as described elsewhere (20). A detailed description of the immunocytofluoresence procedure and scoring method is included in the Supplemental data.
Immunohistochemistry on CWR22 xenograft tissues
The CWR22 xenograft model mimics PCa clinical progression. The serially transplanted AR-positive, androgen-stimulated CWR22 was derived from a primary human PCa. The CWR22 xenograft was maintained in nude mice that are castrated and implanted sc with testosterone (T) pellets [12.5 mg sustained-release T proprionate (TP) pellets] to standardize T serum levels (26, 27). CWR22 cell suspensions were injected sc. Bilateral tumors were allowed to grow to 10 mm maximum diameter in 1–2 months. At that time, animals were castrated by removal of the T pellet. The CWR22 xenograft recurred 4–5 months after castration as recurrent CWR22. CWR22 tissues were harvested before castration and 1, 2, 4, 6, 12, 20, 30, 60, and 90 d after castration and at recurrent disease. At the 6-d, 12-d, and recurrence time points, a fraction of the animals was treated for 48 h with T to further assess androgen-dependence of the parameters to be evaluated. Tissues were fixed in formalin, and a CWR22 tissue microarray (TMA) was built.
TMA sections were deparaffinized, rehydrated through alcohol gradient, and antigen retrieved using 1× Reveal Decloaker (Biocare Medical, Concord, CA) for 30 sec at 125 C in the Decloaking Chamber Plus (Biocare Medical). Cooled sections were immunostained using EnVision G/2 Doublestain System, (DAKO, Glostrup, Denmark). Briefly, the sections were blocked for endogenous peroxidase activity, labeled with RhoA antibody at 1:200 for 1 h at 37 C. Staining was visualized using diaminobenzidine. Sections were counterstained using hematoxylin and mounted using Permount (Fischer Scientific, Pittsburgh, PA).
Images were scored visually by two independent observers. The observers were blinded to the type of CWR22 tumor, and they scored the cytoplasmic areas using the technique developed by Miyamoto et al. (28). For each tissue specimen, 100 cytoplasmic areas were graded for intensity of RhoA staining (0, absent; 1,weak; 2, moderate; or 3, strong) to yield to a visual score ranging from 0–300.
Immunohistochemistry scores were compared between groups using a linear mixed model with the immunohistochemistry score as the response with group and a random rater effect as the model predictors. Tukey pairwise comparisons of the individual groups were done. To assess the association between immunohistochemistry scoring and time after castration, a linear mixed model was fit using immunohistochemistry as the response with time and a random rater effect as the predictors. Effects of T resubstitution (at time point d 6, d 12, and at recurrence) were evaluated using a linear mixed model with immunohistochemical score as the response with group (T or no T) and a random rater effect as the predictors.
Immunohistochemistry on PCa patient specimens
A detailed description of the patient population, RhoA immunostaining procedure, immunostaining quantitation methods, and statistical analysis is included in the Supplemental data.
siRNA transfection
siRNA transfections were performed as described previously (21, 24).
Wound-healing assay
LNCaP cells were transfected with siRNA targeting RhoA or control siRNA as described elsewhere (21, 24). The next day, cells were split and seeded into culture inserts (Ibidi, Martinsried, Germany). Inserts were removed 24 h later, and cells were photographed. Cell migration was documented daily for the next 4 d using Infinity Analyze software (Lumenera Corp., Ottawa, Ontario, Canada).
Replication of results
Results shown are representative of two or more independent experiments.
Results
Androgen regulation of the SRF target gene FHL2 relies on RhoA and RhoA effectors
To explore whether androgen regulation of SRF is mediated through RhoA signaling, the effect of inhibitors of the RhoA effector ROCK (Rho-associated coiled-coil containing protein kinase 1) on androgen stimulation of the SRF target gene FHL2 was investigated. As shown in Fig. 1A, combination of androgen treatment with either Y-27632, hydroxyfasudil, or Ro-318220, three independent ROCK inhibitors, completely prevented or severely attenuated androgen induction of FHL2 expression. Basal expression levels of FHL2 were not altered upon treatment with these compounds (data not shown). In addition, after siRNA-mediated loss of RhoA, androgen exposure of LNCaP cells did not induce FHL2 expression to the level seen in control-transfected condition (Fig. 1B). As an alternative approach to interfere with the activity of the RhoA/actin/MAL signaling axis, androgen treatment was combined with latrunculin B. This compound prevents the actin polymerization that is needed to allow the release of the SRF cofactor MAL from G-actin and its subsequent nuclear relocalization. Consistent with the observations in Fig. 1, A and B, exposure to latrunculin B hampered androgen modulation of FHL2 expression (Fig. 1C).
Fig. 1.
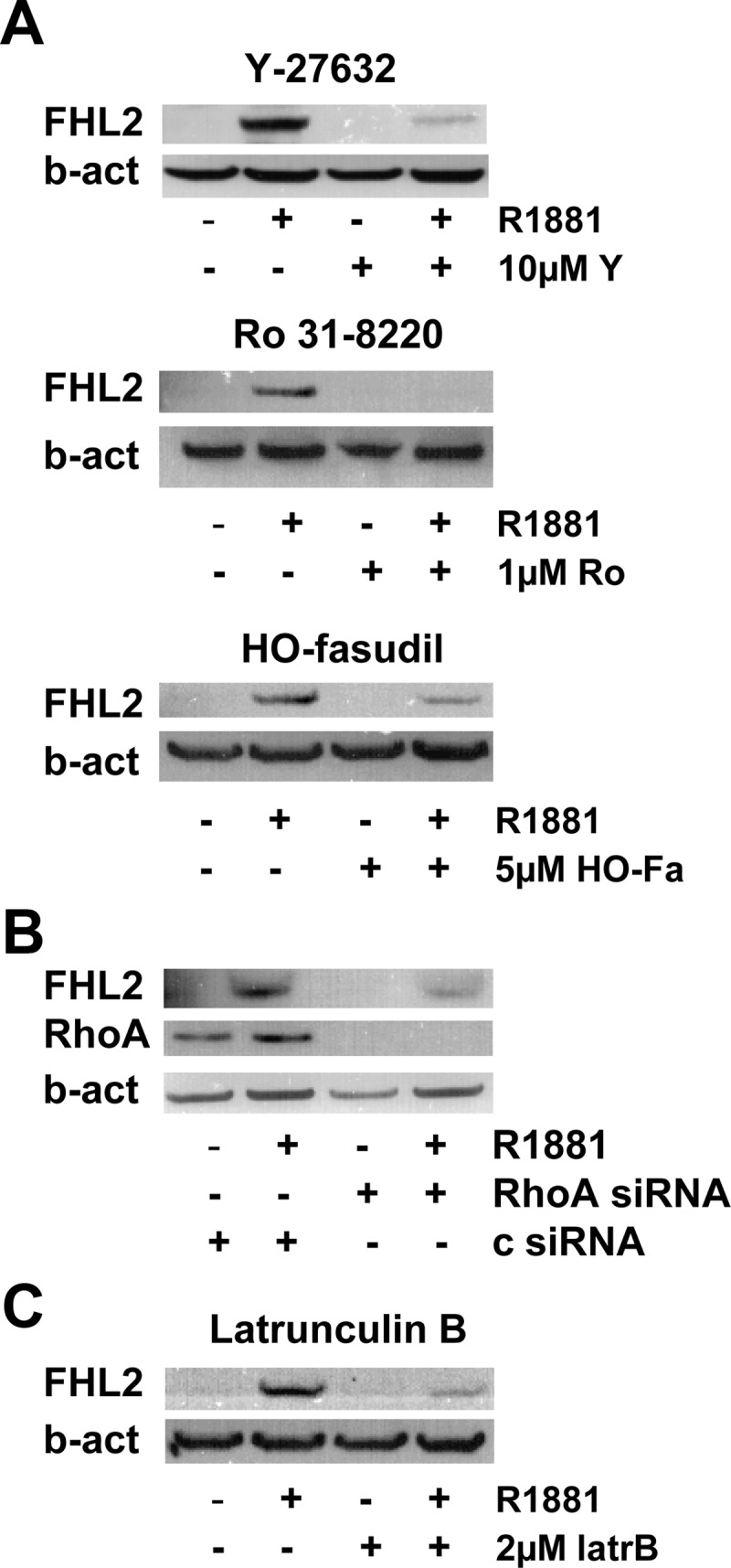
Interference with RhoA signaling axis prevents full androgen induction of FHL2. LNCaP cells were treated with 5 nm R1881 or ethanol vehicle in the presence of Y-27632 (Y, 10 μm), Ro-328220 (Ro, 1 μm), or hydroxyfasudil (HO-Fa, 5 μm) or vehicle for 48 h (A). LNCaP cells were transfected with siRNA targeting RhoA or control siRNA (c). Cells were treated 42 h after transfection with 5 nm R1881 (+) or ethanol vehicle (−) for another 48 h (B). LNCaP cells were treated with 5 nm R1881 or ethanol vehicle in the presence of latrunculin B (latrB, 2 μm) or vehicle for 48 h (C). Western blotting was performed using antibodies directed against FHL2 (A, B, and C) and RhoA (B). To control for loading differences, the blot was stripped and reprobed with an antibody against β-actin (b-act).
Androgen stimulation induces nuclear translocation of the SRF cofactor MAL
To assess further the involvement of RhoA signaling in androgen induction of FHL2, the effect of androgen exposure on the cellular distribution of MAL was investigated. In the absence of antibodies that allow for routine and reliable assessment of endogenous MAL expression, LNCaP cells were electroporated with an expression construct encoding FLAG-tagged MAL or empty vector and cultured in the presence or absence of the synthetic androgen R1881 for 4, 16, or 48 h. Western blot analysis of nuclear extracts demonstrated enrichment of MAL expression in the nucleus under androgen-supplemented conditions. MAL nuclear relocalization was obvious 16 h after stimulation and was more pronounced at later time points (Fig. 2A). These kinetics indicate that relocalization of this critical cofactor of SRF precedes androgen-dependent changes in expression of SRF target genes (20, 21). Androgen-induced nuclear enrichment of AR expression confirmed the efficacy of the cell fractionation procedure. As observed earlier, androgen treatment did not affect the nuclear content of SRF. Validation experiments in which whole cell lysates, cytoplasmic fractions, and nuclear fractions were prepared from the same electroporation experiment showed a modest increase in total cellular MAL levels after 16 h of androgen treatment. This observation is consistent with expression of MAL from a plasmid driven by an androgen-sensitive cytomegalovirus promoter (pCMX). At 16 h, this was reflected in a slight increase in cytoplasmic levels of MAL after androgen exposure and more robust androgen-stimulated recruitment of MAL to the cell nucleus. At 48 h, these effects become more pronounced (Supplemental Fig. 1). Immunocytofluorescence analysis validated the androgen-dependent redistribution of MAL from cytoplasm to cell nucleus (Fig. 2B, left panel). Quantification of the immunocytofluorescence data confirmed that in the majority (70%) of cells MAL staining is predominantly cytoplasmic under androgen-deprived conditions. Androgen exposure led to a reversal of these ratios; in 70% of cells the immunoreactive MAL signal became stronger in the cell nucleus than in the cytoplasm (Fig. 2B, right panel). The relevance of androgen-induced MAL relocalization for SRF-dependent transcription was confirmed by chromatin immunoprecipitation experiments showing enrichment of MAL to SRF recruitment sites in the FHL2 promoter after androgen exposure (Fig. 2C), but not on non-CArG box containing genomic regions. Real-time RT-PCR quantification indicated that MAL enrichment on the FHL2 promoter ranged from 3- to 23-fold in independent experiments (Fig. 2C, bottom panel). Control experiments in which a nonspecific antibody was used or the primary antibody was omitted failed to produce a signal as did studies in which empty vector was transfected (Fig 2., A–C, data not shown). The importance of MAL for androgen- and SRF-dependent regulation of FHL2 expression was verified in experiments in which its expression was silenced using specific siRNA. As shown in Fig. 2, D and E, loss of MAL prevented androgen induction of FHL2 at the protein and mRNA level. Only one out of four antibodies tested yielded a weak immunoreactive signal that is consistent with MAL expression (Fig. 2D). Western blot analysis using this antibody suggested that MAL expression may be subject to modest androgen regulation. The efficacy of the siRNA directed against MAL in silencing the expression of its target gene was validated in the real-time RT-PCR experiments (Fig. 2E). Of note, overexpression of MAL led to an increase in basal FHL2 mRNA expression and enhanced androgen regulation of FHL2 expression (Fig. 2F). Transient transfection studies using a reporter construct under control of the FHL2 promoter yielded similar results and confirmed the involvement of SRF in the effects of MAL overexpression on FLH2 messenger levels (Supplemental Fig. 2).
Fig. 2.

MAL is involved in androgen induction of FHL2. LNCaP cells were electroporated with an expression construct encoding FLAG-tagged MAL. Cells were treated 2 d later with 5 nm R1881 or ethanol vehicle for 4, 16, or 48 h. Nuclear extracts were prepared and subjected to Western blotting using antibodies directed against the FLAG-tag, AR, and SRF (A). LNCaP cells were electroporated and treated as above. Cells were fixed 48 h later and permeabilized, and immunocytochemistry was performed using an antibody against the FLAG tag. Cell images were visualized using a LSM510 confocal microscope. A representative image is shown. Magnification, ×100 (left panel). Quantification of immunocytochemistry results. C>N, cytoplasmic staining stronger than nuclear staining; C = N, cytoplasmic staining equally strong as nuclear staining; N > C, nuclear staining stronger than cytoplasmic staining (right panel) (B). LNCaP cells were electroporated and treated for 48 h as above, and ChIP was performed using antibody directed against the FLAG-tag, SRF, or nonspecific IgG. Top panel: representative agarose gel electrophoresis image obtained after end point PCR on ChIP'ed DNA; bottom panel: quantification of results using q-RT-PCR. FHL2, FHL2 promoter region; FHL2c, non-CArG box-containing FHL2 genomic region (C). LNCaP cells were transfected with siRNAs targeting MAL or control siRNAs (c) and treated as described. Western blotting was performed using antibodies directed against FHL2. Blots were stripped and reprobed for MAL. Asterisk indicates MAL-specific immunoreactive band. To control for loading differences, the blot was stripped and reprobed with an antibody against β-actin (β-act) (D). LNCaP cells were transfected with siRNA targeting MAL or nonspecific control siRNA (c) as above. FHL2 (top panel) and MAL (bottom panel) expression was evaluated using real-time RT-PCR. Target gene mRNA levels were normalized with the values obtained from glyceraldehyde-3-phosphate dehydrogenase expression and are expressed as relative expression values, taking the value obtained from one of the vehicle-treated control-transfected conditions as 1. Columns, means of values obtained from three independent biological replicates; bars, sem values (E). LNCaP cells were electroporated with an expression construct encoding FLAG-tagged MAL (pCMX-MAL) or empty vector (pCMX) and treated as above. Expression of FHL2 was evaluated by real-time RT-PCR as above. Columns, means of values obtained from three independent biological replicates; bars, sem values (F). DAPI, 4′,6-diamidino-2-phenylindole.
Androgen stimulation induces RhoA activity
The involvement of the RhoA/actin/MAL signaling axis in the androgen regulation of the SRF target gene FHL2 suggests that androgens may regulate the expression and/or activity of RhoA. Treatment of LNCaP cells with R1881 for 4, 16, or 48 h did not alter RhoA expression levels (Fig. 3A). RhoA activity, however, was induced robustly after androgen treatment (Fig. 3B). The increase in RhoA activity, which was pronounced 24 h after R1881 exposure (left panel), was obvious 4 h following treatment (right panel), thus preceding androgen stimulation of MAL relocation and induction of SRF target gene expression (20, 21). Activation of RhoA by phorbol 12-myristate 13-acetate, which has been described in LNCaP cells previously (29), serves as validation for use of the RhoA activation assay (Fig. 3B, left panel). To our knowledge, these findings are the first to demonstrate that androgens activate RhoA in PCa cells.
Fig. 3.

Androgen stimulation induces RhoA activity. LNCaP cells were treated with 5 nm R1881 or ethanol vehicle for 4, 16, or 48 h. Western blotting was performed using antibodies directed against RhoA. To control for loading differences, the blot was stripped and reprobed with an antibody against β-actin (b-act) (A). LNCaP cells were treated with 5 nm R1881 or ethanol vehicle for 24 (left panel) or 4 h (right panel), or with 100 nm phorbol 12-myristate 13-acetate (PMA) or dimethylsulfoxide (DMSO) for 1 h (left panel). A RhoA activity assay was done and Western blotting was performed as described (B).
RhoA conveys androgen regulation to a subset of androgen-responsive SRF target genes
The indirect mechanism of androgen action in which the effects of androgens are conveyed to target genes via the secondary TF SRF was identified while studying androgen regulation of the gene encoding FHL2 (20). By means of a microarray-based screening approach, we isolated 158 genes in addition to FHL2 that are androgen regulated in an SRF-dependent manner (21). To verify to what extent the role of RhoA in androgen dependency of FHL2 can be generalized to this 158-gene profile, a representative sample of eight genes was examined. Androgen regulation of these genes was assessed after siRNA-mediated silencing of RhoA using real-time RT-PCR. Figure 4 shows that androgen-responsiveness of five of these eight genes was hampered either completely (IER5, PLEKHH1) or severely (CCL8, CDC25A, SLITRK6) upon knockdown of RhoA. After inclusion of another eight SRF- and AR-responsive genes (Supplemental Figs. 3 and 4) and taking into account the results obtained for FHL2, androgen regulation of 23.5% (four of 17), 47% (eight of 17), and 29.5% (five of 17) of SRF target genes was found to be either lost completely, partially, or not affected, respectively, after silencing of RhoA. Noteworthy, an siRNA-induced decrease in MAL expression affected the androgen modulation of these same genes in a similar manner (Supplemental Fig. 5). These findings indicate that RhoA-dependent androgen modulation of SRF target genes is not limited to FHL2, but affects a substantial segment (>50% of genes under investigation here) of androgen- and SRF-dependent gene expression.
Fig. 4.

RhoA mediates androgen regulation of a subset of SRF target genes. LNCaP cells were transfected with siRNA targeting RhoA or nonspecific control siRNA (c) as described. Androgen-responsiveness of SRF target genes was evaluated by real-time RT-PCR. Target gene mRNA levels were normalized with the values obtained from glyceraldehyde-3-phosphate dehydrogenase expression and are expressed as relative expression values, taking the value obtained from one of the vehicle-treated control-transfected conditions as 1. Columns, Means of values obtained from three independent biological replicates; bars, sem values.
RhoA is overexpressed in PCa
The androgen- and SRF-dependent gene signature is sufficient to separate benign from malignant prostate samples, correlates with aggressive disease, and is associated with poor outcome after prostatectomy (21). Given its role in mediating androgen-responsiveness of SRF target genes, the relevance of RhoA to clinical PCa progression was explored. To this end, expression of RhoA was examined by immunohistochemical analysis of prostate tissues from 91 patients with biopsy-proven diagnosis of PCa who were treated with radical retropubic prostatectomy (RRP) without neoadjuvant hormonal therapy. Information related to patients' clinical and RRP pathological features and outcome after RRP is summarized in Supplemental Table 2. The specificity of the antibody used for this study is shown in Supplemental Fig. 6. By Western blot analysis the RhoA antibody detected a single immunoreactive band, which was lost after transfection with RhoA-specific siRNA. In prostate tissue samples RhoA expression presented as a membraneous cytoplasmic staining pattern, which is consistent with cellular distribution characteristics of activated RhoA (30) (Fig. 5A). RhoA intensity in cancer and benign cells was recorded as absent, focal, moderate, or marked and is summarized in Table 1. Among the 80 specimens that contain both cancer and benign cells, RhoA intensity was higher in cancer cells (P = 0.016; signed rank test). Only 11 specimens showed higher RhoA intensity in benign cells, whereas 29 specimens had higher levels of RhoA intensity in cancer cells.
Fig. 5.

RhoA overexpression in PCa is associated with poor outcome. Representative image of RhoA immunostaining in prostate tissue. Magnification, ×5; inset, ×20 (A). Marked RhoA intensity in PCa cells is associated with decrease in cancer progression-free survival (B).
Table 1.
RhoA intensities in benign prostate epithelial cells and PCa cells
| RhoA intensity | n (%) |
|---|---|
| Cancer cells (n = 87) | |
| Absent | 2 (2.3) |
| Focal | 12 (13.8) |
| Moderate | 39 (44.8) |
| Marked | 34 (39.1) |
| Benign cells (n = 84) | |
| Absent | 1 (1.2) |
| Focal | 17 (20.2) |
| Moderate | 52 (61.9) |
| Marked | 14 (16.7) |
RhoA overexpression in PCa is associated with aggressive disease and poor outcome
Associations of cancer RhoA intensity with clinical and RRP pathological features are summarized in Table 2. Some levels of intensity were combined for analysis. For example, only two specimens did not show any RhoA staining in cancer cells so cancer RhoA intensity was analyzed as absent/focal vs. moderate vs. marked. Increasing levels of cancer RhoA intensity were associated with larger tumors, higher GPSM scores [scoring algorithm that takes into account Gleason score, prostate-specific antigen (PSA) levels, seminal vesicle involvement, and margin status to predict biochemical recurrence after RRP (31)], lymph node involvement, and extraprostatic extension at RRP. For example, none of the specimens with absent/focal cancer RhoA intensity demonstrated extraprostatic extension compared with 9 (23.1%) and 15 (44.1%) specimens with moderate and marked levels of cancer RhoA intensity, respectively (P = 0.006).
Table 2.
Association of RhoA intensity with clinical and RRP pathological features
| Cancer RhoA intensity |
||||
|---|---|---|---|---|
| Absent/focal (n = 14) | Moderate (n = 39) | Marked (n = 34) | P-value | |
| Age | 66 (53–74) | 64 (43–73) | 65.5 (47–76) | 0.275 |
| Preoperative serum PSA (n = 82) | 5.2 (1.9–15.3) | 6.1 (2.0–50.2) | 7.4 (1.4–39.7) | 0.116 |
| Tumor volume (n = 86) | 1.4 (0.1–7.5) | 2.1 (<0.1–40.0) | 5.3 (0.1–131.3) | 0.023 |
| GPSM score (n = 82) | 7.5 (5–10) | 9 (6–15) | 9 (6–15) | 0.007 |
| Gleason score | N (%) | |||
| 5 or 6 | 7 (50) | 20 (51.3) | 14 (41.2) | 0.586 |
| 7 | 7 (50) | 14 (35.9) | 17 (50.0) | |
| 8 or 9 | 0 | 5 (12.8) | 3 (8.8) | |
| Seminal vesicle involvement | 0 | 5 (12.8) | 9 (26.5) | 0.058 |
| Surgical margins | ||||
| negative | 11 (78.6) | 20 (51.3) | 15 (44.1) | 0.091 |
| positive | 3 (21.4) | 19 (48.7) | 19 (55.9) | |
| Lymph node involvement | 0 | 2 (5.1) | 8 (23.5) | 0.025 |
| Extraprostatic extension | 0 | 9 (23.1) | 15 (44.1) | 0.006 |
| Extensive HGPIN (n = 86) | 3 (23.1) | 7 (18.0) | 3 (8.8) | 0.380 |
| DNA ploidy at RRP (n = 86) | ||||
| diploid | 13 (92.9) | 25 (64.1) | 24 (72.7) | 0.414 |
| tetraploid | 1 (7.1) | 10 (25.6) | 7 (21.2) | |
| aneuploid | 0 | 4 (10.3) | 2 (6.1) | |
Numbers preceding parentheses indicate median values. Numbers within parentheses indicate range.
The association of RhoA expression with PCa progression after RRP was assessed also. At last follow-up 30 patients experienced cancer progression at a median of 3.2 yr after RRP (range, 0.2–9.3). The median duration of follow-up among the patients who did not progress was 9.5 yr (range, 2.1–11.2). Analysis of univariate associations of RhoA intensity with cancer progression after RRP demonstrated that patients whose specimens contained marked levels of cancer RhoA intensity are more than twice as likely to progress compared with patients whose specimens contained absent, focal, or moderate levels (risk ratio, 2.59; 95% CI 1.25–5.36; P = 0.011; Fig. 5B).
To further explore the association of RhoA with disease progression, the percentage of benign and malignant prostate epithelial cells that is positive for RhoA was recorded. Of the 80 specimens that contained both benign and malignant cells, 50 had a higher percentage of cancer cells staining positive for RhoA intensity. Intensity and percentage were multiplied to summarize overall expression of RhoA. RhoA expression was higher in cancer cells (P < 0.001; signed rank test). Moreover, increased RhoA expression was associated with extraprostatic extension at RRP (P = 0.021) and a significant increase in risk of progression after RRP (risk ratio 1.31; 95% CI 1.02–1.69; P = 0.035).
Overexpression of RhoA enhances androgen responsiveness of select SRF target genes
In view of the association of elevated RhoA expression with aggressive disease features and progression after RRP, the effect of RhoA overexpression on androgen-responsiveness of SRF target genes was investigated. LNCaP cells were electroporated with an expression construct for RhoA or empty vector and subsequently cultured in the presence or absence of R1881 for 48 h. Under vehicle-treated conditions, a 3-fold increase in RhoA expression was noted upon transfection with the RhoA-expressing construct. Androgen stimulation further elevated RhoA expression, which is common for gene expression derived from the simian virus 40 promoter-driven pSG5 expression vector. As shown in Fig. 6A, increased RhoA expression led to a marked amplification in the androgen-responsiveness of the representative SRF target gene FHL2. Similar effects were seen for CCL8, another gene that relies on RhoA to achieve full androgen response (cfr Fig. 4), but not for SSR3, for which androgen dependency is not affected by RhoA silencing (Fig. 6B). Results were confirmed in LNCaP cells that are stably transfected with a RhoA expression construct and in which the overexpression of RhoA is less pronounced (≤ 2-fold) (Supplemental Fig. 7). These observations indicate that the stimulatory effect of RhoA on androgen regulation of SRF target genes is not merely due to a general induction of (SRF target) gene transcription. Moreover, loss of SRF attenuated the effect of RhoA overexpression on androgen induction of SRF target genes such as FHL2 (Fig. 6C). Whether the observed changes in androgen-regulation of select SRF target genes are merely the result of overexpression of RhoA or rely also on its GTPase activity could not be deduced from these experiments. To discriminate between these possibilities, expression constructs encoding versions of RhoA that have undergone mutations in its GTPase domain and act as constitutively active (V14-RhoA) or dominant-negative RhoA (N19-RhoA) (32) were generated. After verification of the intended mutations using Sanger sequencing, the functional consequences of these mutations on SRF activity were validated in transient transfection studies using a reporter construct driven by four copies of a CArG box (SRE-luc). As shown in Fig. 7A, constitutively active V14-RhoA resulted in higher reporter activity than seen for wild-type RhoA, whereas N19-RhoA did not, or slightly decreased, transcription from the SRE-luc promoter-reporter construct. The involvement of SRF in the stimulatory effects of wild-type RhoA and V14-RhoA on reporter activity was validated using specific siRNA (Supplemental Fig. 8). Similar results were observed when the effects of overexpression of these RhoA forms on androgen-dependent expression of endogenous SRF target genes was examined: V14-RhoA led to a level of androgen regulation of the genes encoding FHL2 and CCL8 that exceeded that obtained for wild-type RhoA, whereas N19-RhoA slightly reduced the androgen regulation of these genes. As before, the extent of androgen regulation of SSR3 was not affected by either version of RhoA (Fig. 7B). The involvement of SRF in the effects of V14-RhoA on androgen sensitivity of CArG box-driven genes such as FHL2 was validated further using CCG-1423, a compound that inhibits interaction between SRF and MAL (33), and a promoter-reporter construct derived from the FHL2 promoter (20). Figure 7C shows that addition of CCG-1423 abolished the androgen overinduction of reporter activity achieved by wild-type RhoA and V14-RhoA. Taken together, these findings demonstrate that overexpression of RhoA alone is not sufficient to explain effects on androgen induction of SRF action and implicate its RhoA GTPase activity in these events.
Fig. 6.

RhoA overexpression enhances androgen regulation of select SRF target genes. LNCaP cells were electroporated with an expression construct for RhoA (pSG5-RhoA) or empty vector (pSG5). Cells were treated 2 d later with 5 nm R1881 or vehicle for 48 h. Real-time RT-PCR was performed as described. RhoA protein expression levels were verified by immunoblotting as described (inset) (A). LNCaP cells were electroporated as above and treated with 5 nm R1881 or vehicle for 48 h. CCL8 and SSR3 mRNA expression levels were evaluated using real-time RT-PCR as before (B). LNCaP cells were electroporated with expression construct for RhoA or empty vector as above. Control siRNA (c siRNA) or siRNA targeting SRF was added to the electroporation reactions. Expression of FHL2 was assessed using real-time RT-PCR performed as before (C).
Fig. 7.
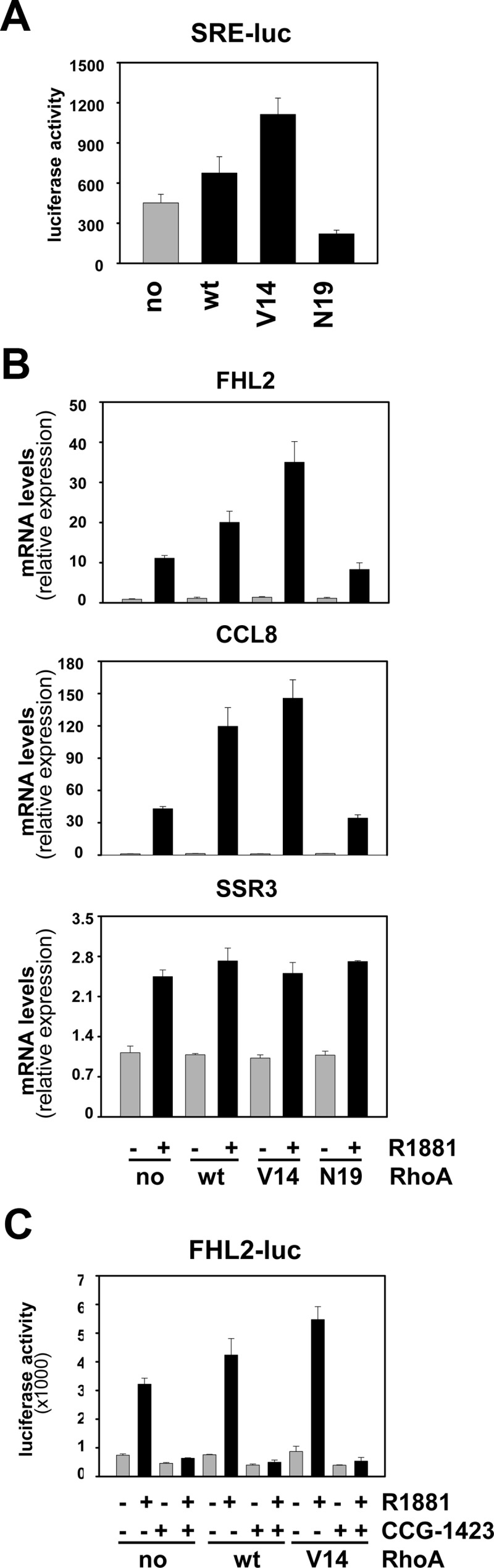
RhoA modulation of androgen-regulated SRF target gene expression relies on its GTPase function. LNCaP cells were transfected with a CArG box-driven promoter reporter construct (SRE-luc) in combination with an expression construct encoding wild-type RhoA (wt), or constructs encoding a RhoA version that has undergone a mutation in the GTPase domain resulting in constitutively active RhoA (V14) or a dominant-negative RhoA (N19), or empty vector (no). Cells were harvested 2 d later, and a luciferase assay was performed. Columns, Mean values from samples in triplicate; bars, sem values (A). LNCaP cells were electroporated with an expression construct encoding wild-type RhoA (wt), V14-RhoA (V14), N19-RhoA (N19), or empty vector (no). Cells were treated 2 d later with 5 nm R1881 (+) or vehicle (−) for 48 h. Expression of FHL2, CCL8, and SSR3 was evaluated using real-time RT-PCR as described (B). LNCaP cells were transfected with a reporter construct driven by a 145-bp FHL2 promoter fragment (FHL2-luc) in combination with an expression construct encoding wild-type RhoA (wt), V14-RhoA, or empty vector (no). The next day, cells were treated with ethanol vehicle or 5 nm R1881 in combination with 10 μm CCG-1423 or dimethylsulfoxide. Cells were harvested 2 d later, and a luciferase assay was performed (C).
RhoA is critical for PCa cell proliferation and migration
The positive correlation between RhoA (over)expression and PCa progression led us to investigate the role of the RhoA signaling axis in PCa cell proliferation and migration. The effect of siRNA-mediated silencing of RhoA was assessed by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assays. Loss of RhoA expression gave rise to a decrease in the number of viable LNCaP cells. This effect was obvious 1 d after transfection and became more pronounced with time (Fig. 8A). To assess the importance of RhoA for androgen-responsive cell proliferation, silencing of RhoA was combined with treatment with a dose-response curve of 0, 0.1, 1, and 10 nm R1881. Androgen treatment increased cell viability 96 h after transfection as reported previously (24). However, at each dose used, silencing of RhoA expression attenuated the number of viable cells, indicating a contribution of RhoA in basal and androgen-dependent cell viability (Fig. 8B). The contribution of androgen-responsive SRF target genes to (androgen-dependent) PCa cell proliferation was assessed because RhoA governs a broad spectrum of cellular functions; its role is not limited to control over SRF action. LNCaP cells were transfected with siRNA directed against five SRF target genes that rely on RhoA for androgen regulation (DGAT2, CYR61, WFS1, SLITRK6, and RAB3IP) and cell viability was assessed. Under normal FBS-supplemented cell culture conditions, loss of four of the five genes that are targeted in these assays (all but CYR61) had little effect on the number of viable cells (Fig. 9A). Similarly, except for CYR61, the androgen-dependent increase in the number of viable LNCaP cells remained unaltered after silencing of these genes (Fig. 9B). These findings suggest a relative small contribution of these AR and SRF target genes to the effects of RhoA on cell proliferation. The specificity of the siRNA for their respective target genes was validated by real-time RT-PCR (Supplemental Fig. 9).
Fig. 8.

RhoA is critical for PCa cell viability. LNCaP cells were transfected with siRNA targeting RhoA or control siRNA. Medium was changed 16 h later, and an MTS assay reading absorbance at 490 nm was performed 24, 48, 72, and 96 h later (A) or cells were treated with 0, 0.1, 1, or 10 nm R1881 for 96 h and cell viability was assessed by MTS assay (B). Columns, Mean values from five individual measurements; bars, se.
Fig. 9.

Minor contribution of androgen-responsive SRF target genes to PCa cell proliferation. LNCaP cells were transfected with siRNA targeting DGAT2, CYR61, WFS1, SLITRK6, RAB3IP, or control siRNA (c). Medium was changed 16 h later, and an MTS assay reading absorbance at 490 nm was performed 96 h later (A) or cells were treated with 5 nm R1881 or ethanol vehicle for 96 h, and cell viability was assessed by MTS assay (B). Columns, Mean values from five individual measurements; bars, se.
Consequently, the relevance of RhoA signaling for PCa cell migration was assessed using wound-healing assays. LNCaP cells were reseeded in culture inserts 1 d after transfection with control siRNA or siRNA targeting RhoA. Culture inserts were removed 24 h later (d 0). Cell migration was documented by daily imaging for another 4 d. As shown in Fig. 10, wound closure was observed earlier and progressed further in control-transfected cells, indicating that RhoA activity is important for PCa cell migration. These effects were seen already at the earliest time point, when effects on cell proliferation were modest. Studies in which siRNA-mediated silencing of RhoA was combined with androgen treatment showed that the androgen-mediated closure of the wound that was observed in control-transfected cell was delayed when RhoA expression was compromised. Under the latter conditions, cell migration was similar to that observed for vehicle-treated and control-transfected cells (Supplemental Fig. 10). The use of CCG-1423 in combination with androgen treatment also blunted androgen-dependent cell migration (Supplemental Fig. 11), which indicated that the SRF-MAL interaction is important for androgen-dependent PCa cell migration.
Fig. 10.

RhoA is critical for PCa cell migration. LNCaP cells were transfected with siRNA targeting RhoA or control siRNA (c). The next day, cells were reseeded for wound healing assay as described. Culture inserts were removed 1 d later (d 0). Cell migration was assessed after 1, 2, 3, and 4 d.
RhoA expression remains unaltered during progression from androgen-stimulated to castration-recurrent PCa
In view of the observation that higher RhoA levels in prostatectomy samples are associated with poorer disease outcome, the possibility that RhoA expression correlates with disease progression was examined. To this end, two isogenic cell line models that represent the progression from AS to CR PCa were used. Comparison of the RhoA protein levels in parental LNCaP cells and its CR sublines C4–2 and LN-Rf, which are derived after long-term ablation of LNCaP cells in vivo or in vitro, respectively (34, 35), showed similar levels of RhoA expression, suggesting no alterations during progression from AS to CR PCa (Fig. 11A). Although RhoA expression (pattern) remains unaltered during the progression from AS to CR PCa, CRPC cells still depend on RhoA, because silencing of RhoA led to marked decreases in the number of viable C4–2 and LN-Rf cells (Fig. 11B). In addition, treatment with the ROCK inhibitor Ro-318220 or with CCG-1423 led to similar decreases in cell viability in LNCaP, C4–2, and LN-Rf cells (Fig. 11, C and D). To validate these findings in a model system that more closely resembles the clinical situation and allows for assessment of RhoA at the AS and CR stage as well as during the transition to CR disease, the CWR22 xenograft model system was used (26, 27). Nude mice were castrated and implanted sc with T pellets [12.5 mg sustained-release T proprionate (TP) pellets] to standardize T serum levels and CWR22 cell suspensions were injected sc. Bilateral tumors were allowed to grow to 10 mm maximum diameter (occurs on average 1–2 months after inoculation), at which time animals were castrated by removal of the T pellet. The CWR22 xenograft recurred as CR disease 4 to 5 months after castration. CWR22 tissues were harvested before castration, 1, 2, 4, 6, 12, 20, 30, 90, and 120 d after castration and at recurrence. At the 6-d, 12-d, and recurrence time points, a fraction of the animals were treated for 48 h with T to further assess androgen dependence of the parameters to be evaluated. Immunohistochemistry was done using the antibody directed against RhoA and a CWR22 xenograft TMA. Immunohistochemistry data were scored by two independent raters and analyzed to assess the effect of short-time and long-time castration as well as the impact of T resubstitution on RhoA expression. Consistent with the findings from the isogenic cell line model, no visual changes in RhoA expression were noted during the progression to CR PCa. Table 3 provides an overview of the animal groups, number of TMA cores assessed for RhoA expression, and the RhoA scoring results. Statistical analysis of the RhoA immunohistochemical scores confirmed that there was no significant association between (duration of) castration and RhoA inmunostaining values. Tukey-adjusted pairwise comparisons showed that only one group (12-d group) was statistically different from the others (Supplemental Table 3). To assess the effect of T resubstitution, a linear mixed model was used with immunohistochemical score as the response with group (T, no T) and a random rater effect as the predictors. This analysis demonstrated that there is no statistically significant difference between T administration or no T administration for the 6-d time point and at recurrent disease (Table 4). However, there was a difference at the 12-d time point (P < 0.001) with the untreated group having higher RhoA immunohistochemistry scores. The reason for the discrepancy in the 12-d time point could not be deduced from this study, but may be related with the location of these cores at the bottom edge of the TMA slide (possibly leading to exposure to more diaminobenzidine reagent than the other tissue cores). Overall, these data provide further evidence that RhoA expression does not change significantly during disease progression. Moreover, they suggest that the level of RhoA that is present at the time of prostatectomy determines the aggressiveness of PCa.
Fig. 11.

RhoA expression is unaltered in CRPC cells and remains essential for CRPC cell proliferation. LNCaP cells and their isogenic CRPC C4–2 and LN-Rf (Rf) cells were seeded in their regular growth medium. Cells were harvested 3 d later, and RhoA expression was assessed using Western blotting as described (A). C4–2 and LN-Rf cells were transfected with siRNA targeting RhoA or nontargeting control siRNA (c). Cells were harvested 96 h after transfection, and RhoA expression was evaluated using Western blotting (top panel). In a parallel experiment, cell viability was determined using an MTS assay reading absorbance at 490 nm (bottom panel) (B). LNCaP, C4–2, and LN-Rf cells were seeded in their regular growth medium and treated with 1 μm Ro-318220 (C) or 10 μm CCG-1423 (D). Cell viability was assessed 96 h after treatment using an MTS assay. b-act, β-Actin.
Table 3.
Summary of RhoA immunohistochemistry results using CWR22 tissue microarray.
| Group | n | Mean (sd) |
|---|---|---|
| Intact | 16 | 139.0 (31.3) |
| 1 d post-CX | 2 | 118.5 (4.9) |
| 2 d post-CX | 4 | 124.5 (30.9) |
| 4 d post-CX | 4 | 125.0 (11.0) |
| 6 d post-CX | 8 | 126.6 (38.0) |
| 6 d post-CX + TP | 8 | 120.5 (12.5) |
| 12 d post-CX | 8 | 189.0 (20.3) |
| 12 d post-CX + TP | 4 | 150.8 (18.5) |
| 20 d post-CX | 4 | 87.8 (19.4) |
| 30 d post-CX | 4 | 111.5 (23.2) |
| 60 d post-CX | 4 | 162.3 (9.2) |
| 90 d post-CX | 4 | 118.3 (14.1) |
| 120 d post-CX | 8 | 132.8 (35.6) |
| Recurrent | 28 | 118.1 (38.3) |
| Recurrent + TP | 8 | 102.1 (20.4) |
An overview of the different animal groups, the number of tissue cores that have been scored for RhoA immunostaining, and the mean (and sd) RhoA scores per group. CX, Castration.
Table 4.
Effect of TP readministration on RhoA immunoscores in the CWR22 model
| Time post-CX | Difference in RhoA score (CX − CX + TP) | P value |
|---|---|---|
| 6 d | 6.13 (se. = 13.64) | 0.6608 |
| 12 d | 38.25 (se. = 8.27) | 0.0007 |
| Recurrent | 16.02 (se. = 14.17) | 0.2664 |
CX, Castration; se, error associated with the estimated difference.
Activity of the AR-RhoA-SRF signaling axis is maintained in CR PCa cells
Our previous work showed that expression of the SRF target gene FHL2 in C4–2 and LN-Rf cells is maintained at levels similar to those observed for the parental cell line LNCaP (20), which suggested that the AR-SRF signaling axis may be active in CR PCa. The mRNA expression of FHL2 and the set of eight SRF target genes that was analyzed in Fig. 4 was assessed in AS and CR CWR22 samples to validate these observations in the CWR22 model of PCa progression. Real time RT-PCR analysis was done starting from RNA derived from the same xenograft samples that were used to construct the CWR22 TMA. No significant changes in the expression levels for these genes were noted between AS and CR tissues (Fig. 12). PSA mRNA levels also did not differ between these two groups. Expression of CCL8 mRNA expression fell below the level of detection for real-time RT-PCR analysis. Real-time RT-PCR studies were extended to include analysis of recurrent samples and recurrent samples derived from animals that had been treated for 48 h with TP to assess whether androgen treatment affects the expression levels of SRF target genes in CR CWR22 tumors. This short-term exposure to androgens was sufficient to reinstate androgen regulation of four of the eight SRF target genes under investigation (SLITRK6, DGAT2, SSR3, IL17RD), which suggested androgen control over SRF action. PSA mRNA levels were not affected notably by renewed androgen exposure (Fig. 13). We have reported that full androgen induction of target gene mRNA expression in the CR cell lines C4–2 and LN-Rf requires longer androgen exposure than needed in the parental LNCaP cell line (24, 36). Longer R1881 treatment (96 h) of the CR C4–2 and LN-Rf PCa cell lines showed androgen regulation of seven of nine SRF target genes (Fig. 14). As observed previously, FHL2 expression was no longer androgen-responsive in the LN-Rf cell line (20). In both CR cell lines, expression of PSA mRNA was induced by androgen exposure. Prolonged periods of androgen treatment could lead to more pronounced effects of RhoA gene silencing on cell proliferation at experimental end points (96 h androgen treatment ends ≈ 6 d after siRNA transfection). Therefore, the involvement of the AR/RhoA/SRF signaling axis in androgen-responsiveness of SRF target genes in CR PCa cells was investigated further using reporter constructs driven by SRF-responsive promoter fragments, for which androgen regulation is detectable at earlier time points. The C4–2 CR model system was chosen because FHL2 retains androgen sensitivity in C4–2 cells. First, cells were transfected with reporter constructs derived from the proximal FHL2 promoter. Loss of AR, RhoA, SRF, or MAL prevented full androgen induction of reporter activity (Fig. 15), which was similar to results obtained for the parental AS LNCaP cells. Studies using an FHL2 promoter-reporter construct in which the SRF binding site is mutated (20) confirmed the involvement of SRF-CArG box interaction in the observed effects. Loss of AR, SRF, and RhoA expression was verified using Western blot (Supplemental Fig. 12). These findings indicate that RhoA control over androgen-responsive SRF target gene expression is maintained. Parallel experiments using a luciferase reporter construct driven by four copies of SRF-binding sites validated the androgen activation of the AR-RhoA-SRF signaling axis in CR cells (Fig. 15). Combination of androgen treatment with ROCK inhibitors or CCG1423 also blunted androgen regulation of reporter gene expression (Supplemental Fig. 13). Moreover, CArG box-driven reporter gene expression was decreased markedly in both C4–2 and LN-Rf cells upon loss of AR, RhoA, SRF, and MAL under regular, FBS-supplemented growth conditions (Supplemental Fig. 14).
Fig. 12.

Androgen-responsive SRF target gene expression is unaltered in intact and recurrent CWR22 xenografts. Expression of AR- and SRF-target genes was evaluated using RNA derived from AS and CR CWR22 xenograft tissues and real-time RT-PCR as described.
Fig. 13.

Short-term androgen readministration restores androgen responsiveness of SRF target genes in recurrent CWR22 xenografts. Real-time RT-PCR analysis of AR- and SRF-target gene expression using RNA derived from CR CWR22 xenograft tissues (−) or CR CWR22 tissues from animals that had been treated for 2 d with TP (+) before euthanasia.
Fig. 14.

Androgen regulation of SRF target gene expression in CR cell line models. C4–2 and LN-Rf cells were treated for 96 h with 5 nm R1881 (+) or ethanol vehicle (−). Androgen-responsiveness of SRF target genes was evaluated by real-time RT-PCR. Target gene mRNA levels were normalized with the values obtained from glyceraldehyde-3-phosphate dehydrogenase expression and are expressed as relative expression values, taking the value obtained from one of the vehicle-treated control-transfected conditions as 1. Columns, Means of values obtained from three independent biological replicates; bars, sem values.
Fig. 15.

Androgen induction of CArG box-dependent reporter gene activity in CR cells relies on the presence of an intact AR-RhoA-SRF signaling axis. LNCaP (A) and C4–2 (B) cells were transfected with FHL2-luc, FHL2-luc in which the CArG box has been mutated (mut FHL2-luc), or SRE-luc and siRNA targeting AR, RhoA, SRF, MAL, or non-targeting control siRNa (c). The next day, cells were treated with ethanol vehicle or 5 nm R1881. Cells were harvested 2 d later, and a luciferase assay was performed. Columns, Mean values from samples in triplicate; bars, sem values.
Discussion
Recently, we have identified, to our knowledge, the first mechanism of androgen action of clinical relevance in PCa (21). By this mechanism, effects of androgens are mediated through the secondary TF SRF (Supplemental Fig. 15). The involvement of an intermediary factor, which constitutes an indirect mechanism of androgen action (37), may be of therapeutic interest because it provides a novel potential target for therapy other than AR. Initial investigation into the molecular mechanism(s) by which androgens regulate SRF activity demonstrated that SRF and AR do not interact directly and that no reciprocal effect on SRF and AR protein expression occurs (data not shown). Moreover, androgen exposure did not induce changes in SRF phosphorylation status, which can affect DNA binding by SRF (38, 39) nor did it affect the cellular localization of SRF [data not shown (Fig. 2 and Supplemental Fig. 1)]. In addition, recruitment of AR to SRF genomic binding sites did not occur under either androgen-deprived or androgen-supplemented conditions (20). Instead, these data supported the established model of SRF action in which SRF is bound constitutively to CArG boxes in regulatory regions of target genes and is activated by the recruitment of cofactors or activation of upstream signaling pathways.
Here, we show that the small GTPase RhoA conveys androgen-responsiveness to more than half of SRF target genes tested. The mechanism of RhoA involvement in androgen regulation of SRF action is supported by several lines of investigation representing diverse experimental approaches. Interference at several levels of the RhoA/actin/MAL signaling axis (RhoA, action of its effector protein ROCK and actin polymerization that leads to release and nuclear translocation of the critical SRF cofactor MAL) prevented androgen regulation of endogenous SRF target genes. These effects are RhoA-specific, because siRNA-mediated loss of the related GTPases Rac1 and cdc42 did not or only partially affect androgen dependency of FHL2 expression (data not shown). Moreover, upon loss of the SRF cofactor MAL, which relies on RhoA to translocate to the nucleus, androgen regulation of the same subset of SRF target genes was hampered. Conversely, overexpression of RhoA, which was observed in clinical specimens where it is associated with aggressive disease features and correlates with poor prognosis, augmented androgen regulation of select SRF target genes, in a manner that relies not only on RhoA's GTPase activity but also on SRF. Similarly, overexpression of MAL, which increases its nuclear content, mimicking the effect of androgens, enhanced androgen induction of FHL2 in an SRF-dependent manner. More importantly, androgen stimulation induced the levels of GTP-bound, active RhoA. Activation of RhoA preceded androgen-dependent nuclear relocalization of MAL and subsequent androgen-dependent modulation of SRF target gene expression. The critical importance of the activation state of RhoA for androgen regulation of SRF action was obvious in experiments using constitutively active and dominant-negative forms of RhoA and assessing expression of both endogenous SRF target genes and reporter constructs driven by SRF-dependent promoters. Androgen induction of SRF-driven reporter genes relied on AR, RhoA, SRF, and MAL, the presence of intact SRF binding sites and interaction between SRF and MAL in both AS and CR PCa cells.
The novel finding that AR acts upstream of RhoA adds more complexity to a growing body of evidence that indicates functional cross-talk between regulators of Rho action and nuclear receptors (e.g Refs. 40–43). Previous studies have reported on the ability of the Rho guanine nucleotide exchange factors (GEF) Vav3 to enhance AR transcriptional activity and simultaneous recruitment of Vav3 and AR to the same transcriptional complexes in AR target gene enhancer after androgen treatment (44–46). In addition, Vav3 expression is up-regulated in CRPC, where it is able to induce AR action in a ligand-independent manner upon stimulation with growth factors. Vav3's GEF function appears dispensable for ligand-dependent AR activation but is required for its ligand-independent activation. Our work identifies that mutual interference between AR and Rho regulators can occur also through control of AR over RhoA activity at select target genes.
RhoA expression is deregulated in several human malignancies (47–49). Our work, however, is the first to report on overexpression of RhoA in PCa and its association with aggressive disease and poor outcome. Our findings are supported by a previous study that, using an antibody unable to discriminate between RhoA, RhoB, and RhoC, noted elevated Rho immunostaining in PCa, which correlated with tumor dedifferentiation (40). The latter study linked Rho overexpression with increased nuclear expression of the SRF target gene FHL2. Our observations of RhoA overexpression at the protein level are corroborated also at the mRNA level. Analysis of oligoarray-derived gene expression profiles, in which clinical relevance of the AR- and SRF-dependent gene signature is confirmed (21), support overexpression of RhoA in PCa compared with benign prostate (data not shown). Our studies using isogenic cell lines and an animal model that mimics the progression from AS to CR PCa indicate that the level of RhoA in primary cancer may dictate disease aggressiveness because RhoA expression is not altered notably during PCa progression. These findings are in agreement with those from an independent study which found that RhoA mRNA expression was higher in patients who suffer systemic progression after biochemical failure after RRP compared with patients that have elevated PSA levels after RRP but whose disease does not progress systemically (50).
Rho family GTPases are molecular switches that control signaling pathways by recruiting effector proteins that induce changes in cellular functions as diverse as cytoskeleton organization, cell polarity, cell cycle progression, and gene transcription (30). RhoA cycles between an active GTP-bound state and an inactive GDP-bound state. GEF catalyze exchange of GDP for GTP, which is induced upon stimulation by growth factor receptors and integrins. GTPase-activating proteins increase the intrinsic GTPase activity, resulting in Rho GTPase inactivation. An additional level of regulation is provided by guanine nucleotide dissociation inhibitors that block the cycling between the GDP- and GTP-bound forms by preventing the exchange of GDP for GTP (30). Several well-known upstream regulators of RhoA activity and multiple downstream effectors are expressed differentially in PCa (e.g. Refs. 51–53), indicating that overexpression of RhoA may be only one contributing factor to facilitate activity of the RhoA signaling axis in PCa. Others such as underexpression of RhoE, which antagonizes RhoA action, may further enhance its action (54). Immunohistochemical assessment of cellular localization of MAL, which serves as another indicator of activity of the RhoA signaling axis, has been hampered by the poor specificity of the antibody directed against this important RhoA-regulated SRF cofactor (data not shown). Similarly, although the expression pattern and cellular localization of RhoA in clinical samples are consistent with the presence of active, GTP-bound RhoA, final validation of RhoA activation status in PCa specimens hinges on the availability of an antibody that can assess reliably RhoA-GTP levels. Activating mutations, which have been identified in other members of the small GTPase family (55), have not been described for RhoA but cannot be ruled out.
The association of higher RhoA expression with larger tumor volumes, higher GPSM scores, lymph node involvement and extraprostatic extension at RRP, and biochemical failure in clinical specimens suggests potential roles for RhoA in (androgen-dependent) PCa cell proliferation and cell migration. We attempted to discriminate between these possibilities using an approach that began with assessing the global cellular role of RhoA, and then narrowed gradually to evaluate the androgen-dependent fraction of RhoA action, androgen-dependent RhoA action that relies on SRF-MAL, and, finally, the contribution of androgen-responsive SRF target genes. Results from these experiments suggest that the fraction of the 158-gene signature that relies on RhoA to achieve androgen-responsiveness may affect preferentially cell migration. These observations are in line with ongoing work in our laboratory that is aimed at identifying the manner by which the SRF-dependent manner of androgen action conveys aggressive behavior to PCa cells. Preliminary results indicate that AR- and SRF target genes predominantly affect cell migration rather than cell proliferation and implicate rearrangements in actin cytoskeleton in cell movement (Verone, A.R., K. Duncan, N. Yadav, A. Godoy, A. Bakin, J-P. Jin, H.V. Heemers, manuscript in preparation). These observations are in agreement with the definition of aggressive disease used in our in silico analysis of AR- and SRF-dependent gene expression (21): increases in Gleason pattern numbers and the presence of lymph nodes metastases at the time of operation.
In addition to RhoA itself, its regulators and effector proteins may represent promising targets for anticancer therapy. Continuous delivery of the ROCK inhibitor Y-27632 considerably reduced the RhoA-mediated dissemination of hepatoma cells implanted in the peritoneal cavity of syngeneic rats (56). In addition, Y-27632 prevents small-cell lung cancer migration through human brain microvascular endothelial cells (57), and intrathecal administration of Y-27632 increased the survival time of rats suffering from neoplastic meningitis (58). Inhibition of the RhoA effector ROCK or interference with SRF-MAL interaction also retards migration and in vivo dissemination of PCa cells (59). The latter studies were limited to AR-negative PC-3 tumor xenografts. Selective inhibition of androgen-dependent RhoA activity will rely on a better understanding of the mechanism(s) by which AR imparts androgen regulation to RhoA or development of drugs specifically targeting RhoA as has been described for Rac (60). The kinetics by which androgens induce nuclear enrichment of MAL and modulate the expression of SRF target genes suggest an ARE-driven event upstream of RhoA. Noteworthy, rapid androgen induction of the putative GEF C-terminal SGEF, which harbors ARE-like motifs, has been reported (61).
Apart from its clinical relevance, our work is the first to demonstrate that the RhoA signaling axis, which harbors drugable targets (e.g. ROCK), mediates androgen action in PCa cells. Ongoing investigations in our laboratory indicate that RhoA control over clinically relevant androgen action is not limited to the LNCaP cell line as representative SRF target genes (e.g. CDC25A) are also androgen-regulated in a RhoA-dependent manner in independent AR-positive VCaP cells (data not shown). Moreover, careful review of the literature further supports androgen control over RhoA action in PCa cells. Treatment of the LuCaP35 PCa xenograft model system with the dual 5-α-reductase inhibitor dutasteride, which blocks the enzymatic conversion of testosterone to its more active metabolite dihydrotestosterone and has been suggested to prevent PCa development, has major effects on the expression of genes involved in Rho signaling (62). A study into the function of protease-activated receptors, which are upstream regulators of RhoA, reported that activity of RhoA is enhanced by dihydrotestosterone in LNCaP cells (63). Similar androgen-activation of RhoA has been reported to occur through a putative membranous AR (64).
RhoA controls the androgen dependency of more than half of SRF target genes tested, which indicates that another molecular mechanism(s) underlies androgen responsiveness of the remaining AR- and SRF target genes. Such a mechanism may also explain partial dependence on RhoA for a number of androgen-responsive SRF target genes (Fig. 4). A hallmark of SRF action is that it controls and toggles between different transcriptional programs by select interaction with a large variety of cofactors at CArG box-containing genomic regions. To date, more than 50 such cofactors have been described (65), several of which are under androgen control in PCa cells (24). We hypothesize that other, as yet unidentified, cofactors are involved in androgen control over SRF target genes that do not, or only partially, rely on RhoA/MAL for androgen regulation. In addition, it is conceivable that signaling cascades other than the one governed by RhoA direct androgen control over a select set of SRF target genes (Supplemental Fig. 15). Studies aimed at identifying these cofactors and/or pathways, their contribution to androgen regulation of SRF action, and the evolution of the expression and functional roles for such factors in the progression from AS to CR PCa, are ongoing in our laboratory.
In conclusion, our work identifies RhoA as a critical mediator of androgen action in PCa. The insights described here may provide the foundation for novel therapeutic approaches that inhibit clinically relevant AR activity in PCa.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants CA121277, CA91956, CA15083, and CA125747 (to D.J.T.), T.J. Martell Foundation (to D.J.T.), Mayo Clinic Prostate Cancer SPORE (to H.V.H.), Department of Defense Prostate Cancer Research Program (to H.V.H.), and by National Cancer Institute grant P30 CA016056.
Disclosure Summary: the authors have nothing to disclose
NURSA Molecule Pages†:
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- ADT
- Androgen deprivation therapy
- AR
- androgen receptor
- ARE
- androgen response element
- AS
- androgen-stimulated
- ChIP
- chromatin immunoprecipitation
- CR
- castration-recurrent
- CRPC
- castration-recurrent PCa
- CSS
- charcoal-stripped serum
- FBS
- fetal bovine serum
- GEF
- guanine nucleotide exchange factor
- MAL
- megakaryocytic acute leukemia
- PCa
- prostate cancer
- PSA
- prostate-specific antigen
- RhoA
- ras homolog family member A
- ROCK
- Rho-associated coiled-coil containing protein kinase 1
- RRP
- radical retropubic prostatectomy
- siRNA
- small interfering RNA
- SRF
- serum response factor
- T
- testosterone
- TF
- transcription factor
- TMA
- tissue microarray
- TP
- testosterone proprionate.
References
- 1. Siegel R, Naishadham D, Jemal A. 2012. Cancer statistics, 2012. CA Cancer J Clin 62:10–29 [DOI] [PubMed] [Google Scholar]
- 2. Klein EA, Ciezki J, Kupelian PA, Mahadevan A. 2009. Outcomes for intermediate risk prostate cancer: are there advantages for surgery, external radiation, or brachytherapy? Urol Oncol 27:67–71 [DOI] [PubMed] [Google Scholar]
- 3. Miyamoto H, Messing EM, Chang C. 2004. Androgen deprivation therapy for prostate cancer: current status and future prospects. Prostate 61:332–353 [DOI] [PubMed] [Google Scholar]
- 4. Litvinov IV, De Marzo AM, Isaacs JT. 2003. Is the Achilles' heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab 88:2972–2982 [DOI] [PubMed] [Google Scholar]
- 5. Debes JD, Tindall DJ. 2004. Mechanisms of androgen-refractory prostate cancer. N Engl J Med 351:1488–1490 [DOI] [PubMed] [Google Scholar]
- 6. Mohler JL. 2008. Castration-recurrent prostate cancer is not androgen-independent. Adv Exp Med Biol 617:223–234 [DOI] [PubMed] [Google Scholar]
- 7. Knudsen KE, Scher HI. 2009. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res 15:4792–4798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G, de Bono JS. 2008. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol 26:4563–4571 [DOI] [PubMed] [Google Scholar]
- 9. Attard G, Reid AH, Olmos D, de Bono JS. 2009. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res 69:4937–4940 [DOI] [PubMed] [Google Scholar]
- 10. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. 2009. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324:787–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Attard G, Cooper CS, de Bono JS. 2009. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell 16:458–462 [DOI] [PubMed] [Google Scholar]
- 12. Chen Y, Clegg NJ, Scher HI. 2009. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol 10:981–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heemers HV, Tindall DJ. 2007. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev 28:778–808 [DOI] [PubMed] [Google Scholar]
- 14. Massie CE, Adryan B, Barbosa-Morais NL, Lynch AG, Tran MG, Neal DE, Mills IG. 2007. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep 8:871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. 2007. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev 21:2005–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Q, Li W, Liu XS, Carroll JS, Jänne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. 2007. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 27:380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jia L, Berman BP, Jariwala U, Yan X, Cogan JP, Walters A, Chen T, Buchanan G, Frenkel B, Coetzee GA. 2008. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS One 3:e3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, Jänne OA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, Kantoff PW, Liu XS, Brown M. 2009. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takayama K, Tsutsumi S, Katayama S, Okayama T, Horie-Inoue K, Ikeda K, Urano T, Kawazu C, Hasegawa A, Ikeo K, Gojyobori T, Ouchi Y, Hayashizaki Y, Aburatani H, Inoue S. 2011. Integration of cap analysis of gene expression and chromatin immunoprecipitation analysis on array reveals genome-wide androgen receptor signaling in prostate cancer cells. Oncogene 30:619–630 [DOI] [PubMed] [Google Scholar]
- 20. Heemers HV, Regan KM, Dehm SM, Tindall DJ. 2007. Androgen induction of the androgen receptor coactivator four and a half LIM domain protein-2: evidence for a role for serum response factor in prostate cancer. Cancer Res 67:10592–10599 [DOI] [PubMed] [Google Scholar]
- 21. Heemers HV, Schmidt LJ, Sun Z, Regan KM, Anderson SK, Duncan K, Wang D, Liu S, Ballman KV, Tindall DJ. 2011. Identification of a clinically relevant androgen-dependent gene signature in prostate cancer. Cancer Res 71:1978–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Posern G, Treisman R. 2006. Actin' together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol 16:588–596 [DOI] [PubMed] [Google Scholar]
- 23. Cen B, Selvaraj A, Prywes R. 2004. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J Cell Biochem 93:74–82 [DOI] [PubMed] [Google Scholar]
- 24. Heemers HV, Regan KM, Schmidt LJ, Anderson SK, Ballman KV, Tindall DJ. 2009. Androgen modulation of coregulator expression in prostate cancer cells. Mol Endocrinol 23:572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heemers HV, Schmidt LJ, Kidd E, Raclaw KA, Regan KM, Tindall DJ. 2010. Differential regulation of steroid nuclear receptor coregulator expression between normal and neoplastic prostate epithelial cells. Prostate 70:959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim D, Gregory CW, French FS, Smith GJ, Mohler JL. 2002. Androgen receptor expression and cellular proliferation during transition from androgen-dependent to recurrent growth after castration in the CWR22 prostate cancer xenograft. Am J Pathol 160:219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karpf AR, Bai S, James SR, Mohler JL, Wilson EM. 2009. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol Cancer Res 7:523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyamoto KK, McSherry SA, Dent GA, Sar M, Wilson EM, French FS, Sharief Y, Mohler JL. 1993. Immunohistochemistry of the androgen receptor in human benign and malignant prostate tissue. J Urol 149:1015–1019 [DOI] [PubMed] [Google Scholar]
- 29. Xiao L, Eto M, Kazanietz MG. 2009. ROCK mediates phorbol ester-induced apoptosis in prostate cancer cells via p21Cip1 up-regulation and JNK. J Biol Chem 284:29365–29375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fritz G, Kaina B. 2006. Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets 6:1–14 [PubMed] [Google Scholar]
- 31. Thompson RH, Blute ML, Slezak JM, Bergstralh EJ, Leibovich BC. 2007. Is the GPSM scoring algorithm for patients with prostate cancer valid in the contemporary era? J Urol [Erratum (2007) 178:2224] 178:459–463;discussion 463 [DOI] [PubMed] [Google Scholar]
- 32. Qiu RG, Chen J, McCormick F, Symons M. 1995. A role for Rho in Ras transformation. Proc Natl Acad Sci USA 92:11781–11785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Evelyn CR, Wade SM, Wang Q, Wu M, Iñiguez-Lluhí JA, Merajver SD, Neubig RR. 2007. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther 6:2249–2260 [DOI] [PubMed] [Google Scholar]
- 34. Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. 1994. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer 57:406–412 [DOI] [PubMed] [Google Scholar]
- 35. Murillo H, Huang H, Schmidt LJ, Smith DI, Tindall DJ. 2001. Role of PI3K signaling in survival and progression of LNCaP prostate cancer cells to the androgen refractory state. Endocrinology 142:4795–4805 [DOI] [PubMed] [Google Scholar]
- 36. Heemers HV, Sebo TJ, Debes JD, Regan KM, Raclaw KA, Murphy LM, Hobisch A, Culig Z, Tindall DJ. 2007. Androgen deprivation increases p300 expression in prostate cancer cells. Cancer Res 67:3422–3430 [DOI] [PubMed] [Google Scholar]
- 37. Verhoeven G, Swinnen JV. 1999. Indirect mechanisms and cascades of androgen action. Mol Cell Endocrinol 151:205–212 [DOI] [PubMed] [Google Scholar]
- 38. Manak JR, de Bisschop N, Kris RM, Prywes R. 1990. Casein kinase II enhances the DNA binding activity of serum response factor. Genes Dev 4:955–967 [DOI] [PubMed] [Google Scholar]
- 39. Janknecht R, Hipskind RA, Houthaeve T, Nordheim A, Stunnenberg HG. 1992. Identification of multiple SRF N-terminal phosphorylation sites affecting DNA binding properties. EMBO J 11:1045–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Müller JM, Metzger E, Greschik H, Bosserhoff AK, Mercep L, Buettner R, Schüle R. 2002. The transcriptional coactivator FHL2 transmits Rho signals from the cell membrane into the nucleus. EMBO J 21:736–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rubino D, Driggers P, Arbit D, Kemp L, Miller B, Coso O, Pagliai K, Gray K, Gutkind S, Segars J. 1998. Characterization of Brx, a novel Dbl family member that modulates estrogen receptor action. Oncogene 16:2513–2526 [DOI] [PubMed] [Google Scholar]
- 42. Su LF, Knoblauch R, Garabedian MJ. 2001. Rho GTPases as modulators of the estrogen receptor transcriptional response. J Biol Chem 276:3231–3237 [DOI] [PubMed] [Google Scholar]
- 43. Kino T, Souvatzoglou E, Charmandari E, Ichijo T, Driggers P, Mayers C, Alatsatianos A, Manoli I, Westphal H, Chrousos GP, Segars JH. 2006. Rho family Guanine nucleotide exchange factor Brx couples extracellular signals to the glucocorticoid signaling system. J Biol Chem 281:9118–9126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lyons LS, Burnstein KL. 2006. Vav3, a Rho GTPase guanine nucleotide exchange factor, increases during progression to androgen independence in prostate cancer cells and potentiates androgen receptor transcriptional activity. Mol Endocrinol 20:1061–1072 [DOI] [PubMed] [Google Scholar]
- 45. Lyons LS, Rao S, Balkan W, Faysal J, Maiorino CA, Burnstein KL. 2008. Ligand-independent activation of androgen receptors by Rho GTPase signaling in prostate cancer. Mol Endocrinol 22:597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rao S, Lyons LS, Fahrenholtz CD, Wu F, Farooq A, Balkan W, Burnstein KL. 2012. A novel nuclear role for the Vav3 nucleotide exchange factor in androgen receptor coactivation in prostate cancer. Oncogene 31:716–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kamai T, Arai K, Tsujii T, Honda M, Yoshida K. 2001. Overexpression of RhoA mRNA is associated with advanced stage in testicular germ cell tumour. BJU Int 87:227–231 [DOI] [PubMed] [Google Scholar]
- 48. Fritz G, Just I, Kaina B. 1999. Rho GTPases are over-expressed in human tumors. Int J Cancer 81:682–687 [DOI] [PubMed] [Google Scholar]
- 49. Abraham MT, Kuriakose MA, Sacks PG, Yee H, Chiriboga L, Bearer EL, Delacure MD. 2001. Motility-related proteins as markers for head and neck squamous cell cancer. Laryngoscope 111:1285–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nakagawa T, Kollmeyer TM, Morlan BW, Anderson SK, Bergstralh EJ, Davis BJ, Asmann YW, Klee GG, Ballman KV, Jenkins RB. 2008. A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PLoS One 3:e2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kelly P, Stemmle LN, Madden JF, Fields TA, Daaka Y, Casey PJ. 2006. A role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J Biol Chem 281:26483–26490 [DOI] [PubMed] [Google Scholar]
- 52. Nie D, Guo Y, Yang D, Tang Y, Chen Y, Wang MT, Zacharek A, Qiao Y, Che M, Honn KV. 2008. Thromboxane A2 receptors in prostate carcinoma: expression and its role in regulating cell motility via small GTPase Rho. Cancer Res 68:115–121 [DOI] [PubMed] [Google Scholar]
- 53. Metzger E, Müller JM, Ferrari S, Buettner R, Schüle R. 2003. A novel inducible transactivation domain in the androgen receptor: implications for PRK in prostate cancer. EMBO J 22:270–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bektic J, Pfeil K, Berger AP, Ramoner R, Pelzer A, Schäfer G, Kofler K, Bartsch G, Klocker H. 2005. Small G-protein RhoE is underexpressed in prostate cancer and induces cell cycle arrest and apoptosis. Prostate 64:332–340 [DOI] [PubMed] [Google Scholar]
- 55. Schubbert S, Shannon K, Bollag G. 2007. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7:295–308 [DOI] [PubMed] [Google Scholar]
- 56. Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S. 1999. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat Med 5:221–225 [DOI] [PubMed] [Google Scholar]
- 57. Li B, Zhao WD, Tan ZM, Fang WG, Zhu L, Chen YH. 2006. Involvement of Rho/ROCK signalling in small cell lung cancer migration through human brain microvascular endothelial cells. FEBS Lett 580:4252–4260 [DOI] [PubMed] [Google Scholar]
- 58. Nakagawa H, Yoshioka K, Miyahara E, Fukushima Y, Tamura M, Itoh K. 2005. Intrathecal administration of Y-27632, a specific rho-associated kinase inhibitor, for rat neoplastic meningitis. Mol Cancer Res 3:425–433 [DOI] [PubMed] [Google Scholar]
- 59. Somlyo AV, Bradshaw D, Ramos S, Murphy C, Myers CE, Somlyo AP. 2000. Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem Biophys Res Commun 269:652–659 [DOI] [PubMed] [Google Scholar]
- 60. Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. 2004. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA 101:7618–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qi H, Fournier A, Grenier J, Fillion C, Labrie Y, Labrie C. 2003. Isolation of the novel human guanine nucleotide exchange factor Src homology 3 domain-containing guanine nucleotide exchange factor (SGEF) and of C-terminal SGEF, an N-terminally truncated form of SGEF, the expression of which is regulated by androgen in prostate cancer cells. Endocrinology 144:1742–1752 [DOI] [PubMed] [Google Scholar]
- 62. Schmidt LJ, Regan KM, Anderson SK, Sun Z, Ballman KV, Tindall DJ. 2009. Effects of the 5 α-reductase inhibitor dutasteride on gene expression in prostate cancer xenografts. Prostate 69:1730–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Greenberg DL, Mize GJ, Takayama TK. 2003. Protease-activated receptor mediated RhoA signaling and cytoskeletal reorganization in LNCaP cells. Biochemistry 42:702–709 [DOI] [PubMed] [Google Scholar]
- 64. Papadopoulou N, Charalampopoulos I, Alevizopoulos K, Gravanis A, Stournaras C. 2008. Rho/ROCK/actin signaling regulates membrane androgen receptor induced apoptosis in prostate cancer cells. Exp Cell Res 314:3162–3174 [DOI] [PubMed] [Google Scholar]
- 65. Miano JM, Long X, Fujiwara K. 2007. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol 292:C70–C81 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.