

After a decade’s worth of rapid research advances, we begin to understand in detail how a normal mammalian cell decides whether it should grow. Those interested in curing cancer have consumed this information avidly. They believe that an understanding of normal cell cycle control provides them with the key to developing new generations of antitumor therapeutics.
At the end of the decade, we now know that the runaway proliferation of a malignant cell is due to minor defects in the machinery that governs normal cell growth and division. On the one hand, this is reassuring, because the root causes of cancer can be pinpointed with precision, ascribed to defective components operating in one or another location in the cellular growth-regulating circuitry. But on the other, the prospects for anticancer drug development have become all the more daunting, since the differences between malignant and normal proliferative control appear to be so small, and thus seem to provide few opportunities for selectively targeting cancer cells while leaving their normal counterparts untouched.
The report by Chen et al. (1) in this issue describes one of the first efforts to exploit our newly gained information on mammalian cell cycle control to kill cancer cells. The fact that this is a pioneering effort in this area is itself surprising. In the face of so much information, relatively few researchers have found windows of opportunity for drug development. They have looked for the Achilles heels of cancer cells and been hard-pressed to identify attractive targets. As Chen and coworkers now show, creative interpretation of recent discoveries about the cell cycle clock has revealed a vulnerability, a chink in the armor of the cancer cell.
Machinery: The Cell Cycle Clock.
In one sense, the metaphor of a clock is unfortunate: the cell cycle clock does not measure elapsed time. Instead, it operates as a complex circuitry that continuously acquires growth-regulating signals from many sources, integrates and processes this information, makes some major decisions, and then dispatches commands to the distant corners of the cell, thereby choreographing cellular behavior. Any resemblance to an actual clock derives from the fact that this behavior includes the orderly movement of the cell through discrete phases of the cell cycle, each phase executed according to a precise, predetermined timetable (2).
The core components of the cell cycle clock are the cyclins and cyclin-dependent kinases (cdks). The cyclins associate with cdks, activate them, and direct them to specific protein substrates. Once modified, the resulting phosphorylated substrates take on new lives. For example, the well-studied retinoblastoma protein (pRB) inhibits cell cycle advance when underphosphorylated; once phosphorylated by two cyclin/cdk complexes, it permits the cell to progress from the G1 phase of its growth cycle into S phase. In a similar manner, the schedule of activation of other cyclin/cdk complexes determines the timing of other transitions in the cell cycle.
The critical decisions about cell growth vs. quiescence are made in the G1 phase of the cell cycle. During most of this phase, the cell is responsive to extracellular signals, including those conveyed by mitogenic growth factors and those carried by anti-proliferative signalers such as transforming growth factor β (TGF-β). These extracellular signals are funneled through the cytoplasm to the nucleus, where the clock operates. They determine whether pRB become phosphorylated, and pRB, in turn issues a GO or NO-GO signal to the cell. The cell is blocked in G1, forced to retreat into a quiescent G0 state, or permitted to advance into S phase (3).
pRB, for its part, issues commands through its ability to control the activities of certain transcription factors and thus responding genes. The most prominent and apparently important of these are factors of the E2F family (4). When active, these E2F factors enable the expression of a large cohort of target genes whose products are required for S-phase entrance and advance. This leaves us with a linear chain-of-command that looks like this:
 |
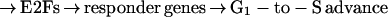 |
This signaling cascade is overlaid with a complex array of governors, including cdk inhibitors that may block the activities of the cyclin/cdk complexes (5), phosphatases that control the cdks directly, and some feedback controls that ensure proper execution of certain steps. A further dimension of complexity is created by the existence of multiple E2Fs with distinct functions and two pRB-related proteins, p107 and p130, which operate in distinct ways to control E2Fs and their downstream responder genes (4).
Action in a Narrow Window of Time.
The E2Fs seem designed to stimulate active transcription in only a narrow window of time. The beginning of this time window is demarcated by the phosphorylation of pRB, the retinoblastoma protein. Throughout most of G1, pRB is hypophosphorylated, and in this state, binds avidly to E2Fs-1, -2, and -3. pRB binding masks the transcription-activating domain of the E2Fs (6). Because this association does not inhibit the E2Fs’ ability to bind to their cognate DNA sequence, pRB/E2F complexes are thought to roost on the promoters of E2F target genes. Once recruited to the promoters in this manner, pRB can block the basal transcription of these genes (7).
Late in G1, as indicated in the above scheme, pRB phosphorylation is provoked by cyclin/cdk complexes and the hyperphosphorylated pRB relaxes its hold on E2Fs. This may then have two molecular consequences (4). The dissociation of pRB from E2Fs will liberate the promoter from the potent transcription-inhibitory effect exerted by pRB. In addition, the uncovering of the transcription-activating domain of the E2Fs will lead to a burst of E2F-activated transcription. In this manner, the phosphorylation of pRB can switch E2F-responsive genes from the fully repressed to the fully induced state.
The end of the time window is established by a second molecular switch. When cells pass through the G1/S transition and enter S phase, cyclin A/cdk2 complexes are activated, in large part to enable S-phase progression. But these cyclin/CDK complexes have an additional function: they phosphorylate E2F-1, -2, and –3, causing them to dissociate from their binding sites on the DNA (8, 9). This inactivation demarcates the end of E2F transcription-promoting activity in the normal cell cycle. Hence E2F-1, -2, and –3 are given free rein only for the short period of time—at most a few hours—intervening between the activation of the cyclin E/cdk2 (which completes pRB phosphorylation in late G1) and the activation of cyclin A/cdk2 (which begins at the G1/S transition).
Paradoxical Mice.
The scheme depicted above indicates that these three E2F factors (1, 2, and 3) function as effectors that are used by the cell cycle clock to execute progression through the end of the G1 phase of the cell cycle; they do so by mobilizing the expression of genes required for S-phase entrance. However, the results of experiments with genetically altered mice, completed several years ago, are hard to reconcile with this scheme (10, 11).
Those who created mice lacking the E2F-1 factor (through germ-line gene inactivation) expected that these animals would show either embryonic lethality or hypoplasia because of the lack of this important proliferative factor. Although some tissue atrophy was observed, the E2f1−/− mice yielded a major surprise: they developed a broad spectrum of tumors, particularly reproductive tract sarcomas and lymphomas (11).
These observations indicated that the E2F-1 gene has, at least in a genetic sense, the properties of a tumor suppressor gene. As for others of this class, the loss or inactivation of this gene favors the appearance of tumors. In their normal incarnations, such genes are presumed to block tumorigenesis through their ability to impede cell cycle advance, to eliminate premalignant cells through apoptosis, or to act in yet other ways to impede tumor progression. None of these functions was easily reconciled with the simple scheme in which E2F-1 acts to facilitate the forward march of cells through their growth cycle.
When Things Go Wrong.
A possible solution to this conundrum has come from studies of cultured cells in which E2F-1 expression has been forced through introduction of an expression vector. When E2F-1 was over-expressed in quiescent G0 cells through use of such a vector, these cells were driven to enter into the active growth cycle and to progress into S phase (12). This was a stunning demonstration that a single transcription factor could push cells all the way from G0 through G1 into S phase, and validated the idea that E2Fs play a vital role in executing the G1 cell cycle program.
However, the endpoint of this experiment yielded a surprise. After entering S phase, these E2F-1-expressing cells died, victims of apoptosis (13–15). This finding yielded a speculation: perhaps the continued activity of E2F-1 extending into S phase, which might occur in cells that express E2F-1 at unusually high levels, was incompatible with normal cell cycle advance, and therefore triggered the alarm signals leading to apoptosis.
This speculation was strongly supported by an experiment in which a mutant E2F-1 was expressed in quiescent cells. This mutant E2F-1 carried an alteration in its cyclin A/cdk2 binding site that rendered it immune to inactivation by this cyclin/cdk complex (16). (As mentioned above, such inactivation, achieved through phosphorylation of E2Fs, normally occurs in S phase and terminates further E2F activity.) Ectopic expression of this mutant E2F-1 resulted in a strong potentiation of apoptosis in S phase. Together, these observations indicated that high levels of E2F-1 favor transit through the G1 phase of the cell cycle, but its continued activity during S phase triggers apoptosis.
These experiments leave a critical question unanswered: can inappropriately activated E2F-1 trigger apoptosis at physiologic levels? A recent study indicates that it can do exactly that (17). Homozygous Rb mutant mice die during embryogenesis as a result of inappropriate S-phase entry and apoptosis in several tissues. The above models would suggest that in the absence of its pRB controller, E2F-1 activity may well be deregulated. And indeed, by crossing the Rb and E2f-1 mutant mice, Tsai et al. showed that apoptosis in these mouse tissues was almost completely abolished when E2F-1 was deleted from these mice. Hence, as speculated above, deregulated E2F-1 activity can act as an agent for triggering apoptosis.
The control circuitry of mammalian cells is wired to ensure that when things go awry, alarm bells sound, including those that provoke apoptosis. The intent is to quickly eliminate cells whose growth-regulating machinery is malfunctioning. Viewed from this perspective, E2F-1-induced apoptosis makes perfect sense: in many human tumors, the upstream controllers of E2Fs (including cyclins, cdk inhibitors, and pRB) are disrupted through a variety of genetic and epigenetic mechanisms (18). The resulting deregulation of E2Fs should then serve as an ideal bellwether, revealing whether its upstream controllers are operating as they should. If the upstream pRB circuitry is damaged, the resulting E2F deregulation can be used to quickly sound the apoptotic alarm, enabling elimination of the aberrant cell.
But E2F-1-provoked apoptosis may have been co-opted by the opportunistic hand of evolution for yet another purpose. Perhaps in some cell types, as part of their normal developmental programs, E2F-1 activity is purposefully deregulated to trigger apoptotic death. For example, certain types of lymphocytes might physiologically up-regulate E2F-1 expression in S phase to activate their own death program. This would explain the unexpected tumors in the E2f1−/− mice. The inability of their lymphocytes to wield the E2F-1 suicide weapon would result in a pathological expansion of the lymphoid compartment and, eventually, in lymphomas.
Killing Cancer Cells.
All this forms the background for the paper by Chen et al. in this issue (1). The authors reasoned that by inhibiting cyclin A/cdk2 activity, they might allow E2F-1 activity to survive into S phase and trigger apoptosis, and this is precisely what they have found. They succeeded in inhibiting cyclin A/cdk activity by using specific oligopeptides that are able to freely enter cells and block the interaction of this cyclin/cdk complex with E2Fs.
Provocatively, these oligopeptides are quite selective, being able to kill certain tumor cells in culture while sparing nontumorigenic counterparts. Over-expression of E2F-1 sensitizes a normal cell to this oligopeptide-induced cell death. All this is consistent with the idea that the observed apoptosis is caused by the inappropriate expression of E2F-1 and that tumor cells, having higher levels of active E2Fs, are more hard-pressed than their normal counterparts to inactivate E2Fs at the critical juncture in early S phase. The authors’ data suggest that a minor reduction in the cyclin A/cdk2-mediated shutdown of E2Fs will push such tumor cells over the edge of the cliff into the apoptotic abyss. Most importantly, these data suggest that cdk2 inhibitors may be very effective weapons in the fight against cancer.
We are still a long way from converting this cleverness into drugs that are effective in the oncology clinic. Oligopeptides are relatively bulky molecules and have not proven to be useful drugs when administered systemically. However, a new cycle of rational drug design will likely be triggered by these findings, in which relatively low molecular weight compounds are synthesized that mimic the steric and functional attributes of the oligopeptides. Strategies like these give great hope to those who would like to translate our now-substantial knowledge about cell cycle control into real changes in the treatment of cancer.
Footnotes
A commentary on this article begins on page 4325.
References
- 1.Chen Y-N P, Sharma S K, Ramsey T M, Jiang L, Martin M S, Baker K, Adams P D, Bair K W, Kaelin W G., Jr Proc Natl Acad Sci USA. 1999;96:4325–4329. doi: 10.1073/pnas.96.8.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nasymth K. Science. 1996;274:1643–1645. doi: 10.1126/science.274.5293.1643. [DOI] [PubMed] [Google Scholar]
- 3.Planas-Silva M D, Weinberg R A. Curr Opin Cell Biol. 1997;9:768–772. doi: 10.1016/s0955-0674(97)80076-2. [DOI] [PubMed] [Google Scholar]
- 4.Dyson N. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 5.Sherr C J, Roberts J M. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 6.Helin K, Lees J A, Vidal M, Dyson N, Harlow E, Fattaey A. Cell. 1992;70:337–350. doi: 10.1016/0092-8674(92)90107-n. [DOI] [PubMed] [Google Scholar]
- 7.DePinho R A. Nature (London) 1998;391:533. doi: 10.1038/35257. , 535–536. [DOI] [PubMed] [Google Scholar]
- 8.Dynlacht B D, Flores O, Lees J A, Harlow E. Genes Dev. 1994;8:1772–1786. doi: 10.1101/gad.8.15.1772. [DOI] [PubMed] [Google Scholar]
- 9.Krek W, Ewen M E, Shirodkar S, Arany Z, Kaelin W G, Jr, Livingston D M. Cell. 1994;78:161–172. doi: 10.1016/0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 10.Field S J, Tsai F Y, Kuo F, Zubiaga A M, Kaelin W G, Jr, Livingston D M, Orkin S H, Greenberg M E. Cell. 1996;85:549–461. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- 11.Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson N J. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- 12.Johnson D G, Schwartz J K, Cress W D, Nevins J R. Nature (London) 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- 13.Shan B, Lee W H. Mol Cell Biol. 1994;14:299–309. doi: 10.1128/mcb.14.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin X Q, Livingston D M, Kaelin W G, Jr, Adams P D. Proc Natl Acad Sci USA. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Levine A J. Proc Natl Acad Sci USA. 1994;91:3602–3606. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krek W, Xu G, Livingston D M. Cell. 1995;83:1149–1158. doi: 10.1016/0092-8674(95)90141-8. [DOI] [PubMed] [Google Scholar]
- 17.Tsai K Y, Hu Y, Macleod K F, Crowley D, Yamasaki L, Jacks T. Mol Cell. 1998;2:293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- 18.Sherr C J. Science. 1996;6:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]