

Abstract
CDK9 is the kinase of positive transcription elongation factor b and facilitates the transition of paused RNA polymerase II to processive transcription elongation. CDK9 is a validated target for the treatment of cancer, cardiac hypertrophy, and human immunodeficiency virus. Here we analyze different CDK9/cyclin T variants to identify a form of the complex amenable to use in inhibitor design. To demonstrate the utility of this system, we have determined the crystal structures of CDK9/cyclin T and CDK2/cyclin A bound to the CDK9-specific inhibitor CAN508. Comparison of the structures reveals CDK9-specific conformational changes and identifies a CDK9-specific hydrophobic pocket, adjacent to the αC-helix. By comparison with a previously published structure of CDK9/cyclin T/human immunodeficiency virus TAT we find that the CDK9 αC-helix has a degree of conformational variability that has the potential to be exploited for inhibitor design.
Many cancer cells rely on the production of anti-apoptotic factors for their survival. Inhibition of mRNA synthesis and the consequent down regulation of anti-apoptotic factors is therefore an attractive strategy for cancer treatment. mRNA synthesis by RNA polymerase II (pol II) is regulated by the phosphorylation of its C-terminal domain (CTD) by a range of cyclin-dependent kinases (CDKs) CDK7–CDK9, CDK12, and CDK13. Since the discovery that the CDK inhibitor flavopiridol induces apoptosis by inhibiting CDK9,1 the enzyme has been a target for anticancer drug design. Subsequent studies established that other CDK inhibitors exploit the same mechanism.2−8 In addition to oncology, CDK9 has also been validated as a drug target in virology and cardiology.9,10 While there are several promising compounds with high affinity for CDK9, their selectivity toward this CDK family member is limited. To exploit CDK9 inhibition for the treatment of these diseases, it is crucial to obtain CDK inhibitors with a high degree of selectivity and potency for CDK9.
CAN508 is an arylazopyrazole compound that inhibits CDK9 with an IC50 of 0.35 μM and exhibits a 38-fold selectivity for CDK9/cyclin T over other CDK/cyclin complexes.3 As is consistent with these properties, CAN508 treatment inhibits the growth of various cancer cell lines and induces apoptosis through mechanisms that include inhibition of pol II CTD phosphorylation and mRNA synthesis, and induction of the tumor suppressor protein p53.3,11 More recently, CAN508 has also been shown to inhibit angiogenesis through a CDK9-dependent mechanism.11
Structure-aided drug design requires readily crystallizable protein that can routinely generate inhibitor co-crystals diffracting to sufficient resolution. To date, structures of the apo form of CDK9/cyclin T as well as complexes bound to ATP and CDK9-selective inhibitors have been determined.12,13 In addition the structure of a CDK9/cyclin T/HIV TAT complex has been reported.14
Here we evaluate the usefulness of the existing CDK9/cyclinT crystal systems for inhibitor design studies. A comparison of the different crystal forms reveals that CDK9/cyclin T has a degree of conformational flexibility that can be exploited for inhibitor design. Serendipitous mutations in CDK9/cyclin T resulted in a form of the complex that has produced crystals that diffract up to 2.5 Å resolution.12 We show that this mutated form has largely unaltered kinetic, structural, and inhibitor binding properties but offers a number of advantages over the wild-type protein for inhibitor design studies. To demonstrate the advantages of comparative CDK structural studies for the design of selective CDK9 inhibitors, we have solved the crystal structures of active CDK9/cyclin T and CDK2/cyclin A bound to the inhibitor CAN508. Comparison of the CDK9/cyclin T- and CDK2/cyclin A-CAN508 co-crystal structures reveals that the inhibitor adopts slightly different orientations within the ATP binding sites of the two kinases. Our results identify a CDK9-specific hydrophobic pocket that may be exploited to design inhibitors with increased selectivity toward CDK9.
The CDK9/cyclin T crystal form used for inhibitor binding studies to date was first reported by Baumli and colleagues.12 This crystal form offers a number of advantages for inhibitor studies, namely: (i) reproducible crystallization conditions that generate crystals that diffract to 2.5–3.0 Å resolution, (ii) a CDK9 ATP binding site that is freely accessible for inhibitor soaking experiments, and (iii) a lack of crystal contacts at the ATP binding site that would preclude inhibitor-induced conformational changes. These crystals result from a CDK9/cyclin T complex that contains three point mutations in the cyclin T sequence at residues Q77R, E96G, and F241L,12 and we therefore designate the triple mutant CDK9/cyclin TQ77R/E96G/F241L.
While CDK9/cyclin TQ77R/E96G/F241L crystallizes reliably, attempts to crystallize wild-type cyclin T in complex with CDK9 resulted in very small crystals that proved unsuitable for structural analysis. We therefore investigated the locations of the three mutations in the crystal structure. F241L is located in the C-terminal cyclin helix that is involved in crystal contacts. The mutation might contribute to the improved crystallization properties of CDK9/cyclin TQ77R/E96G/F241L. Q77R lies on a surface loop on cyclin T and is a natural variant of the cyclin sequence (UniProt entry: O60563). E96G is located at the CDK/cyclin interface, and mutation to a glycine residue might influence the interaction of cyclin T with CDK9.
To dissect the contributions that each mutation makes to the structure of CDK9/cyclin TQ77R/E96G/F241L, we crystallized a complex that is wild-type at cyclin T positions Gln77 and Glu96 but retains the F241L mutation to aid crystallization. We solved the structure of this complex at 3.2 Å resolution (Table 1). In CDK9/cyclin TF241L Glu96 of cyclin T hydrogen bonds to and reorients the side chain of CDK9 Arg65. Both cyclin T Glu96 and CDK9 Arg65 participate in the same hydrogen bonding network that is observed in the CDK9/cyclin T/TAT structure (PDB id: 3MI9).14 Despite the additional contact between the CDK and cyclin subunits the structure is very similar to that of CDK9/cyclin TQ77R/E96G/F241L (rmsd 0.93 Å over all CDK9 atoms compared to 3BLH and rmsd 1.3 Å compared to 3MI9; Supplementary Figure S1, Supplementary Table 1). The side chain of Gln77, like Arg77 in the CDK9/cyclin TQ77R/E96G/F241L structure, points out into solvent and makes no intra- or intermolecular contacts. Taken together these results indicate that the mutations present in CDK9/cyclin TQ77R/E96G/F241L do not influence the overall conformations of CDK9, cyclin T, or the CDK9/cyclin T complex. In particular we can exclude the possibility that conformational differences between PDB id's 3MI9 and 3BLH result from the E96G mutation in cyclin T present in PDB id 3BLH.
Table 1. Crystallographic Parameters.
| CDK9/cyclin TQ77R/E96G/F241L/CAN508 | CDK9/cyclin TF241L/CAN508 | CDK9/cyclin TF241L | CDK2/cyclin A/CAN508 | |
|---|---|---|---|---|
| Data Collection | ||||
| beamline | ESRF ID29 | DLS I24 | DLS I24 | DLS I02 |
| space group | H3 | H3 | H3 | P212121 |
| unit cell (Å) | a = b = 173.42;c = 97.9 | a = b = 173.30;c = 97.22 | a = b = 173.6;c = 97.47 | a = 74.05; b = 134.57; c = 148.28 |
| α = β = 90°; γ = 120° | α = β = 90°; γ = 120° | α = β = 90°; γ = 120° | α = β = γ = 90° | |
| resolution (highest resolution shell) (Å) | 43.36–2.95 (3.11–2.95) | 49.00–3.20 (3.29–3.20) | 59.52–3.23 (3.41–3.23) | 44.86–2.00 (2.11–2.00) |
| total observations | 84542 (12557) | 61839 (4669) | 58769 (8830) | 397326 (56907) |
| unique | 23081 (3394) | 17892 (1327) | 17300 (2557) | 100421 (14505) |
| Rmerge | 0.072 (0.59) | 0.051 (0.54) | 0.070 (0.49) | 10.3 (0.57) |
| multiplicity | 3.7 (3.7) | 3.5 (3.5) | 3.4 (3.5) | 4.0 (3.9) |
| mean I/σI | 13.3 (1.9) | 15.1 (2.0) | 9.9 (2.4) | 8.8 (2.2) |
| completeness | 99.8% (100%) | 99.8% (99.9%) | 98.8% (99.7%) | 99.8% (97.0%) |
| Refinement Statistics | ||||
| highest resolution shell (Å) | 3.08–2.95 | 3.40–3.20 | 3.44–3.23 | 2.02–2.00 |
| total no. of atoms | 4587 | 4358 | 4489 | 18970 |
| no. of waters | 8 | NA | NA | 913 |
| R | 0.176 (0.314) | 0.177 (0.259) | 0.163 (0.242) | 0.1877 (0.269) |
| Rfree (highest resolution shell) | 0.221 (0.358) | 0.226 (0.318) | 0.210 (0.310) | 0.2266 (0.301) |
| rms bonds | 0.005 | 0.013 | 0.009 | 0.007 |
| rms angles | 0.842 | 1.526 | 1.344 | 1.038 |
Although they do not introduce a significant structural perturbation, the mutations present in CDK9/cyclin TQ77R/E96G/F241L might affect CDK9/cyclin T stability and kinetic parameters. To test this hypothesis, we first analyzed the thermal stability of the CDK9/cyclin T variants. While CDK/cyclin complexes composed of either the wild-type proteins or cyclin TF241L have very similar melting temperatures (Tm) of 52.6 ± 0.16 and 51.8 ± 1.1 °C, respectively, the triple mutant complex, CDK9/cyclin TQ77R/E96G/F241L denatures at a lower temperature of 48.25 ± 0.58 °C (Supplementary Figure S2a). The lower Tm of this complex most likely results, at least in part, from the loss of the hydrogen bonding network in which Glu96 participates.
To determine whether this lowered Tm has an impact on inhibitor binding to the mutant CDK9/cyclin T, we employed differential scanning fluorimetry (DSF), a technique that exploits the fact that ligand binding to a protein increases its thermal stability and hence its apparent melting temperature (ΔTm). The ΔTm measured is related to the binding affinity of the ligand for the protein and is dependent upon the ligand concentration. It also reflects the constituent thermodynamic properties of the ligand binding event that includes ΔS and ΔCp.15 A set of ΔTm values measured for a given enzyme bound to a diverse panel of ligands can, therefore, provide a fingerprint that is characteristic of the ligand binding properties of that enzyme. The fingerprints of two different enzymes can be quantitatively compared by evaluating the R2 linear correlation coefficient between sets of inhibitor-induced ΔTm values measured for those enzymes.
To analyze the effect of the cyclin T mutations on the interactions between CDK9 and ATP-competitive inhibitors, we tested the ability of known CDK9 inhibitors (DRB, CAN508, (S)-CR8, Roscovitine, Staurosporine, AMPPNP, and Wang compound 82) to stabilize the different CDK9/cyclin T complexes. The profiles of inhibitor-induced Tm shifts for wild-type CDK9/cyclin T and the mutant complexes are very similar (R2 > 0.93 for all combinations of CDK9/cyclin T variants tested (Figure 1a)). For comparison, the R2 value obtained when comparing the ΔTm values measured for CDK2/cyclin A with those obtained for wild-type CDK9/cyclin T is significantly lower (R2 = 0.33), as expected (Figure 1a). These results indicate that, despite slightly lowering the enzyme's inherent Tm, the three mutations within CDK9/cyclin TQ77R/E96G/F241L do not result in detectable changes to the inhibitor binding properties of the CDK9 ATP-binding site.
Figure 1.

Comparison of different CDK9/cyclin T variants. (a) Correlation between inhibition profiles of CDK9/cyclin T variants and CDK2/cyclin A. Each data point represents the average ΔTm of three curves. The internal correlation within a data set is R2 > 0.9. Individual denaturation profiles can be found in Supplementary Figure S2a. (b) Activity of CDK9/cyclin T and CDK9/cyclin TQ77R/E96G/F241L in the presence of increasing amounts of ATP. Measurements were done in triplicates and confirmed in independent experiments. Error bars indicate standard errors.
Finally we analyzed whether the three cyclin T mutations together affect the catalytic activity of the CDK9/cyclin TQ77R/E96G/F241L complex. To this end we determined the kinetic properties of both the wild-type and triple mutant complexes with respect to the ATP substrate and the inhibitor CAN508. Both complexes have a KMapp, ATP of 95 μM (Figure 1b), indicating that ATP binding is not affected by the presence of the mutations. This result is in agreement with the results of the DSF experiments. However, we did observe a decrease in the Vmax for the CDK9/cyclin TQ77R/E96G/F241L complex (Figure 1b). CAN508 inhibits both kinase complexes with an IC50 of 0.75 μM (Supplementary Figure S2b), in good agreement with previously published results.3
The close agreement between the ΔTm, KMapp, ATP, and CAN508 IC50 values for the wild-type and mutant complexes indicates that the mutations do not affect ATP or inhibitor binding to CDK9 or the complex’s catalytic activity. Taken together, our structural, thermodynamic, and enzymatic experiments validate the use of CDK9/cyclin TQ77R/E96G/F241L for structural analysis.
To rationalize the observed selectivity of CAN508 toward CDK9 over other CDKs, we determined structures of CAN508 bound to CDK9/cyclin T and to CDK2/cyclin A (Figure 2). We used both CDK9/cyclin TF241L and CDK9/cyclin TQ77R/E96G/F241L variants for our crystallographic studies. In agreement with our observations described above, both structures are highly similar, and the inhibitor electron density within both CDK9 active sites supports a shared binding mode (Supplementary Figure S3).
Figure 2.

Structures of CDK2/cyclin A/CAN508 and CDK9/cyclin T/CAN508. (a) CDK2 (orange) and CDK9 (green) are shown as ribbon representations, with CAN508 molecules as stick models. The glycine-rich loop is indicated. (b) Chemical structure of CAN508. (c, d) The CDK9/cyclin T (c) and CDK2/cyclin A (d) ATP binding sites complexed with CAN508. Contacting residues within 3.5 Å are drawn as stick models, and hydrogen bonds are indicated by dotted lines. (e) Superposition of selected ATP binding site residues in active CDK2 (orange) and CDK9 (green). CDK9 residues are labeled. Figures were prepared using PyMOL.
CAN508 binds to the ATP binding site located between the N- and C-terminal lobes of the CDK9 fold and is sandwiched between Ala46 in the N-terminal and Leu156 in the C-terminal lobe. As is observed for most ATP-competitive inhibitors, CAN508 makes hydrogen bonds to the CDK9 backbone in the hinge region.16 N16 and N14 of the diaminopyrazole ring are a hydrogen bond donor and acceptor, respectively, to the main chain oxygen of CDK9 Asp104 and the main chain nitrogen of Cys106, mimicking the interactions made by N6 and N1 of the purine ring of ATP (Figure 2b).12 In addition N13 contacts the main chain oxygen of Cys106. All of these inhibitor–CDK interactions are conserved in the CDK2-bound structure.
In a previously determined structure of CAN508 bound to monomeric CDK2, the inhibitor has been described as adopting a different and markedly strained conformation.3 Our structure of CAN508 bound to active CDK2/cyclin A is likely to provide a more useful model by which to understand the selectivity displayed by the inhibitor.
Although CAN508 forms similar interactions when bound to CDK9/cyclin T and CDK2/cyclin A, there is an additional interaction between CAN508 N12 and the carbonyl group of Ile25 of CDK9, made possible by a movement of the glycine-rich loop toward the ATP binding pocket (Figure 2a, c). This rearrangement of the active site does not occur in CDK2 upon CAN508 binding but has been observed to accompany the binding to CDK9 of a number of ATP-competitive inhibitors with very different chemical scaffolds.12,13 In all these structures the glycine-rich loop is restrained in its downward position by a concomitant downward movement of the β3-αC loop. These movements cause the CDK9 glycine-rich loop residues to contribute to the formation of a solvent-excluded channel into which inhibitors are sequestered and are qualitatively different to inhibitor-induced changes in CDK2. We note, however, that the glycine-rich loop is relatively flexible in both apo and inhibitor bound conformations, as judged from high temperature factors.
The higher conformational variability of CDK9 as compared to other CDK/cyclin complexes may offer an opportunity to enhance inhibitor potency and selectivity. In both CDK9/cyclin T/CAN508 structures, conformational changes result in optimal accommodation of the inhibitor in the ATP binding site. The ability to induce these CDK9-specific conformational changes may be a general characteristic of inhibitors that show good selectivity toward CDK9.
The binding mode of CAN508 to CDK2/cyclin A and to CDK9/cyclin T is very similar, but the inhibitor does adopt somewhat different orientations within the two ATP binding sites. The inhibitor phenolic moiety forms an aromatic contact with the CDK9 gate-keeper residue Phe103 (CDK2 Phe80) (d = 3.1 Å, interplanar angle = 120°) and exploits the hydrophobic pocket lined by Phe168 at the back of the ATP binding site. Within the CDK9-bound structure, the OH-group of the CAN508 phenolic moiety engages in a network of hydrogen bonds with residues Glu66, Lys48, and the backbone nitrogen of Phe168 (Figure 2c). In CDK2 this network only extends to include Glu51 and Lys33 (Figure 2d). This difference results from the displacement of CDK9 Glu66 by approximately 1 Å with respect to the CDK2 structure to generate a larger binding pocket close to Phe168 that can better accommodate the CAN508 phenolic group (Figure 2e, Supplementary Figure S3d). Interestingly, a CAN508 derivative lacking the OH group on its phenyl moiety still inhibits CDK9 but not the other CDKs tested.3 While all phenyl derivatives tested showed activity toward CDK9, substitution on the phenyl ring with an o-NO2 or m-OH moiety recovered some activity toward the other CDKs possibly by forming equivalent hydrogen bonds to those made by the phenolic OH of CAN508.
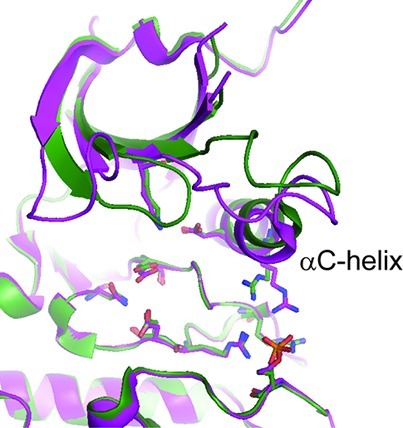
The hydrophobic pocket in CDK9 exploited by CAN508 results from a displaced αC-helix. αC is the only helix in the N-terminal lobe of a canonical eukaryotic kinase fold and has been shown to adopt different conformations dependent on the enzyme’s activation state.17 To determine whether αC-helix displacement is apparent in other CDK9 structures, we compared the structures of CDK9/cyclin T (PDB id: 3BLQ(12)) and CDK9/cyclin T/TAT (PDB id: 3MIA(14)). To analyze potential differences in the position of the αC-helix we superimposed the two complexes on their C-terminal kinase domains. As expected, we observed very little difference in the conformations of the kinase C-terminal lobes including the activation segment (rmsd 1.25 Å over all atoms (Figure 3)). While the relative orientations of the N- and C-terminal kinase lobes vary slightly between the two structures, all catalytically important residues superpose well and adopt the functionally relevant conformations as seen for other kinases,18,19 indicating that both crystal structures represent active kinase conformations. The N-terminal kinase lobes vary in the conformations of the flexible regions (i.e., the glycine-rich loop and the β3-αC loop) as well as in the position of the αC-helix (Figure 3). As cyclin T associates with CDK9 through the N-terminal kinase lobe and the αC-helix, the repositioning of the αC-helix leads to a shift in the position of the cyclin as described.14 Taken together, these observations suggest that the different conformations observed for the αC-helix likely reflect the inherent flexibility of this region of the protein.
Figure 3.

Superposition of CDK9/cyclin TQ77R/E96G/F241L (PDB id: 3BLQ, green and brown) and CDK9/cyclin T/TAT (PDB id: 3MIA, lilac and rose). Structures are represented as cartoon models, with catalytically important residues as stick models. The kinase subunits were superposed on their C-terminal kinase domains (residues 111–315). TAT is omitted for clarity.
Our studies validate the use of CDK9/cyclin TQ77R/E96G/F241L for the determination of inhibitor co-crystal structures. Our data suggest that the increased overall flexibility of CDK9, particularly in respect to the glycine-rich loop and the αC-helix conformations, together with its more extended ATP binding pocket, offer an opportunity to develop CDK9-specific inhibitors. Repositioning of the αC-helix is a widely exploited mechanism by which the activities of protein kinases are modulated.18 However, it appears that the particular flexibility apparent in CDK9 allows it to adopt a conformation that optimizes the interactions that can be made by CAN508. It will be important to determine if this mechanism for specificity also discriminates between CDK9 and functionally related kinases such as CDK12 and CDK13. Such discrimination would allow studies to dissect the individual contributions of these CDKs in regulating transcription and further the design of specific inhibitors to modulate the cellular levels of anti-apoptotic factors.
Methods
Protein Purification, Crystallization, and Structure Determination
CDK9 (residues 1–330)/cyclin T (residues 1–259) and CDK2/cyclin A complexes were prepared and crystallized as previously described.12,19 For structure determination of CDK9/cyclin T/CAN508 complexes, crystals were soaked for 1 h to overnight in the crystallization condition saturated with CAN508 containing 18% glycerol and then cryo-cooled in liquid nitrogen. Purified CDK2/cyclin A was incubated with 7.5 mM CAN508 for 30 min on ice, and the inhibitor complex was co-crystallized. All structures were solved as described previously.13
Kinetic Analysis
To determine the IC50 values toward CAN508, 10 ng of CDK9/cyclin T or CDK9/cyclin TF241L complex were incubated with 24 μg of GST-CTD in 10 μL reactions containing 10 mM MgCl2, 50 mM HEPES pH 7.5, 100 μM ATP, 5 mM DTT, 0.2 μCi γ-32P-labeled ATP, and increasing amounts of CAN508. Reactions were incubated for 5 min at 30 °C and terminated by the addition of SDS sample buffer. KMapp toward the ATP substrate were determined under the same reaction conditions using 26 μg/mL GST-CTD. Samples were analyzed by SDS-PAGE and autoradiography. Data were fitted with GraphPad Prism version 5.02 (www.graphpad.com). Differential scanning fluorimetry experiments were done as described previously.13
Acknowledgments
We thank V. Krystof for his kind gift of CAN508. We thank V. Krystof and L. Johnson for informal discussions. We thank the European Synchrotron Radiation Facility and the Diamond Light Source for providing excellent facilities and E. Lowe for data collection management. This work was supported by the Medical Research Council (S.B., M.E.M.N., and J.A.E.) and The Wellcome Trust (A.J.H.).
Glossary
Abbreviations
- CDK
cyclin-dependent kinase
- P-TEFb
positive transcription elongation factor b
Supporting Information Available
This material is available free via the Internet at http://pubs.acs.org.
Accession Codes
PDB id codes: 3TNI (CDK9/cyclin TF241L), 3TNH (CDK9/cyclin TF241L/CAN508), 3TN8 (CDK9/cyclin TQ77R/E96G/F241L/CAN508), 3TNW (CDK2/cyclin A/CAN508)
The authors declare no competing financial interest.
Supplementary Material
References
- Lam L. T.; Pickeral O. K.; Peng A. C.; Rosenwald A.; Hurt E. M.; Giltnane J. M.; Averett L. M.; Zhao H.; Davis R. E.; Sathyamoorthy M.; Wahl L. M.; Harris E. D.; Mikovits J. A.; Monks A. P.; Hollingshead M. G.; Sausville E. A.; Staudt L. M. (2001) Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2, RESEARCH0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. D.; Griffiths G.; Midgley C. A.; Barnett A. L.; Cooper M.; Grabarek J.; Ingram L.; Jackson W.; Kontopidis G.; McClue S. J.; McInnes C.; McLachlan J.; Meades C.; Mezna M.; Stuart I.; Thomas M. P.; Zheleva D. I.; Lane D. P.; Jackson R. C.; Glover D. M.; Blake D. G.; Fischer P. M. (2010) Discovery and characterization of 2-anilino-4-(thiazol-5-yl)pyrimidine transcriptional CDK inhibitors as anticancer agents. Chem. Biol. 17, 1111–1121. [DOI] [PubMed] [Google Scholar]
- Krystof V.; Cankar P.; Frysova I.; Slouka J.; Kontopidis G.; Dzubak P.; Hajduch M.; Srovnal J.; de Azevedo W. F.; Orsag M.; Paprskarova M.; Rolcik J.; Latr A.; Fischer P. M.; Strnad M. (2006) 4-Arylazo-3,5-diamino-1H-pyrazole CDK inhibitors: SAR study, crystal structure in complex with CDK2, selectivity, and cellular effects. J. Med. Chem. 49, 6500–6509. [DOI] [PubMed] [Google Scholar]
- Liu X.; Shi S.; Lam F.; Pepper C.; Fischer P. M.; Wang S. (2012) CDKI-71, a novel CDK9 inhibitor, is preferentially cytotoxic to cancer cells compared to flavopiridol. Int. J. Cancer 130, 1216–1226. [DOI] [PubMed] [Google Scholar]
- Nekhai S.; Bhat U. G.; Ammosova T.; Radhakrishnan S. K.; Jerebtsova M.; Niu X.; Foster A.; Layden T. J.; Gartel A. L. (2007) A novel anticancer agent ARC antagonizes HIV-1 and HCV. Oncogene 26, 3899–3903. [DOI] [PubMed] [Google Scholar]
- Chen R.; Wierda W. G.; Chubb S.; Hawtin R. E.; Fox J. A.; Keating M. J.; Gandhi V.; Plunkett W. (2009) Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 113, 4637–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettayeb K.; Baunbæk D.; Delehouze C.; Loaëc N.; Hole A. J.; Baumli S.; Endicott J. A.; Douc-Rasy S.; Bénard J.; Oumata N.; Galons H.; Meijer L. (2010) CDK inhibitors Roscovitine and CR8 trigger Mcl-1 down-regulation and apoptotic cell death in neuroblastoma Cells. Genes Cancer 1, 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar S. M.; Rathos M. J.; Sonawane V.; Rao S. V.; Joshi K. S. (2011) Cyclin-dependent kinase inhibitor, P276-00 induces apoptosis in multiple myeloma cells by inhibition of Cdk9-T1 and RNA polymerase II-dependent transcription. Leukemia Res. 35, 821–830. [DOI] [PubMed] [Google Scholar]
- Wang S.; Fischer P. M. (2008) Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 29, 302–313. [DOI] [PubMed] [Google Scholar]
- Krystof V.; Chamrad I.; Jorda R.; Kohoutek J. (2010) Pharmacological targeting of CDK9 in cardiac hypertrophy. Med. Res. Rev. 30, 646–666. [DOI] [PubMed] [Google Scholar]
- Krystof V.; Rarova L.; Liebl J.; Zahler S.; Jorda R.; Voller J.; Cankar P. (2011) The selective P-TEFb inhibitor CAN508 targets angiogenesis. Eur. J. Med. Chem. 46, 4289–4294. [DOI] [PubMed] [Google Scholar]
- Baumli S.; Lolli G.; Lowe E. D.; Troiani S.; Rusconi L.; Bullock A. N.; Debreczeni J. E.; Knapp S.; Johnson L. N. (2008) The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 27, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumli S.; Endicott J. A.; Johnson L. N. (2010) Halogen bonds form the basis for selective P-TEFb inhibition by DRB. Chem. Biol. 17, 931–936. [DOI] [PubMed] [Google Scholar]
- Tahirov T. H.; Babayeva N. D.; Varzavand K.; Cooper J. J.; Sedore S. C.; Price D. H. (2010) Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 465, 747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M.; Niesen F. H.; Allali-Hassani A.; Fedorov O. Y.; Finerty P. J. Jr.; Wasney G. A.; Yeung R.; Arrowsmith C.; Ball L. J.; Berglund H.; Hui R.; Marsden B. D.; Nordlund P.; Sundstrom M.; Weigelt J.; Edwards A. M. (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U.S.A. 103, 15835–15840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura N.; Zhang X.; Endres N. F.; Seeliger M. A.; Schindler T.; Kuriyan J. (2011) Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M.; Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282. [DOI] [PubMed] [Google Scholar]
- Brown N. R.; Noble M. E.; Endicott J. A.; Johnson L. N. (1999) The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1, 438–443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.