

Abstract
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, is a highly evolved human pathogen characterized by its formidable cell wall. Many unique lipids and glycolipids from the Mtb cell wall are thought to be virulence factors that mediate host–pathogen interactions. An intriguing example is Sulfolipid-1 (SL-1), a sulfated glycolipid that has been implicated in Mtb pathogenesis, although no direct role for SL-1 in virulence has been established. Previously, we described the biochemical activity of the sulfotransferase Stf0 that initiates SL-1 biosynthesis. Here we show that a stf0-deletion mutant exhibits augmented survival in human but not murine macrophages, suggesting that SL-1 negatively regulates the intracellular growth of Mtb in a species-specific manner. Furthermore, we demonstrate that SL-1 plays a role in mediating the susceptibility of Mtb to a human cationic antimicrobial peptide in vitro, despite being dispensable for maintaining overall cell envelope integrity. Thus, we hypothesize that the species-specific phenotype of the stf0 mutant is reflective of differences in antimycobacterial effector mechanisms of macrophages.
Microbial pathogens have engineered their cell surfaces to serve as protective barriers against host degradative enzymes, radical ion species, and antibiotics. This is especially true of the human pathogen Mycobacterium tuberculosis (Mtb), which possesses a formidable cell wall composed of many natural products important to its lifecycle.1 Distinctive features of the mycobacterial cell wall include long chain mycolic acids (C60–90) covalently linked to an arabinogalactan core that serve to noncovalently anchor an outer membrane of lipids and glycolipids.1 In addition to functioning as a protective barrier, many components of the Mtb cell wall are implicated in modulating host–pathogen interactions. For example, glycolipids such as phenolic glycolipids (PGL), lipoarabinomannan (LAM), and trehalose dimycolate (TDM) interact with immune cells to modulate the host response.2−4
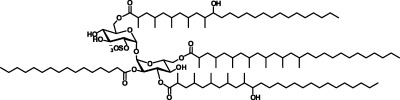
The sequencing of the Mtb genome led to the identification of genes involved in lipid biosynthesis, enabling their targeted disruption and subsequent evaluation of roles in Mtb pathogenesis.5 Results from these experiments demonstrated that cell wall metabolites can contribute to Mtb virulence.2,6,7 Despite this progress, the biological function of one class of cell wall glycolipids, the sulfatides, remains elusive. Sulfolipid-1 (SL-1), the most abundant sulfatide, is a tetra-acylated trehalose-based glycolipid located on the mycobacterial outer membrane of Mtb (see Figure 1).8 Provocatively, SL-1 is uniquely expressed in pathogenic mycobacteria, and its amounts have been positively correlated with strain virulence.9,10 The linkage between SL-1 and Mtb virulence inspired a search for functions of SL-1, which over the span of 50 years has resulted in attribution of numerous, sometimes conflicting, biological activities to this sulfatide. For example, in cell culture models purified SL-1 has been proposed to alter phagosome–lysosome fusion, disrupt mitochondrial oxidative phosphorylation, and activate as well as suppress the production of cytokines and reactive oxygen species produced by human leukocytes.11−17 More recently, knowledge of the genes involved in SL-1 biosynthesis has allowed the generation of Mtb strains deficient in SL-1 production.18−22 However, strains of Mtb lacking fully elaborated SL-1 do not appear to have consistent phenotypes or phenotypes distinguishable from wild-type Mtb in animal models of infection. This observation calls into question both the physiological relevance of the in vitro activities ascribed to SL-1 and the ability of animal infection to legitimately model the role of SL-1 in human tuberculosis.19−21,23
Figure 1.

Structure of SL-1 and a synthetic analogue. The synthetic analogue of SL-1 is composed of a T2S core esterified with fatty acyl groups at the 2′, 3′, 6′, and 6 positions, similar to SL-1. However, due to the complex synthesis of the fatty acyl substituents found on the natural product, SL-A lacks the hydroxyl groups and multimethyl branched units found on SL-1.
To directly address the role of SL-1 in Mtb pathogenesis, we utilized SL-1 purified as a natural product from Mtb whole cells as well as a fully synthetic analogue of SL-1, termed SL-A (see Figure 1),24 to probe the previously reported ability of SL-1 to modulate leukocyte function. Interestingly, we find that SL-1 and SL-A elicit a unique transcriptional signature distinct from known pro-inflammatory agonists. To evaluate the role of SL-1 in the context of its native environment, the Mtb cell envelope, we engineered a strain of Mtb in which SL-1 biosynthesis is abolished at its first committed step,22 sulfation of trehalose by the sulfotransferase Stf0 (see entire pathway elaborated in Supplementary Figure 1). Mtb strains deficient in SL-1 production demonstrated augmented survival in human macrophages. Furthermore, the stf0-deletion mutant exhibited increased resistance in vitro to a human cationic antimicrobial peptide, LL-37, which suggests a mechanism for the improved intracellular survival of Δstf0. Both murine macrophages and a murine model of tuberculosis failed to recapitulate this phenotype, indicating that SL-1 may contribute to pathogenesis in a species-specific manner during infection.
Results and Discussion
SL-1 Stimulates a Unique Transcriptional Signature in Host Cells
SL-1 is situated on the outer membrane of Mtb where it is poised to interact with host cells during the course of Mtb infection. To test this possibility, we purified SL-1 directly from Mtb whole cells (Supplementary Figure 2) and monitored its effect on human leukocytes using whole-genome expression profiling. Since any metabolite purified from cells may contain trace levels of bioactive contaminants, we also tested the activity of a fully synthetic analogue of SL-1,24 termed SL-A, in these assays (see Figure 1). Human primary immature dendritic cells (hiDC) were derived from peripheral blood monocytes of four independent donors and treated with either SL-1, SL-A, vehicle only, or the well-characterized Toll-like receptor (TLR) lipoprotein agonist Pam3Cys-Ser-(Lys)4 (P3K) as a positive control. First, we analyzed our microarray data to identify the top 50 statistically significant (p < 0.02) transcripts upregulated by SL-1, SL-A, or P3K as compared to vehicle treated hiDCs. As expected, P3K induced a robust pro-inflammatory gene expression profile in hiDCs, with the majority of induced transcripts involved in mounting an immune response (Figure 2 and Supplementary Table 1). SL-1 and SL-A, conversely, did not induce a similar pattern of pro-inflammatory gene expression in hiDCs like P3K; instead, SL-1 and SL-A stimulated the upregulation of a unique set of transcripts with 24% concordance in genes upregulated in both SL-1 and SL-A treated hiDCs (Figure 2 and Supplementary Table 1). Thus, our microarray data suggests that SL-1 is not acting as a pro-inflammatory stimulus as previously suggested.13 To further validate the observation that purified SL-1 plays little role in mediating a pro-inflammatory immune response, we measured by ELISA amounts of the pro-inflammatory cytokine tumor necrosis factor (TNF) induced by SL-1 and SL-A in a variety of cell types, including hiDCs, human primary macrophages, the human monocyte THP-1 cell line, and the murine macrophage RAW 264.7 cell line. Neither SL-1 nor SL-A stimulated production of TNF in any of the cell types we examined (Supplementary Figure 3), which corroborated with our microarray data to provide further evidence that purified SL-1 is not a classical pro-inflammatory stimulus. Reasons for these discrepancies with previous data could include the presence of small levels of impurities in previous SL-1 preparations, differences in the concentrations of SL-1 used, or differences in the culture conditions of mammalian cells.
Figure 2.

SL-1 induces a unique transcriptional signature in human leukocytes. The transcript profiles of hiDCs stimulated with P3K, SL-1, SL-A, or vehicle only were assessed by microarray. The 50 most highly upregulated and statistically significant transcripts (p < 0.02) induced by SL-1, SL-A, or P3K as compared to vehicle only treated cells were organized according to function. Due to limited overlap in genes upregulated by each stimulus, different functional categories were chosen. The percent overlap of gene identities between P3K vs SL-1, P3K vs SL-A, and SL-1 vs SL-A is highlighted. The identities of individual genes are listed in Supplementary Table 1.
To determine a broad subset of genes whose transcription is influenced by SL-1, we further expanded the analysis of our microarray data to include all of the statistically significant (p < 0.02) genes that are transcriptionally regulated (i.e., genes induced and repressed) by SL-1 and SL-A. This analysis revealed 789 genes reproducibly regulated by SL-1 and 432 genes regulated by SL-A with 172 genes commonly regulated by both SL-1 and SL-A in hiDCs (Supplementary Table 2). While we feel that the concordance of genes regulated by SL-1 and SL-A is strong, it is important to note that SL-1 and SL-A are not structurally identical. Due to synthetic challenges, SL-A is composed of straight chain lipid moieties at its 3′, 6, and 6′ positions instead of the fully elaborated phthioceranoyl and hydroxyphthioceranoyl substituents of SL-1 (see Figure 1).24 Nonetheless, to be rigorous we defined a set of SL-1-regulated genes as those whose expression is seen to be influenced by both SL-1 and SL-A in these experiments (Supplementary Table 2). Notably, in this set of SL-1-regulated genes we observed SL-1-dependent upregulation of CD1D, a gene involved in activation of NKT cells during Mtb infection,25 as well as SL-1-dependent downregulation of IL8, a chemokine important for neutrophil chemotaxis.26 Also, SL-1 and SL-A caused upregulation of transcripts involved in phosphatase/kinase activation, PTPRE, PTPRN, PPP1R11, and PPP1R14A, which could influence unidentified signaling pathways in mycobacterial infection (Supplementary Table 1). Overall, we did not see modulation of classic pro- and anti-inflammatory genes such as TNF, MIP2, IL10, NFKB, CD83, CD86, or CD80, which have been previously reported to be regulated by the mycobacterial lipids TDM and LAM.3,4 Interestingly, many of the SL-1-regulated genes are of unknown function, and thus these genes will guide future studies to improve our knowledge of key host–pathogen interactions involved in Mtb pathogenesis.
Disruption of stf0 Creates a Strain of Mtb for Evaluating the Contribution of SL-1 to Virulence
With evidence of an SL-1 induced transcriptional response in immune cells, we decided to evaluate the role of SL-1 during pathogenesis in the context of its natural environment, the Mtb cell envelope. Using bioinformatics methods, we previously identified the sulfotransferase Stf0 (CAA17370; Rv0295c) in the Mtb strain H37Rv22 that installs a sulfate group onto trehalose to form trehalose-2-sulfate (T2S) as the first committed step in SL-1 biosynthesis (Figure 3, panel A). In order to address the role of SL-1 biosynthesis in Mtb pathogenesis, we constructed an stf0 knockout in the Erdman strain of Mtb, because it has recently been reported that the H37Rv strain can undergo spontaneous loss of the well-established virulence factor lipid phthiocerol dimycocerosate (PDIM) during in vitro culture.27 Indeed, when we constructed an Erdman strain of Mtb in which stf0 was disrupted, it no longer biosynthesized T2S or SL-1, but PDIM biosynthesis remained intact (Figure 3, panel B; Supplementary Figure 4, panel a, b). SL-1 production was restored by complementing the Δstf0 mutant with a plasmid expressing stf0 under the control of its native promoter (Δstf0 + pstf0) (Figure 3, panel B).
Figure 3.

Sulfation of trehalose by Stf0 is the first committed step in SL-1 biosynthesis. (A) The sulfotransferase Stf0 sulfates free trehalose to initiate SL-1 biosynthesis. (B) SL-1 is absent in the Δstf0 strain. Surface exposed lipids were removed by hexanes extraction of WT, Δstf0, and complemented (Δstf0 + pstf0) Mtb strains and analyzed by FT-ICR MS. SL-1 is observed as a collection of lipoforms that vary in the lengths of their acyl groups (±14 mass units).
Loss of SL-1, which makes up approximately 1% of Mtb’s dry weight,28 could alter growth, cell envelope permeability and integrity, or surface lipid composition. We observed that the growth of Mtb in liquid culture was not affected by disruption of stf0 or complementation of the Δstf0 strain (Supplementary Figure 5). To assess cell envelope integrity and permeability, we challenged the bacteria with detergents or antibiotics, respectively. As shown in Table 1, the minimal concentration of the detergents sodium dodecyl sulfate (SDS) and deoxycholate (DOC) that inhibited 50% of bacterial growth (MIC50) were similar for the Δstf0 mutant and WT Mtb. Likewise, the MIC50 values of the antibiotics isoniazid, rifampicin, amikacin, and kanamycin were similar between Δstf0 and WT Mtb (Table 1). These data suggest that loss of the major cell envelope component SL-1 does not compromise the overall integrity or permeability of the Mtb cell envelope in vitro. Additionally, production of other surface extractable lipids including polyacyltrehalose (PAT)29 and phthiocerol dimycocerosate (PDIM) remained robust as determined by qualitative FT-ICR MS analysis of the stf0 mutant (Supplementary Figure 4, panel b).
Table 1. Δstf0 Does Not Exhibit Global Defects in Cell Wall Integritya.
| WT | Δstf0 | Δstf0 + pstf0 | ||
|---|---|---|---|---|
| detergents (% w/v) | SDS | 0.02 | 0.02 | 0.02 |
| DOC | 0.010 | 0.007 | 0.008 | |
| antibiotics (μg/mL) | rifampicin | 0.025 | 0.0125 | 0.0125 |
| isoniazid | 0.025 | 0.027 | 0.030 | |
| amikacin | 0.30 | 0.30 | 0.40 | |
| kanamycin | 1.1 | 1.5 | >7.5b |
Mtb was grown in the presence of each detergent or antibiotic. The minimal inhibitory concentration that inhibited 50% of Mtb growth was noted after 7 days. These data are representative of at least two independent experiments each performed in duplicate.
Δstf0 + pstf0 contains a kanamycin resistance gene.
SL-1 Biosynthesis Enhances Mtb’s Susceptibility to the Antimicrobial Peptide LL-37
Because SL-1 is an anionic species found in the mycobacterial outer membrane, we reasoned that it could facilitate interactions with cationic antimicrobial peptides (AMP), as has been demonstrated for other negatively charged bacterial lipid species including LPS in the outer membrane of Gram-negative bacteria and phosphatidylglycerol (PG) in the plasma membrane of Mtb.30,31 AMPs are small, cationic peptides that are important components of the innate immune defense against invading pathogens, including Mtb,32 and exert antimicrobial effects by inserting into and disrupting bacterial cell membranes.33 Therefore, we examined the in vitro susceptibility of the Δstf0 and WT strains as well as another SL-1-deficient strain (ΔpapA2, Supplemental Figure 1) to LL-37/CAP-18, a human AMP expressed in macrophages with known antimicrobial activity against Mtb.32,34 As previously demonstrated, LL-37 reduced the viability of WT Mtb (Figure 4). Conversely, stf0 and papA2 mutants lacking SL-1 proved more resistant to LL-37-mediated killing in vitro, a phenotype that could be complemented by expressing stf0 or papA2 (Figure 4 and Supplemental Figure 6). The resistance of the stf0 and papA2 mutants to LL-37 was not concentration-dependent in the range of concentrations tested, suggesting that the interaction of Mtb with LL-37 is dependent on SL-1 as well as other negatively charged components that may reside in the Mtb cell envelope. Thus, we conclude that the susceptibility of Mtb to LL-37 is mediated in part by the highly abundant, anionic Mtb cell envelope component SL-1.
Figure 4.

Δstf0 is more resistant to LL-37 compared to WT Mtb. The indicated strains of Mtb were exposed to increasing concentrations (0, 6.5, and 65 μg/mL) of LL-37. After 3 days, Mtb viability was measured by plating bacteria on solid agar to enumerate cfu counts. Data are representative of one of three independent experiments each performed in triplicate. * P = 0.74 (not significant) for the comparison of Δstf0 versus WT; ** P = 0.012 for the comparison of Δstf0 versus WT. Error bars correspond to the means (±SD); an error of less than ∼2% is not visible on this graph (SD of Δstf0 at 65 μg/mL = 1.0%; SD of Δstf0 + pstf0 at 65 μg/mL = 1.27%).
Stf0 Negatively Regulates the Intracellular Growth of Mtb in Human Macrophages
The intracellular growth of Mtb in human macrophages is limited by antimicrobial peptides such as LL-37, in contrast to murine macrophages that rely mostly on the production of nitric oxide radicals for bactericidal activity.32,34,35 Thus, we hypothesized that the Δstf0 strain lacking the negatively charged SL-1 may be less susceptible than WT Mtb to antimicrobial agents produced by human macrophages thereby enhancing its intracellular survival. To test this idea, we infected human THP-1 and murine RAW264.7 macrophage cell lines with WT, Δstf0, or Δstf0 + pstf0 strains and evaluated their intracellular growth. Interestingly, the stf0 mutant displayed increased intracellular survival in THP-1 cells as demonstrated by a 2.5-fold increase in cfu counts 6 days post infection (Figure 5, panel A). This growth phenotype could be partly complemented by expression of stf0 in the mutant strain (Δstf0 + pstf0) (Figure 5, panel A). Partial complementation of the stf0 mutant phenotype could result from use of an integrating plasmid containing the putative native promoter for stf0 that may not recapitulate wild type transcriptional regulation of stf0. The augmented survival of Δstf0 was likely not due to enhanced infectivity of this mutant strain, as no significant difference in cfu counts was seen 4 h post infection (day = 0) (Figure 5, panel A). In contrast to their different growth phenotypes in human macrophages, the Δstf0 and WT strains had indistinguishable growth phenotypes in RAW264.7 murine macrophages up to 6 days post infection (Figure 5, panel B). Using a murine model of tuberculosis, we found that Δstf0 and WT cells proliferated at similar rates in the lungs, livers, and spleens of BALB/c mice infected via aerosolization (Figure 5, panel C and Supplementary Figure 7). In addition, we observed no change in the average time to death for mice infected with Δstf0 as compared to WT (Figure 5, panel D). These results suggest that SL-1 biosynthesis affects Mtb survival in human but not murine macrophages or murine in vivo infection models.
Figure 5.

Stf0 restricts the intracellular growth of Mtb in human macrophages. (A) Human THP-1 macrophages or (B) murine RAW macrophages were infected with the indicated strain of Mtb. After 4 h (day 0) and 6 days post infection, adherent macrophages were lysed and plated on solid agar to determine the number of viable intracellular bacteria by cfu counts. Data represent the average cfu count obtained from three independent experiments performed in triplicate (±SD). * P < 4 × 10–5 for the comparison of Δstf0 versus WT; ** P < 0.03 for the comparison of Δstf0 versus Δstf0 + pstf0 (A) or one of three independent experiments performed in triplicate (B) (±SD). Δstf0 behaves similarly to WT Mtb in a murine model of infection. (C) Lung cfu counts for BALB/c mice infected via aerosol with WT or Δstf0 Mtb. Each data point represents the average cfu count from 4–5 mice, and error bars indicate the standard deviation from the mean. (D) Time-to-death for mice infected with WT or Δstf0 Mtb.
The augmented growth of Δstf0 in human macrophages places stf0 in the small but expanding class of Mtb genes that have been observed to negatively regulate growth in animal or cell culture models of infection.36 Interestingly, included in this family is another Mtb sulfotransferase gene, stf3 (CAB00968, Rv2267c), that produces a sulfated form of menaquinone.37,38 Strains of Mtb deficient for stf3 demonstrate a higher bacterial burden and an increased time to death in mice.37 In addition to Mtb, Xanthomonas oryzae was recently found to produce a sulfated factor that compromises the bacteria’s ability to cause infection.39 This rice blight-causing pathogen secretes a sulfated peptide that interacts with a host receptor, in a sulfate-dependent manner, to activate innate immunity.39 These examples indicate that prokaryotic sulfated metabolites can mediate host–pathogen interactions that are beneficial to the host. However, virulence factors that appear to negatively influence the pathogenicity of an organism likely have been retained to serve additional biological functions that benefit the biology or virulence of the pathogen.
Conclusion
SL-1 is an abundant glycolipid located in the mycobacterial outer membrane of Mtb that is unique to the pathogenic Mtb complex. While amounts of SL-1 have been positively correlated with strain virulence, the molecular mechanism by which SL-1 influences pathogenesis remained unclear. In this work, we begin to reconcile a large body of data that ascribe a plethora of biological functions to SL-1. First, we probed the transcriptome of human leukocytes stimulated with highly pure SL-1, as well as a fully synthetic analogue of SL-1, to find that SL-1 induces a unique transcriptional signature. Interestingly, many of the genes induced by SL-1 and its synthetic analogue are of unknown function, but distinct from those known to be induced by well-characterized pro-inflammatory mediators. Thus, we created an Erdman strain of Mtb completely lacking SL-1 (Δstf0), by deletion of the enzyme involved in the first committed step of SL-1 biosynthesis, to probe the role of SL-1 during host pathogenesis in the context of its natural environment: the Mtb cell envelope. Strains lacking SL-1 exhibited enhanced intracellular survival in human but not murine macrophages, placing SL-1 in the small but expanding class of components that negatively regulate Mtb virulence. In addition, we observed no difference in growth or average time to death for mice infected with Δstf0 as compared to WT cells. These results suggest that SL-1 biosynthesis affects Mtb survival in human but not murine macrophages. In vitro, the stf0 mutant was more resistant to killing by the antimicrobial peptide LL-37, revealing a potential mechanism of increased survival for Δstf0 in human macrophages. These results provide the first molecular evidence for the role of SL-1 in Mtb infection and suggest a Mtb virulence determinant that could provide host specificity.
Given the success of the latent phase in Mtb pathogenesis, it is not surprising that Mtb has retained mechanisms that may regulate its intracellular growth. Nonetheless, it is interesting to speculate why Mtb has retained genes for SL-1 biosynthesis that seemingly make it more susceptible to antimicrobial activity, and whether this indicates that SL-1 may serve additional functions that are beneficial to Mtb pathogenesis. Many bacterial pathogens have evolved means to limit their susceptibility to AMPs by modulating the net charge or abundance of their cell wall lipids.40 Mtb has recently been shown to modify PG with lysine to provide protection from killing by cationic antibiotics and AMPs.30 Thus, Mtb may also have means of altering levels of SL-1 by tightly controlling its biosynthesis or localization to the outer cell wall. In fact, transcriptional and metabolic mechanisms do exist that appear to regulate SL-1 production.41−44 It will be interesting to further examine the environmental and bacterial cues that control SL-1 biosynthesis, molecular structure, and cell wall localization in the context of Mtb infection. It could prove that expression of SL-1 during certain phases of the Mtb infection cycle is important for resistance to host defense mechanisms that are independent of AMP activity. Furthermore, production of SL-1 may have been retained by Mtb to serve as a molecular determinant uniquely sensed by human cells. Ultimately, understanding which Mtb virulence determinants and host factors confer species-specificity will improve vaccine and drug development.
Methods
Bacterial Strains and Culture
Mtb cells (Erdman strain) were grown at 37 °C in Middlebrook 7H9 broth or 7H11 agar supplemented with 0.5% glycerol, 0.05% Tween-80, and 10% OADC (v/v). Δstf0 in the Erdman background was constructed as previously described.29 For complementation of Δstf0, the stf0 gene was cloned from Mtb into the integrating mycobacterial expression vector pMV306 under the control of its putative endogenous promoter.45 This plasmid was electroporated into Δstf0, and transformants were selected on kanamycin-containing plates.
Microarray
Purified SL-1 or the synthetic compound SL-A (synthesis reported in ref (24)) were each dissolved in petroleum ether and added to a tissue culture dish to give a final concentration of 25 μg mL–1. The dishes were incubated at 37 °C to allow the solvent to evaporate. P3K (EMC Microcollections) is water-soluble and was added directly to cell culture media for a final concentration of 20 ng mL–1. Each treatment condition (SL-1, SL-A, vehicle control, or P3K) was tested in quadruplicate using 1 × 106 human peripheral blood-derived iDCs derived from four different donors. After 4 h of co-incubation total RNA was isolated using the RNAqueous kit (Ambion). Genomic DNA was removed using Turbo DNA-free (Ambion). Microarray sample preparation, labeling, and array hybridizations were performed as described in the Supporting Information.
Mass Spectrometry
For MS analysis, actively growing cultures were grown to early log phase, washed with 10% glycerol (v/v), and grown in 7H9 media lacking Tween for 2 days. Surface lipids were extracted with hexanes, while total lipids were extracted with chloroform/methanol (1:1, v/v) as described.19 All spectra were obtained on an Apex II Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR MS) equipped with a 7 T actively shielded superconducting magnet (Bruker Daltonics). Ions were introduced into the ion source via direct injection at a rate of 1 μL min–1. Ions were generated with an Apollo pneumatically assisted electrospray ionization source (Analytica) operating in the negative ion mode to detect sulfolipids or the positive ion mode to detect PDIM and were accumulated in an RF-only external hexapole for 1 s before being transferred to the ICR cell for mass analysis. Mass spectra consist of 512,000 data points and are an average of either 16 or 50 scans. The spectra were acquired using XMASS version 7.0.8 (Bruker Daltonics). All spectra were internally calibrated with at least four known compounds.
Minimal Inhibitory Concentration (MIC) Assays
Susceptibility of Mtb to various in vitro stress conditions was determined as described by identifying the minimal inhibitory concentration of a given compound that inhibited 50% of bacterial growth after 7 days as monitored by measuring the growth of Mtb at OD600. Strains of Mtb were grown to late log phase, washed twice in PBS, and pelleted by centrifugation at low speed to remove clumps. The OD600 of the cultures was adjusted to 0.02 in a 96-well plate format, and Mtb was exposed to 1:1 serial dilutions of SDS (0–0.1%), sodium DC (0–0.2%), kanamycin (0–7 μg mL–1), isoniazid (0–0.2 μg mL–1), amikacin (0–7 μg mL–1), or rifampicin (0–0.2 μg mL–1).
Antimicrobial Peptide Susceptibility
Sensitivity of Mtb to antimicrobial peptides was assayed as previously described.32 Briefly, Mtb was grown to late log phase, washed in PBS, and pelleted by centrifugation at low speed to remove clumps. Cells (2 × 106) of each strain were exposed to the indicated concentration of LL-37 (AnaSpec) dissolved in RPMI: water (1:4, v/v) at 37 °C. After 3 days, bacteria were plated on solid agar to enumerate the number of viable bacteria by cfu counts.
Macrophage Infections
THP-1 (ATCC TIB-202) cells were cultured in RPMI media supplemented with 10% FBS (v/v), 10 mM HEPES, 1 mM sodium pyruvate, 0.05 mM β-mercaptoethanol, and penicillin/streptomycin. RAW264.7 cells (ATCC TIB-71) were cultured in RPMI media supplemented with 10% FBS (v/v) and penicillin/streptomycin. THP-1 and RAW cells were infected with the indicated strain of Mtb at a multiplicity of infection (MOI) of 0.5 using infection media: RPMI + 10% (v/v) horse serum. To determine viable bacteria, cfu counts were enumerated at the indicated time points by lysing adherent cells and plating serial dilutions on 7H11 agar.
Mouse Infections
Bacteria were grown to log phase, cup-sonicated by using a Branson Sonicator 250 (Danbury, CT) at 90% for 15 s, spun for 5 min at 40 × g to remove clumps, and diluted to the desired inoculum in PBS. Bacteria were administered to BALB/c mice via nebulization for 15 min using a custom-built aerosolization chamber (Mechanical Engineering Shops, University of Wisconsin, Madison, WI). For infections, bacterial suspensions with an OD600 = 0.1 were used, resulting in an initial seeding of ∼250 bacteria per mouse. Organs from infected mice were homogenized and plated on solid media to enumerate colony forming units (cfu) at 0, 10, 23, and 44 days post infection. For the time to death study, mice were monitored for weight loss and sacrificed after 15% loss of body weight. All mice were housed and treated humanely as described in an animal care protocol approved by the University of California, San Francisco, Institutional Animal Care and Use Committee.
Statistics
All data were analyzed by Student’s t test for normally distributed data with equal variances. A P value < 0.05 was considered statistically significant.
Acknowledgments
H37Rv whole cells were provided by the National Institutes of Health, NIAID Contract No. HHSN266200400091C, “Tuberculosis Vaccine Testing and Research Materials” (Colorado State University). We gratefully acknowledge support from the Sandler Asthma Basic Research (SABRE) Center Functional Genomics Core Facility at University of California, San Francisco and would like to thank R. Barbeau, C. Eisley, A. Barczak, and D. Erle for assistance with microarrays. We also thank K. Sogi, P. Drake, A. Iavarone, J. Koerber, and members of the Bertozzi lab for helpful discussions and technical contributions. This work was supported by a National Institutes of Health Grant AI51622 (to CRB & J.A.L.).
Supporting Information Available
Complete materials and methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kaur D.; Guerin M. E.; Skovierova H.; Brennan P. J.; Jackson M. (2009) Chapter 2: Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69, 23–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed M. B.; Domenech P.; Manca C.; Su H.; Barczak A. K.; Kreiswirth B. N.; Kaplan G.; Barry C. E. 3rd. (2004) A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431, 84–87. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek T. B.; Van Vliet S. J.; Koppel E. A.; Sanchez-Hernandez M.; Vandenbroucke-Grauls C. M.; Appelmelk B.; Van Kooyk Y. (2003) Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowdish D. M.; Sakamoto K.; Kim M. J.; Kroos M.; Mukhopadhyay S.; Leifer C. A.; Tryggvason K.; Gordon S.; Russell D. G. (2009) MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathog. 5, e1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S. T.; Brosch R.; Parkhill J.; Garnier T.; Churcher C.; Harris D.; Gordon S. V.; Eiglmeier K.; Gas S.; Barry C. E. 3rd; Tekaia F.; Badcock K.; Basham D.; Brown D.; Chillingworth T.; Connor R.; Davies R.; Devlin K.; Feltwell T.; Gentles S.; Hamlin N.; Holroyd S.; Hornsby T.; Jagels K.; Barrell B. G.; Krogh A; Mclean J; Moule S; Murphy L; Oliver K; Osborne J; Quail M. A.; Rajandream M. A.; Rogers J; Rutter S; Seeger K; Skelton J; Squares R; Squares S; Sulston J. E.; Taylor K; Whitehead S; Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. [DOI] [PubMed] [Google Scholar]
- Glickman M. S.; Cox J. S.; Jacobs W. R. Jr. (2000) A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol. Cell 5, 717–727. [DOI] [PubMed] [Google Scholar]
- Cox J. S.; Chen B.; McNeil M.; Jacobs W. R. Jr. (1999) Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402, 79–83. [DOI] [PubMed] [Google Scholar]
- Middlebrook G.; Coleman C. M.; Schaefer W. B. (1959) Sulfolipid from virulent tubercle bacilli. Proc. Natl. Acad. Sci. U.S.A. 45, 1801–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnikin D. E.; Kremer L.; Dover L. G.; Besra G. S. (2002) The methyl-branched fortifications of Mycobacterium tuberculosis. Chem. Biol. 9, 545–553. [DOI] [PubMed] [Google Scholar]
- Gangadharam P. R.; Cohn M. L.; Middlebrook G. (1963) Infectivity, pathogenicity and sulpholipid fraction of some Indian and British strains of tubercle bacilli. Tubercle 44, 452–455. [DOI] [PubMed] [Google Scholar]
- Goren M. B.; D’Arcy Hart P.; Young M. R.; Armstrong J. A. (1976) Prevention of phagosome-lysosome fusion in cultured macrophages by sulfatides of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 73, 2510–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst M. J.; Gross J. M.; Brozna J. P.; Goren M. B. (1988) Inhibition of macrophage priming by sulfatide from Mycobacterium tuberculosis. J. Immunol. 140, 634–640. [PubMed] [Google Scholar]
- Brozna J. P.; Horan M.; Rademacher J. M.; Pabst K. M.; Pabst M. J. (1991) Monocyte responses to sulfatide from Mycobacterium tuberculosis: inhibition of priming for enhanced release of superoxide, associated with increased secretion of interleukin-1 and tumor necrosis factor alpha, and altered protein phosphorylation. Infect. Immun. 59, 2542–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; English D.; Andersen B. R. (1991) Activation of human neutrophils by Mycobacterium tuberculosis-derived sulfolipid-1. J. Immunol. 146, 2730–2736. [PubMed] [Google Scholar]
- Kato M.; Goren M. B. (1974) Synergistic action of cord factor and mycobacterial sulfatides on mitochondria. Infect. Immun. 10, 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Goren M. B.; Holzer T. J.; Andersen B. R. (1988) Effect of Mycobacterium tuberculosis-derived sulfolipid I on human phagocytic cells. Infect. Immun. 56, 2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P.; Poquet Y.; Levillain F.; Peguillet I.; Larrouy-Maumus G.; Gilleron M.; Ewann F.; Christophe T.; Fenistein D.; Jang J.; Jang M. S.; Park S. J.; Rauzier J.; Carralot J. P.; Shrimpton R.; Genovesio A.; Gonzalo-Asensio J. A.; Puzo G.; Martin C.; Brosch R.; Stewart G. R.; Gicquel B.; Neyrolles O. (2010) High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog. 6, e1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirakova T. D.; Thirumala A. K.; Dubey V. S.; Sprecher H.; Kolattukudy P. E. (2001) The Mycobacterium tuberculosis pks2 gene encodes the synthase for the hepta- and octamethyl-branched fatty acids required for sulfolipid synthesis. J. Biol. Chem. 276, 16833–16839. [DOI] [PubMed] [Google Scholar]
- Converse S. E.; Mougous J. D.; Leavell M. D.; Leary J. A.; Bertozzi C. R.; Cox J. S. (2003) MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc. Natl. Acad. Sci. U.S.A. 100, 6121–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Schelle M. W.; Jain M.; Lin F. L.; Petzold C. J.; Leavell M. D.; Leary J. A.; Cox J. S.; Bertozzi C. R. (2007) PapA1 and PapA2 are acyltransferases essential for the biosynthesis of the Mycobacterium tuberculosis virulence factor sulfolipid-1. Proc. Natl. Acad. Sci. U.S.A. 104, 11221–11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech P.; Reed M. B.; Dowd C. S.; Manca C.; Kaplan G.; Barry C. E. 3rd. (2004) The role of MmpL8 in sulfatide biogenesis and virulence of Mycobacterium tuberculosis. J. Biol. Chem. 279, 21257–21265. [DOI] [PubMed] [Google Scholar]
- Mougous J. D.; Petzold C. J.; Senaratne R. H.; Lee D. H.; Akey D. L.; Lin F. L.; Munchel S. E.; Pratt M. R.; Riley L. W.; Leary J. A.; Berger J. M.; Bertozzi C. R. (2004) Identification, function and structure of the mycobacterial sulfotransferase that initiates sulfolipid-1 biosynthesis. Nat. Struct. Mol. Biol. 11, 721–729. [DOI] [PubMed] [Google Scholar]
- Rousseau C.; Turner O. C.; Rush E.; Bordat Y.; Sirakova T. D.; Kolattukudy P. E.; Ritter S.; Orme I. M.; Gicquel B.; Jackson M. (2003) Sulfolipid deficiency does not affect the virulence of Mycobacterium tuberculosis H37Rv in mice and guinea pigs. Infect. Immun. 71, 4684–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh C. D.; Bertozzi C. R. (2008) Synthetic studies toward Mycobacterium tuberculosis sulfolipid-I. J. Org Chem. 73, 1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sada-Ovalle I.; Chiba A.; Gonzales A.; Brenner M. B.; Behar S. M. (2008) Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog. 4, e1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggiolini M.; Dewald B.; Moser B. (1997) Human chemokines: an update. Annu. Rev. Immunol. 15, 675–705. [DOI] [PubMed] [Google Scholar]
- Domenech P.; Reed M. B. (2009) Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: implications for virulence studies. Microbiology 155, 3532–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren M. B. (1970) Sulfolipid I of Mycobacterium tuberculosis, strain H37Rv. I. Purification and properties. Biochim. Biophys. Acta 210, 116–126. [DOI] [PubMed] [Google Scholar]
- Hatzios S. K.; Schelle M. W.; Holsclaw C. M.; Behrens C. R.; Botyanszki Z.; Lin F. L.; Carlson B. L.; Kumar P.; Leary J. A.; Bertozzi C. R. (2009) PapA3 is an acyltransferase required for polyacyltrehalose biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 284, 12745–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney E.; Stankowska D.; Zhang J.; Fol M.; Cheng Q. J.; Lun S.; Bishai W. R.; Rajagopalan M.; Chatterjee D.; Madiraju M. V. (2009) The two-domain LysX protein of Mycobacterium tuberculosis is required for production of lysinylated phosphatidylglycerol and resistance to cationic antimicrobial peptides. PLoS Pathog. 5, e1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer W. M.; Casey S. G.; Spitznagel J. K. (1984) Lipid A and resistance of Salmonella typhimurium to antimicrobial granule proteins of human neutrophil granulocytes. Infect. Immun. 43, 834–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P. T.; Stenger S.; Li H.; Wenzel L.; Tan B. H.; Krutzik S. R.; Ochoa M. T.; Schauber J.; Wu K.; Meinken C.; Kamen D. L.; Wagner M.; Bals R.; Steinmeyer A.; Zugel U.; Gallo R. L.; Eisenberg D.; Hewison M.; Hollis B. W.; Adams J. S.; Bloom B. R.; Modlin R. L. (2006) Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311, 1770–1773. [DOI] [PubMed] [Google Scholar]
- Radek K.; Gallo R. (2007) Antimicrobial peptides: natural effectors of the innate immune system. Semin. Immunopathol. 29, 27–43. [DOI] [PubMed] [Google Scholar]
- Liu P. T.; Stenger S.; Tang D. H.; Modlin R. L. (2007) Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J. Immunol. 179, 2060–2063. [DOI] [PubMed] [Google Scholar]
- Thoma-Uszynski S.; Stenger S.; Takeuchi O.; Ochoa M. T.; Engele M.; Sieling P. A.; Barnes P. F.; Rollinghoff M.; Bolcskei P. L.; Wagner M.; Akira S.; Norgard M. V.; Belisle J. T.; Godowski P. J.; Bloom B. R.; Modlin R. L. (2001) Induction of direct antimicrobial activity through mammalian toll-like receptors. Science 291, 1544–1547. [DOI] [PubMed] [Google Scholar]
- ten Bokum A. M.; Movahedzadeh F.; Frita R.; Bancroft G. J.; Stoker N. G. (2008) The case for hypervirulence through gene deletion in Mycobacterium tuberculosis. Trends Microbiol. 16, 436–441. [DOI] [PubMed] [Google Scholar]
- Mougous J. D.; Senaratne R. H.; Petzold C. J.; Jain M.; Lee D. H.; Schelle M. W.; Leavell M. D.; Cox J. S.; Leary J. A.; Riley L. W.; Bertozzi C. R. (2006) A sulfated metabolite produced by stf3 negatively regulates the virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 4258–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsclaw C. M.; Sogi K. M.; Gilmore S. A.; Schelle M. W.; Leavell M. D.; Bertozzi C. R.; Leary J. A. (2008) Structural characterization of a novel sulfated menaquinone produced by stf3 from Mycobacterium tuberculosis. ACS Chem. Biol. 3, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. W.; Han S. W.; Sririyanum M.; Park C. J.; Seo Y. S.; Ronald P. C. (2009) A type I-secreted, sulfated peptide triggers XA21-mediated innate immunity. Science 326, 850–853. [DOI] [PubMed] [Google Scholar]
- Peschel A.; Sahl H. G. (2006) The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4, 529–536. [DOI] [PubMed] [Google Scholar]
- Singh A.; Crossman D. K.; Mai D.; Guidry L.; Voskuil M. I.; Renfrow M. B.; Steyn A. J. (2009) Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 5, e1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo Asensio J.; Maia C.; Ferrer N. L.; Barilone N.; Laval F.; Soto C. Y.; Winter N.; Daffe M.; Gicquel B.; Martin C.; Jackson M. (2006) The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J. Biol. Chem. 281, 1313–1316. [DOI] [PubMed] [Google Scholar]
- Walters S. B.; Dubnau E.; Kolesnikova I.; Laval F.; Daffe M.; Smith I. (2006) The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microbiol. 60, 312–330. [DOI] [PubMed] [Google Scholar]
- Jain M.; Petzold C. J.; Schelle M. W.; Leavell M. D.; Mougous J. D.; Bertozzi C. R.; Leary J. A.; Cox J. S. (2007) Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc. Natl. Acad. Sci. U.S.A. 104, 5133–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover C. K.; de la Cruz V. F.; Fuerst T. R.; Burlein J. E.; Benson L. A.; Bennett L. T.; Bansal G. P.; Young J. F.; Lee M. H.; Hatfull G. F.; Snapper S. B.; Barletta R. G.; Jacobs W. R. Jr.; Bloom B. R. (1991) New use of BCG for recombinant vaccines. Nature 351, 456–460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.