

Abstract
Background
Emerging evidence suggests that ‘adaptive’ induction of autophagy (the cellular process responsible for the degradation and recycling of proteins and organelles) may confer a cardioprotective phenotype and represent a novel strategy to limit ischemia-reperfusion injury. Our aim was to test this paradigm in a clinically relevant, large animal model of acute myocardial infarction.
Methods and Results
Anesthetized pigs underwent 45 min of coronary artery occlusion and 3 hours of reperfusion. In the first component of the study, pigs received chloramphenicol succinate (CAPS: an agent that purportedly up-regulates autophagy; 20 mg/kg) or saline at 10 min before ischemia. Infarct size was delineated by tetrazolium staining and expressed as a % of the at-risk myocardium. In separate animals, myocardial samples were harvested at baseline and 10 min following CAPS treatment and assayed (by immunoblotting) for two proteins involved in autophagomsome formation: Beclin-1 and light chain (LC) 3B-II. To investigate whether the efficacy of CAPS was maintained with ‘delayed’ treatment, additional pigs received CAPS (20 mg/kg) at 30 min post-occlusion. Expression of Beclin-1 and LC3B-II, as well as infarct size, were assessed at end-reperfusion. CAPS was cardioprotective: infarct size was 25±5% and 41±4% in the CAPS-pretreated and CAPS-delayed treatment groups versus 56±5% in saline-controls (p<0.01 and p<0.05 versus control). Moreover, administration of CAPS was associated with increased expression of both proteins.
Conclusion
Our results demonstrate attenuation of ischemia-reperfusion injury with CAPS, and are consistent with the concept that induction of autophagy may provide a novel strategy to confer cardioprotection.
Keywords: myocardial infarction, ischemia, reperfusion, autophagy
Autophagy is the cellular process responsible for the degradation and removal of damaged organelles and protein aggregates.1-3 Although the role of autophagy in cardiac pathophysiology is complex and poorly understood,1-5 emerging data suggests that an ‘adaptive’ induction of autophagy may confer a cardioprotective phenotype.1-4, 6-11 This concept raises the intriguing possibility that pharmacologic up-regulation of autophagy may represent a novel therapeutic strategy to protect the heart against ischemia-reperfusion injury.
Three discrete pieces of evidence have identified chloramphenicol, a cytochrome P450 monooxygenase inhibitor and bacteriostatic antimicrobial, as a potential candidate agent to test this paradigm. First, P450 monooxygenase inhibitors, including chloramphenicol, have been reported to limit infarct size in small animal (rat and rabbit) models of ischemia-reperfusion.12 Second, chloramphenicol has been shown to induce an autophagic response in osteosarcoma cells.13 Moreover, recent preliminary data from our laboratory revealed a similar response in rat myocardium following in vivo administration of the succinated form of the agent: i.e., a rapid up-regulation in molecular markers of autophagosome formation. Our aim in the current study was to extend these observations and interrogate the paradigm of ‘adaptive’ autophagy in a clinically relevant, in vivo large animal model of myocardial ischemia-reperfusion injury. Specifically, we sought to establish: (i) whether pre-ischemic administration of chloramphenicol succinate (CAPS) reduces infarct size in the anesthetized porcine model of coronary artery occlusion-reperfusion; (ii) whether cardioprotection can be achieved if administration of CAPS is ‘delayed’ (i.e., given before reperfusion, rather than as a pretreatment); and (ii) whether administration of CAPS was associated with induction of autophagy.
Methods
This study was approved by the Institutional Animal Care and Use Committee of Wayne State University and was performed in accordance with the Guide for the Care and Use of Laboratory Animals from the Institute of Laboratory Animals Resources (NIH Publication Vol. 25 No. 28, revised 1996).
Surgical Preparation
Twenty-one domestic swine (10 male; 11 female; 26±2 kg [mean ± SD]) were pre-medicated with ketamine (33 mg/kg intramuscular) and an intravenous (IV) catheter was positioned in an ear vein. All pigs were then anesthetized with sodium pentobarbital (20-40 mg/kg IV), intubated and ventilated with room air. Core body temperature was monitored with a rectal temperature probe and maintained between 37-38°C throughout the experiment. Fluid-filled catheters were positioned in the right carotid artery for continuous monitoring and measurement of systemic hemodynamic parameters (heart rate; arterial pressure), and in the right external jugular vein for administration of fluids and supplemental anesthesia. The electrocardiogram was continuously monitored throughout each experiment.
The heart was exposed via a median sternotomy and suspended in pericardial cradle. A fluid-filled catheter was inserted into the left atrial appendage, and a segment of the left anterior descending coronary artery (LAD) was isolated, usually mid-way along its course.
CAPS: Pretreatment
Infarct size
For the first 12 pigs enrolled into the study, our aim was to establish whether prophylactic administration of CAPS was cardioprotective. After stabilization, baseline values of body temperature, heart rate and arterial pressures were tabulated. Each pig was then assigned to receive CAPS (20 mg/kg dissolved in 12 mL phosphate-buffered saline [PBS]; n=6), or a matched volume of PBS (n=6). CAPS (rather than chloramphenicol) was utilized, as the succinated form is soluble in aqueous media and thus more suitable for in vivo administration. Treatment was administered in a blinded manner and given as a slow bolus (over 2-3 minutes) into the left atrial appendage.
Ten minutes after treatment, pigs underwent 45 minutes of coronary occlusion followed by 3 hours of reperfusion (achieved by placement/removal of an atraumatic vascular clamp on the isolated LAD segment). To minimize the possible confounding effects of occlusive thrombosis at the site of the vascular clamp, animals received heparin (2000 units IV) prior to coronary artery occlusion. In addition, to limit the incidence of lethal ventricular fibrillation (VF), lidocaine was administered to all animals (2 mg/kg IV bolus) immediately before restoration of blood flow. Pigs that developed VF either during ischemia or upon reflow were resuscitated by applying low-energy DC countershocks directly to the heart (energy of 25 Joules; maximum of 4 attempts).
At the conclusion of the 3 hour reperfusion period, the LAD was ligated, the ascending aorta was briefly clamped and Evan's blue dye was injected into the coronary circulation via the left atrial catheter to delineate the area at risk of infarction. All pigs were then euthanized under deep pentobarbital anesthesia by intracardiac injection of KCl. The heart was immediately excised, sliced into 5-6 transverse slices and digital images acquired. To differentiate necrotic versus viable myocardium, the heart slices were incubated in triphenyltetrazolium chloride for 10 minutes at 37°C and re-photographed.14, 15
Our primary endpoint in this component of the study was infarct size. Area at risk (AR) and area of necrosis (AN) in each heart slice were quantified from the digital photographs using image analysis software (SigmaScan Pro, SPSS Inc., Chicago IL), corrected for tissue weight, and summed for each heart. AR was then expressed as a % of the total left ventricular (LV) weight, while AN was expressed as a % of the AR.14, 15 All analysis of risk region and infarct size was done in a blinded manner, without knowledge of the study group. Secondary endpoints included hemodynamics (heart rate, arterial pressure) assessed repeatedly throughout each experiment and, for each pig, the incidence of VF.
Molecular markers of autophagy
Two additional pigs were instrumented as described previously. After stabilization, myocardial biopsies were obtained from the LV free wall, and CAPS (20 mg/kg) was administered as a slow bolus into the left atrium. At 10 minutes after the onset of treatment (the time corresponding to the onset of coronary occlusion in the previous infarct size experiments), the pigs were euthanized and LV tissue blocks were rapidly excised. All tissue samples were immediately snap-frozen and stored at -80°C until processed.
Our aim was to assess the expression of two pivotal proteins involved in autophagomsome formation: Beclin-1 and light chain (LC) 3-II.1-5 Frozen myocardial samples (50-100 mg) were minced and homogenized in an extraction buffer containing 50 mM Tris-HCl (pH 7.5), 5mM EDTA, 10mM EGTA, 10mM benzamidine, 0.3% β-mercaptoethanol and 1X Complete Mini EDTA-Free Protease Inhibitor Cocktail (Roche, Mannheim, Germany). Homogenates were centrifuged at 1000xg for 10 minutes, the supernatant was decanted, and protein concentration was determined by Bradford Assay (Thermo Scientific, Rockford, IL). Proteins (20 μg per lane) were separated by SDS-polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA) and probed with primary antibodies for Beclin-1 and the B-isoform of LC3-II (Cell Signaling Technology, Beverly, MA and Novus Bio, Littleton, CO). Immunoblotting for GAPDH (Sigma Aldrich, St. Louis MO) was conducted to control for protein loading. After washing, the blots were incubated with an HRP-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), and developed with the Pierce ECL chemiluminescence kit (Thermo Scientific, Rockford, IL). Blots were scanned and quantified using image analysis software (SigmaScan Pro, SPSS Inc., Chicago IL).
CAPS: ‘Delayed’ treatment
Infarct size
The final 7 pigs enrolled in the study underwent 45 min of LAD occlusion and 3 hours of reperfusion as described previously. Our objective in these supplemental experiments was to determine whether cardioprotection can be achieved if CAPS is given before reperfusion, rather than as a pretreatment; accordingly animals received CAPS (20 mg/kg dissolved in 12 mL PBS: n=6) or PBS alone (n=1) at 30 min post-occlusion. AR and AN were quantified as described above, and hemodynamics were assessed throughout each experiment.
Molecular markers of autophagy
LV samples were obtained at 3 hours post-reperfusion (3.25 hours after administering CAPS or PBS), before delineation of infarct size. Tissue from remote, normally perfused myocardium (i.e., not confounded by the presence of necrosis or the effect of ischemia-reperfusion per se on autophagy11) was processed and probed for expression of Beclin-1, LC3B-II and, as a loading control, GAPDH.
Statistics
AR/LV and AN/AR were compared among the control, CAPS-pretreated and CAPS-delayed treatment groups by analysis of variance (ANOVA) followed by the Newman-Keuls post-hoc test. Comparisons of heart rate and mean arterial pressure among groups were made using generalized estimating equations (GEE). The decision to use parametric (rather than non-parametric) statistics and present the data as mean ± SEM (rather than as medians, first and third quartiles) was made after applying the Kolmogorov-Smirnov normality test and obtaining p-values >0.10. Incidence of VF among groups was compared using an extension of Fisher's exact test. Expression of Beclin-1 and LC3B-II following CAPS treatment, corrected for GAPDH expression and normalized to either baseline values (for CAPS-pretreatment) or control values (for CAPS-delayed treatment) is reported but, because of the limited number of samples obtained, no conclusions regarding statistical significance are made.
Results
Incidence of ventricular fibrillation
Of the 19 pigs that underwent coronary artery occlusion-reperfusion, 8 developed VF during ischemia and/or at the time of reflow: 4/7 in the control group, 3/6 pretreated with CAPS and 1/6 that received CAPS-delayed treatment (p=0.38 [not significant]). There were no premature deaths: all animals were successfully resuscitated and completed the protocol.
Hemodynamics (Figure 1)
Figure 1.

Heart rate and mean arterial pressure (mean ± SEM), measured at baseline, immediately before occlusion (Pre), and throughout coronary occlusion-reperfusion.
There were no significant differences in heart rate (p=0.33) or mean arterial pressure (p=0.65) among control, CAPS-pretreated and CAPS-delayed treatment groups.
Infarct size (Figure 2)
Figure 2.

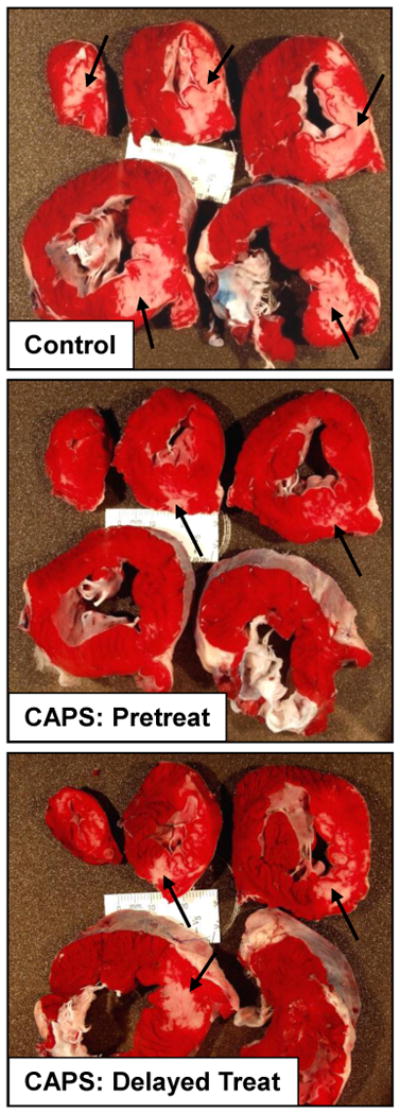
Reduction of infarct sized with CAPS. Top panel: Infarct size, expressed as a % of the myocardium at risk (mean ± SEM) for Control, CAPS-pretreated and CAPS-delayed treatment cohorts. Bottom panel: Images of heart slices obtained from one Control, one CAPS-pretreated pig and one pig that received CAPS-delayed treatment. Heart slices were incubated in triphenyltetrazolium chloride; using this method, viable myocardium stains red while areas of necrosis (denoted by arrows) are unstained and thus appear pale.
Area at risk was comparable in control, CAPS-pretreated and CAPS-delayed treatment groups, averaging, 23±2%, 21±1% and 23±1% of the total LV weight, respectively. Mean infarct size in the control cohort was 56±5% of the risk region. In contrast, AN/AR was significantly reduced with CAPS (p=0.009 by ANOVA): area of necrosis averaged 25±5% and 41±4% of the myocardium at risk in the CAPS-pretreated and CAPS-delayed treatment cohorts, respectively (p<0.01 and p<0.05 versus control).
Molecular markers of autophagy (Figures 3 and 4)
Figure 3.
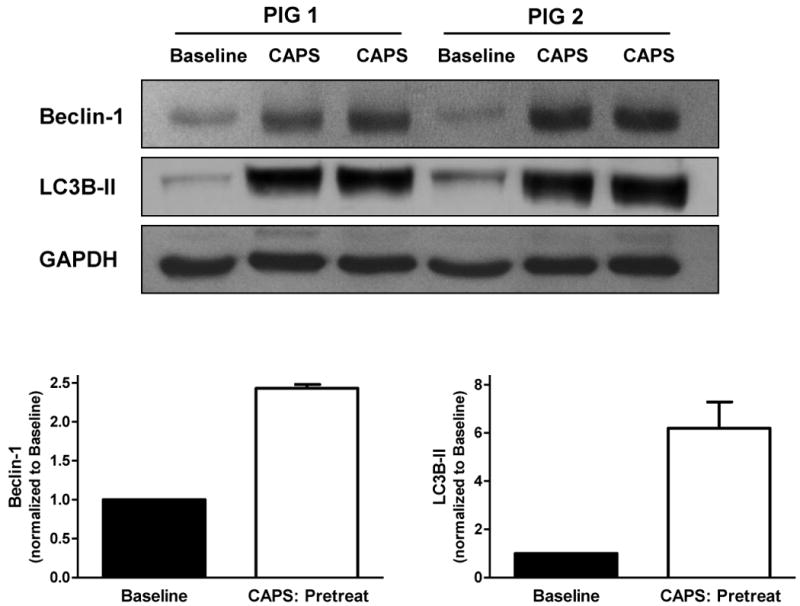
Expression of Beclin-1 and LC3B-II with CAPS-pretreatment. Myocardial samples were obtained at baseline and 10 minutes following administration of CAPS. Top panel: Original immunoblots. Bottom panel: Immunoreactivity of Beclin-1 and LC3B-II, corrected for GAPDH expression and normalized to baseline values (mean ± SEM).
Figure 4.
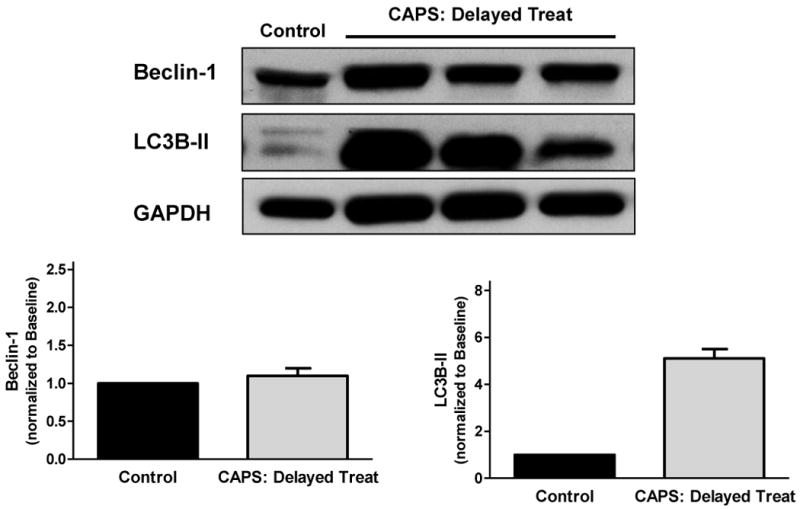
Expression of Beclin-1 and LC3B-II with CAPS-delayed treatment. Samples were obtained from remote, normally perfused myocardium at end-reperfusion, 3.25hours following CAPS administration. Top panel: Original immunoblots. Bottom panel: Immunoreactivity of Beclin-1 and LC3B-II, corrected for GAPDH expression and normalized to control values (mean ± SEM).
In pigs pretreated with CAPS, myocardial expression of Beclin-1 and LC3B-II was 2.4- and 6.2-fold higher at 10 min following treatment versus baseline (Figure 3). Delayed administration of CAPS was associated with increased expression of LC3B-II (5.1-fold greater than control), but not Beclin-1, at 3 hours following relief of ischemia (Figure 4).
Discussion
We report that CAPS, administered as a pre-treatment or before reperfusion, rendered the porcine heart resistant to ischemia-reperfusion injury. Moreover, our results suggest that the infarct-sparing effect of CAPS is associated with increased myocardial expression of molecular markers of autophagy.
Autophagy: beneficial versus detrimental?
Autophagy is the highly conserved cellular process whereby subcellular components (including damaged organelles and protein aggregates) are sequestered in autophagosomes for delivery to lysosomes and subsequent degradation by lysosomal enzymes.1-4 There is compelling evidence that constitutive autophagy is crucial for the maintenance of cardiomyocyte structure and function under basal conditions.1, 16 In addition, it is well-documented that acute and chronic stressors – including ischemia-reperfusion and ischemia per se, hypoxia, starvation, hypertrophy and heart failure – induce an up-regulation in autophagy.1-5, 8, 16, 17 However, the role of autophagy in the setting of stress (protective versus detrimental) is complex, poorly understood, and in all likelihood dependent upon the severity and duration of the insult.
Recent attention has focused on the concept that pre-ischemic induction of autophagy may confer a cardioprotective phenotype and render the heart resistant to lethal ischemia-reperfusion injury, possibly via the removal of damaged and dysfunctional mitochondria, disposal of misfolded and aggregated proteins, and subsequent recycling of amino acids and fatty acids to provide a compensatory source of energy production during the sustained ischemic insult.2-4, 7, 11 In support of this concept, induction of autophagy has been implicated to play a role in the protection achieved with ischemic preconditioning and administration of the adenosine A1 receptor agonist and classic ‘preconditioning-mimetic’ 2-chloro-N(6)-cyclopentyl- adenosine (CCPA).6, 7, 9 However, in apparent contrast, other studies have reported an attenuation (rather than exacerbation) of ischemia-reperfusion injury in hearts from mice in which autophagy is down-regulated via knock-down of Beclin-1.18, 19 Our current results, demonstrating reduction of infarct size and increased expression of Beclin-1and LC3B-II with CAPS-pretreatment, are consistent with the proposed association between pre-ischemic induction of autophagy and cardioprotection. Moreover, we extend this concept and show that prophylactic administration of CAPS is not a prerequisite for protection; infarct size reduction was maintained (albeit lesser in magnitude) when CAPS was given during, rather than before, sustained ischemia. Addition of chloramphenicol at the time of reperfusion has been reported to limit infarct size in isolated buffer-perfused hearts;12 however, our study provides the first evidence of infarct size reduction with ‘delayed’ CAPS treatment in an in vivo, large animal model.
Reduction of infarct size with CAPS
Our aim was to investigate the paradigm of ‘adaptive’ autophagy by establishing whether a pharmacologic agent capable of initiating an autophagic response would limit myocardial infarct size. In this regard, our results provide qualitative evidence that CAPS triggers an increase in myocardial expression of Beclin-1 and LC3B-II. Furthermore, data obtained with delayed administration of CAPS imply a persistent increase in markers of autophagy (specifically, LC3B-II) at 3.25 hours post-treatment.
Beclin-1 and LC3-II are crucial proteins required for autophagosome formation.2, 3, 20, 21 Moreover, although the mechanisms of autophagy are highly integrated with phosphatidylinositol-3 kinase (PI3 kinase)/Akt signaling2, 3 and recent data have identified the involvement of protein kinase C (PKC),22 the two proteins are not, based on present knowledge, components of conventional, cardioprotective signaling pathways.23 Rather, Beclin-1 and LC3-II are considered specific markers of autophagy and autophagosome formation; thus, the increased in expression of Beclin-1 and LC3B-II seen with administration of CAPS imply induction of autophagy by this agent.
It appears implausible that the increased expression of autophagic markers seen at 10 minutes after CAPS treatment can reflect de novo protein synthesis. A brisk increase in expression of LC3B-II within this time-frame is feasible, as LC3-II is generated from LC3-I via C-terminal cleavage and conjugation to phosphatidylethanolamine.2, 3 In contrast, the increase in Beclin-1 is more difficult to reconcile and may reflect an as-yet unexplained, rapid attenuation in degradation (rather than increased synthesis) of the protein. Resolution of this question will require future, prospective investigation.
Limitations and future directions
The results of the current study demonstrate reduction of infarct sized with CAPS, an effect that was associated with qualitative evidence of increased expression of molecular markers of autophagy. The data do not, however, provide insight into the molecular mechanisms responsible for initiating this apparent increase in expression of Beclin-1 and LC3B-II, and do not provide evidence of cause-and-effect. Indeed, we cannot exclude the possibility that other mechanisms, including purported protective effects of succinate,24 may contribute to the infarct-sparing effect of CAPS. Efforts to obtain definitive proof that up-regulation of autophagy is necessary and sufficient for CAPS-induced cardioprotection are confounded by the lack of selectivity of current pharmacologic tools: i.e., wortmannin and 3-methyladenine inhibit the initiation of autophagy, but also inhibit the classic, PI3 kinase/Akt ‘survival’ pathway.2, 3 Moreover, despite the clinical relevance afforded by the use of the in vivo porcine model, application of selective molecular tools, such as suppression of autophagy by point mutation of the essential autophagy gene Atg5,7, 22 is not feasible in this preparation.
Although our data imply an up-regulation of Beclin-1 and LC3B-II with CAPS, expression of autophagic markers was assessed in a small number of pigs (thereby precluding statistical comparisons) and at limited time-points following drug administration. Indeed, it could be argued that, in the CAPS-delayed treatment group, a more meaningful approach may have been to obtain tissue samples during the early minutes post-reflow rather than at end-reperfusion. Given the propensity of the pig to develop reperfusion-induced VF, we elected not to undermine the successful completion of these experiments and assessment of our primary endpoint (infarct size) by obtaining biopsies at this vulnerable time-point. In addition, we did not quantify autophagosome abundance or autophagic flux (that is, the dynamic formation and clearance of autophagomes) in CAPS-treated hearts versus controls.2, 3, 21
In addition to the resolution of these mechanistic issues, future studies are required to establish whether alterations in the dose, timing or regimen of CAPS treatment (bolus + infusion rather than bolus alone) might yield even greater cardioprotective efficacy, and whether addition of CAPS to cardioplegic solution confers benefit in the setting of cardiopulmonary bypass surgery. Nonetheless, our results demonstrate attenuation of ischemia-reperfusion injury with CAPS, and are consistent with the concept that induction of autophagy may provide a novel therapeutic strategy to confer cardioprotection.
Acknowledgments
Funding Sources: Supported in part by NIH R43 HL095199 to Radical Therapeutix, Inc.
Footnotes
Presented in part at the Scientific Sessions 2009 of the American Heart Association, Orlando FL, November 2009
Disclosures: Roberta A. Gottlieb, MD is Founder and CEO of Radical Therapeutix, Inc., and Robert M. Mentzer Jr., MD is on the Scientific Advisory Board of Radical Therapeutix, Inc.
References
- 1.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009;16:31–38. doi: 10.1038/cdd.2008.163. [DOI] [PubMed] [Google Scholar]
- 2.Gottlieb RA, Finley KD, Mentzer RM., Jr Cardioprotection requires taking out the trash. Basic Res Cardiol. 2009;104:169–180. doi: 10.1007/s00395-009-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong Y, Undyala VV, Gottlieb RA, Mentzer RM, Jr, Przyklenk K. Autophagy: Definition, molecular machinery and potential role in myocardial ischemia-reperfusion injury. J Cardiovasc Pharm Ther. 2010 doi: 10.1177/1074248410370327. in press. [DOI] [PubMed] [Google Scholar]
- 4.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–661. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13:373–387. doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson AB, Mentzer RM, Jr, Gottlieb RA. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol. 2009;104:157–167. doi: 10.1007/s00395-009-0006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dosenko VE, Nagibin VS, Tumanovska LV, Moibenko AA. Protective effect of autophagy in anoxia-reoxygenation of isolated cardiomyocyte? Autophagy. 2006;2:305–306. doi: 10.4161/auto.2946. [DOI] [PubMed] [Google Scholar]
- 9.Yan L, Sadoshima J, Vatner DE, Vatner SF. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy. 2009;5:709–712. doi: 10.4161/auto.5.5.8510. [DOI] [PubMed] [Google Scholar]
- 10.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 12.Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth P, Jr, Yeager M, Gottlieb RA. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci U S A. 2004;101:1321–1326. doi: 10.1073/pnas.0308185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prigione A, Cortopassi G. Mitochondrial DNA deletions and chloramphenicol treatment stimulate the autophagic transcript ATG12. Autophagy. 2007;3:377–380. doi: 10.4161/auto.4239. [DOI] [PubMed] [Google Scholar]
- 14.Colantonio DA, Van Eyk JE, Przyklenk K. Stunned peri-infarct canine myocardium is characterized by degradation of troponin T, not troponin I. Cardiovasc Res. 2004;63:217–225. doi: 10.1016/j.cardiores.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Kristo G, Yoshimura Y, Ferraris SP, Jahania SA, Mentzer RM, Jr, Lasley RD. The preischemic combination of the sodium-hydrogen exchanger inhibitor cariporide and the adenosine agonist AMP579 acts additively to reduce porcine myocardial infarct size. J Am Coll Surg. 2004;199:586–594. doi: 10.1016/j.jamcollsurg.2004.05.274. [DOI] [PubMed] [Google Scholar]
- 16.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 17.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 18.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 19.Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS, Stephanou A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006;40:846–852. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- 20.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perry CN, Kyoi S, Hariharan N, Takagi H, Sadoshima J, Gottlieb RA. Novel methods for measuring cardiac autophagy in vivo. Methods Enzymol. 2009;453:325–342. doi: 10.1016/S0076-6879(08)04016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, Tsukada YT, Hamacher-Brady A, Mentzer RM, Jr, Gottlieb RA. Autophagy and protein kinase C are required for cardioprotection with sulfaphenazole. Am J Physiol. 2010:H570–579. doi: 10.1152/ajpheart.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118:1915–1919. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]
- 24.Sakamoto M, Takeshige K, Yasui H, Tokunaga K. Cardioprotective effect of succinate against ischemia/reperfusion injury. Surg Today. 1998;28:522–528. doi: 10.1007/s005950050177. [DOI] [PubMed] [Google Scholar]