

Abstract
Four mixtures of four fluorous-tagged quasiisomers have been synthesized, demixed and detagged to make all 16 stereoisomers of the macrocyclic lactone natural product Sch725674. A new bare-minimum tagging pattern needs only two tags— one fluorous and one non-fluorous—to encode four isomers. The structure of Sch725674 is assigned as (5R,6S,8R,14R,E)-5,6,8-trihydroxy-14-pentyloxacyclotetradec-3-en-2-one. Various comparisons of spectra of 32 lactones (16 with tags, 16 without) and 16 ester precursors (8 with tags, 8 without) provide insights into when and why related compounds have the same or different spectra.
Introduction
Fluorous mixture synthesis (FMS) is proving to be a powerful tool to make stereoisomer libraries of natural products.1 In a simple view of this process, molecules are labeled instead of vials or flasks. Different fluorous tags are attached to true isomers to make quasiisomers2 (the labeled molecules) with encoded stereochemical information.3 The quasiisomers can then be mixed and carried through multiple steps until a final demixing, which is also controlled by the fluorous tags.
Three of the most important themes in fluorous mixture synthesis have been tagging strategies,4 expedited natural product structure assignment,5 and comparison of spectra and properties of stereoisomer libraries.6 Here we report a total synthesis of the complete stereoisomer library of the macrolactone Sch725674 that advances all three of these themes. We introduce a new tagging strategy that uses fewer total fluorine atoms than any other to date, we confirm the two-dimensional structure of Sch725674 and assign the three-dimensional structure, and we derive interesting information from comparison of spectra of true stereoisomers and quasiisomers both before and after macrolactonization. This is the first time that a natural product stereostructure has been assigned from scratch by making a complete stereoisomer library by FMS.
Macrolactones and related macrolactams are large and important classes of natural products that are often thought to strike a balance between preorganization and conformational flexibility in binding to their biological targets.7 Because of these features, diversity oriented synthesis based on macrolactone templates is a lively area of research.8
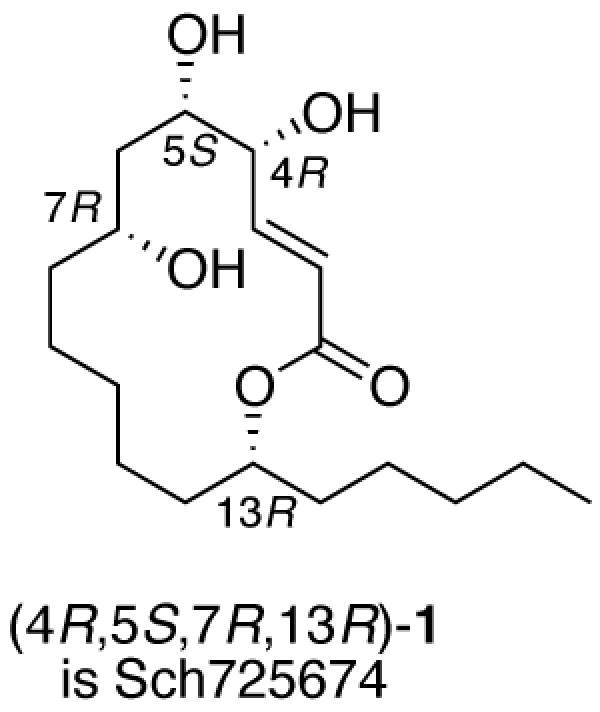
Sch725674 is a macrolactone that was isolated by Yang and coworkers from a culture of Aspergillus sp.,9 and it displayed antifungal activity against Saccharomyces cerevisiae and Candida albicans. The two dimensional structure was assigned as (E)-5,6,8-trihydroxy-14-pentyloxacyclotetradec-3-en-2-one 1 based on a battery of 2D NMR experiments. We follow the numbering system of the isolation paper, which puts stereocenters at C4, C5, C7, and C13, as shown in Figure 1.
Figure 1.
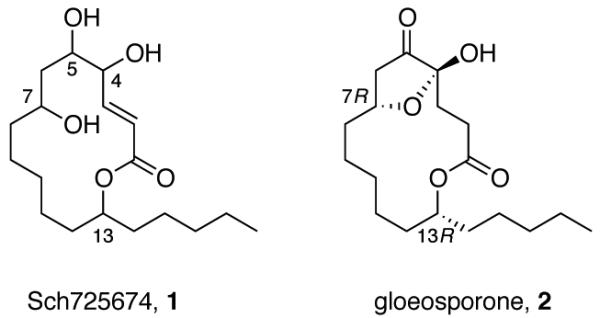
Two-dimensional structure of Sch725674 and complete structure of gloeosporone.
14-Membered macrolactone templates like 1 that lack methyl groups on the backbone are rare, with the popular synthetic target10 gloeosporone11 2 being the closest relative of 1. All known 14-membered (and related 18-membered ring) macrolactone natural products have the (13R)-configuration as established by Celmer,12 Seebach and Schreiber.11a Those that have a hydroxyl group at C7 are also usually (7R), again like gloeosporone.
Results and Discussion
We describe here a second-generation synthesis of the Sch725674 stereoisomer library. The first-generation synthesis, described in the thesis of Dr. X. Wang,13 produced eight isomers (including Sch725674) on a sub-milligram scale. Improvements to make the synthesis robust enough to produce all sixteen isomers on multi-milligram scale and model tagging experiments are discussed in the thesis of Dr. J. Moretti.14 Here we focus exclusively on the successful second-generation fluorous mixture synthesis.
Tagging plan
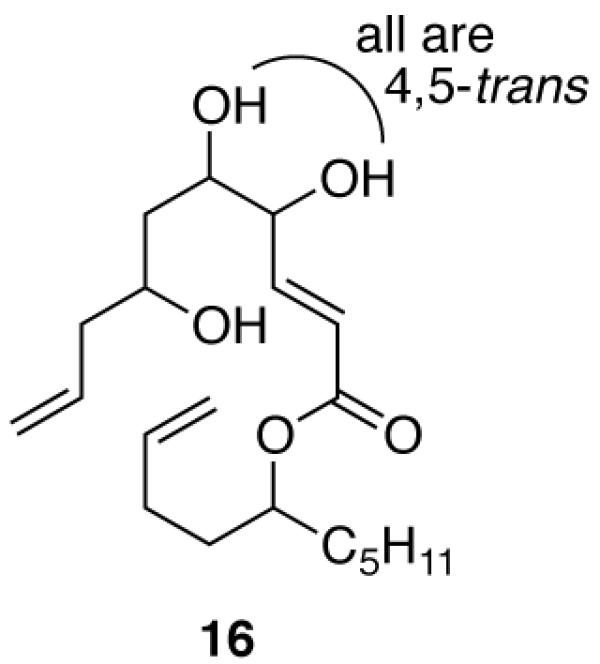
We set out to make all 16 stereoisomers of Sch725674 1, and the plan that evolved for the FMS is shown in Figure 2. Late stage coupling of a pair of enantiopure fragments (R)-3 and (S)-3 with mixtures M-415 followed by ring-closing metathesis, hydrogenation, demixing and detagging would give the final products. In turn, the preparation of the 4,5-trans (4R,5R and 4S,5S) series and 4,5-cis-series (4R,5S and 4S,5R) isomers needed different chemistry, so two four-compound mixtures of 4 were prepared.
Figure 2.
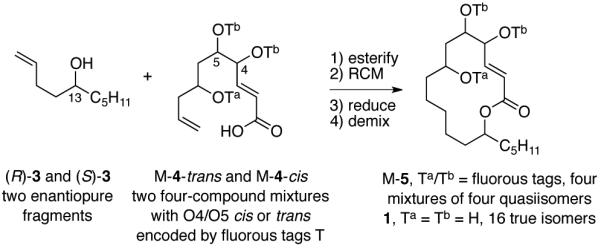
The tagging plan calls for one principal fragment 3 as two enantiomers and the other principal fragment 4 as two mixtures of four quasiisomers. One of these mixtures is the 4,5-trans series and the other is the 4,5-cis series. Coupling of the two enantiomers of 3 with the two mixtures of 4 gives four mixtures of four tagged quasiisomers M-5 as penultimate precursors of the 16 true isomers of 1.
The tagging pattern for each of the four-compound mixtures needs to encode two pieces of information (configurations at C4,5 and at C7) to allow for orderly demixing of the four components. Information and separation (demixing) aspects of several possible tagging strategies are shown in Figure 3. Here the numbers (#) on the tags T# serve as proxies for fluorine content, so the prospect for demixing of a mixture can be assessed simply by summing the tag numbers of each component. After the tagging is complete, each component of the mixture should be a quasiisomer of all the other components, having different configuration and constitution (specifically, fluorine content) from all the other components.
Figure 3.
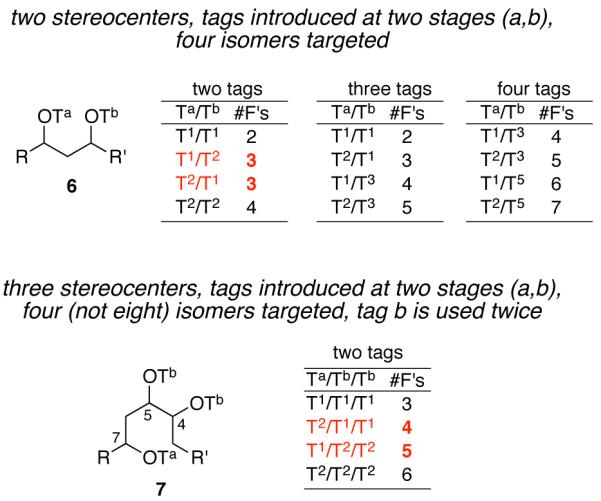
Tagging patterns for making four quasiisomers of compounds with two (6) and three (7) stereocenters. Tags “a” and “b” encode stereochemical information. The superscripted numbers are proxies for the fluorine content. Notice the redundancy when two tags encode two stereocenters that is broken when two tags encode three stereocenters.
Consider first a compound 6 (Figure 3) with two tagged stereocenters. From the information standpoint, only two tags (T1 and T2) are needed to uniquely encode four bits of information, here the four possible configurations of the two stereocenters. However, summing the tag numbers of the four different components that are generated with two tags shows that two of the components will be constitutional isomers (not quasiisomers). These have the same fluorine content (T1 + T2 = T2 + T1), so demixing (which is based primarily of fluorine content) will not be reliable.
The use of four different tags (in the sequence T1, T2, T3, T5) solves this redundancy problem for 6. Less obvious but also reliable is the use of only three tags.4 Here, the first stereocenter gets T1 and T2. However, instead of using T3 and T5 to tag the second stereocenter (the four-tag method), T1 is reused along with a T3. This gives products with different fluorine count; in other words, all are quasiisomers and none are constitutional isomers.
We recognized that the redundancy problem that is apparently inherent in the two-tag approach could be lifted by putting one set of tags (here “a”) on once and the other set of tags (“b”) on twice. This is shown in Figure 3 with a triol 7 as used in the Sch725674 setting.
Stereocenter C7 gets one tag that encodes its absolute configuration, for example (7R) gets T1 and (7S) gets T2. In contrast, stereocenters C4 and C5 receive tags at the same time and accordingly get the same tag. These tags again encode absolute configuration, but of only one of the possible pairs of relative configurations (two cis or two trans).
For example, the two possible trans configurations of 7, (4R,5R) and (4S,5S), are again encoded with T1 and T2 respectively. These are the same tags as on C7; however, each isomer needs two tags at this point, so the fluorine content of the second set of tags is doubled and the redundancy problem is lifted. Four quasiisomers result. The up side of this strategy is that four quasiisomers can be tagged with two tags, but there is also a down side; there are eight total isomers, so the other four cis-isomers are not provided for at all. However, this fits the Sch725674 plan well since the trans- and cis-series of precursors will be made by different chemistry.
To summarize, the sixteen-compound library of isomers 1 will be made from four mixtures of four quasiisomers each. The absolute configuration at C13 and the relative configuration at C4/C5 are known by which mixture the sample comes from (in other words, the labels on the flasks), while the identities of each of the four quasiisomers that constitute those mixtures are known by the fluorous tags (in other words, the labels on the molecules). For the first time, the encoding and separation of four quasiisomers is enabled with only two tags. And only one of the two tags needs to be fluorous.
Stereoisomer library synthesis
Prior to starting the fluorous mixture synthesis phase, the needed stereoisomeric precursors were made as summarized in Schemes 1 and 2. In the 4,5-trans series, Scheme 1, Sharpless asymmetric dihydroxylation16 of diene 8 with Admix-α produced diol (S,S)-9 as a 96/4 ratio of enantiomers as assessed by Mosher ester analysis17 (see Supporting Information). The (S,S)-configuration was encoded by attachment of two standard TIPS groups (triisopropylsilyl, Si(iPr)3, hereafter shown as TH) to give (S,S)-10 in 100% yield.
Scheme 1. Synthesis of trans-series quasiisomers 13a-d.

Minor products 12 from allylboration were separated prior to silylation. TH is –Si(iPr)3; TF is –Si(iPr)2CH2CH2C2F5.
Standard removal of the PMB group and Swern oxidation provided aldehyde (S,S)-11, which was divided in half and exposed to both enantiomers of the Brown/Ramachandran reagent,18 Ipc2B(allyl). Each reaction produced a major diastereomer, (SSR)-12 or (SSS)-12, in about 4/1 ratio along with the C7 epimer. To ensure maximum stereopurity of the tagged isomers, these mixtures were separated to give the two pure isomers in 59% and 67% yield, respectively. Then the configurations at C7 were encoded with a standard TIPS group (TH) for (7R) and a fluorous TIPS group bearing a pentafluoroethyl substituent (Si(iPr)2CH2CH2C2F5, hereafter FTIPS or simply TF) for (7S). This gives (S,S,R)-13a with zero fluorines and (S,S,S)-13b with five.
Members of the (4R,5R) series isomers were made from 8 and Admix-β, then encoded with the same two fluorous TIPS groups (TF).19 Brown/Ramachandran allylation and isomer purification as before provided the RRR and RRS isomers of 12 (not shown), which were again encoded with TIPS for (7R) and FTIPS for (7S) to give (R,R,R,)-13c (10 F’s) and (R,R,S)-13d (15 F’s). The resulting four quasiisomers were then mixed in equimolar amounts to make the first trans-series mixture M-13a-d.
The 4,5-cis-series quasiisomers were made by a similar series of reactions starting from both enantiomers of 2-deoxyribose, as detailed fully in the Supporting Information. The key allylation results are summarized in Scheme 2.
Scheme 2. Synthesis of cis-series quasiisomers 13e-h.
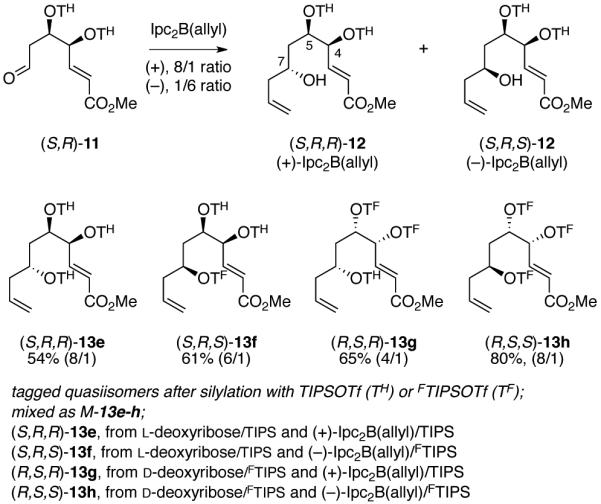
The minor allylation products could not be separated so the products 12 were silylated directly. Numbers in parentheses are epimer ratios of the tagged quasiisomers at C7.
While better diastereoselectivities (up to 8/1) were achieved in the Brown/Ramachandran allylations, the minor C7 diastereomers of 12 could not be separated from the major ones in any case, either before or after silylation. Contrast this to the trans-series where the minor products for the allylation were conveniently separated before silylation in every case. Lacking a better option, we tagged these mixtures to give 13e-h and moved ahead. The same tagging pattern was used in the cis-series as in the trans-series (a/e, 0 F; b/f, 5 F; c/g, 10 F; d/h 15 F). The four quasiisomers were mixed in equimolar amounts to make the first cis-series mixture M-13e-h.
Prior to starting the fluorous mixture syntheses in earnest, we tested the demixing of M-13a-d and M-13e-f by analytical fluorous HPLC. The chromatogram resulting from the trans series isomers is representative and is shown in Figure 4. The quasiisomers eluted in order of fluorine content and four wellseparated peaks were observed.
Figure 4.
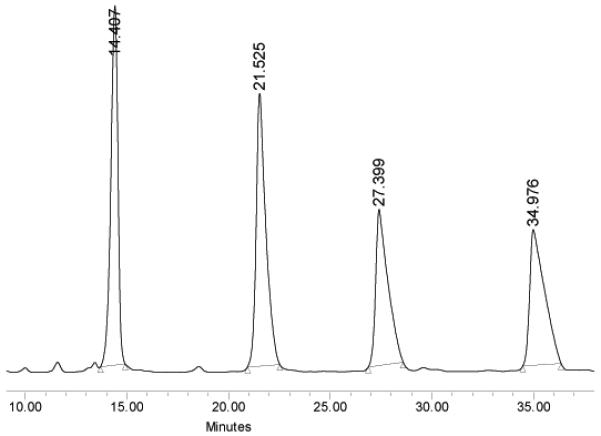
Trial demixing of M-13a-d. Compounds elute in order of fluorine content: 13a (0 F), 13b (5 F), 13c (10 F), 13d (15 F). FluoroFlash PF-C8 column, 90/10 MeCN/water for 10 min, then 100% MeCN
The mixture synthesis phase of the work is summarized in Scheme 3. Again taking the 4,5-trans-series as representative, the methyl ester of M-13a-d was cleaved with TMSOK,20 then the acid was divided in half and each portion was coupled with either (R)-3 or (S)-3 under Yamaguchi conditions21 to give esters M-(R)-14a-d and M-(S)-14a-d in good yields (80% and 84%). Ring closing metathesis with the second-generation Grubbs catalyst22 followed by hydrogenation with a poisoned Pd-catalyst gave the final tagged quasiisomer mixtures M-(R)-15a-d and M-(S)-15a-d (88% and 87%). The SrCO3 poison was important since other conditions often gave side products in which both double bonds were saturated.
Scheme 3. Fluorous mixture synthesis provides the penultimate precursors 15 of the final 16 stereoisomers of 1 as four mixtures of four quasiisomers.
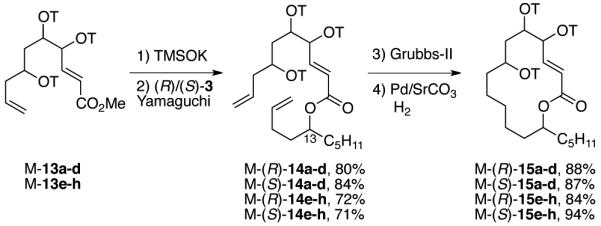
(R)/(S) represents the configuration at C13; a-d (4,5-trans series) or e-h (4,5-cis series) represents the various quasiisomers at C4/C5, and C7.
Conditions: 1) TMSOK, Et2O, rt, 16 h; 2) 2,4,6-trichlorobenzoyl chloride, DMAP, Et3N, toluene, rt, 3 h; 3) 2nd generation Grubbs catalyst, CH2Cl2, reflux, 48 h; 4) Pd/SrCO3, hydrogen balloon, EtOH, rt, 1 h.
The same reactions were conducted on the cis-series M-13e-h with similar yields to provide the other two mixtures of four quasiisomers, M-(R)-15e-h and M-(S)-15e-h. The final four mixtures of four quasiisomers were produced in substantial quantities (>200 mg each).
Demixing and detagging
The results of the demixing and detagging are summarized globally in Tables 1 and 2. In total, 24 individual, pure samples were produced after detagging. Eight ester stereoisomers 16 in the trans-series were isolated for comparison of spectra of pairs of ring-open and ring-closed analogs with the same configurations. And all 16 Sch725674 stereoisomers 1 were isolated to compare with each other and with the natural product. This necessitated six preparative demixings (two ester mixtures and four macrolactone mixtures), which are detailed in the Supporting Information.
Table 1. Summary of demixing and detagging of open esters M-(R)-14a-d and M-(S)-14a-d to give eight open esters 16 in the 4,5-trans series.
 | |||
|---|---|---|---|
| isomer of 16 | precursor | amount | yielda |
| (4S,5S,7R,13R) | (R)−14a | 17 mg | 65% |
| (4S,5S,7S,13R) | (R)−14b | 16 mg | 60% |
| (4R,5R,7R,13R) | (R)−14c | 26 mg | 69% |
| (4R,5R,7S,13R) | (R)−14d | 11 mg | 63% |
| (4S,5S,7R,13S) | (S)−14a | 24 mg | 67% |
| (4S,5S,7S,13S) | (S)−14b | 30 mg | 85% |
| (4R,5R,7R,13S) | (S)−14c | 24 mg | 77% |
| (4R,5R,7S,13S) | (S)−14d | 29 mg | 88% |
includes demixing, detagging and flash chromatography.
Table 2. Summary of demixing and detagging of lactones M-(R)-15a-h and M-(S)-15a-h to give the 16 final isomers of 1. The assigned structure of Sch725674 is shown as an example.
 | |||
|---|---|---|---|
| isomer of 1 | precursor | amount | yielda |
| (4S,5S,7R,13R) | (R)−15a | 6 mg | 19%b |
| (4S,5S,7S,13R) | (R)−15b | 15 mg | 51% |
| (4R,5R,7R,13R) | (R)−15c | 13 mg | 60% |
| (4R,5R,7S,13R) | (R)−15d | 8 mg | 51% |
| (4S,5S,7R,13S) | (S)−15a | 5 mg | 15%b |
| (4S,5S,7S,13S) | (S)−15b | 17 mg | 67% |
| (4R,5R,7R,13S) | (S)−15c | 3 mg | 14%b |
| (4R,5R,7S,13S) | (S)−15d | 11 mg | 29% |
| (4S,5R,7R,13R) | (R)−15e | 11 mg | 29%b |
| (4S,5R,7S,13R) | (R)−15f | 12 mg | 37%b,c |
| (4R,5S,7R,13R) | (R)−15g | 11 mg | 32%b |
| (4R,5S,7S,13R) | (R)−15h | 5 mg | 40%b |
| (4S,5R,7R,13S) | (S)−15e | 13 mg | 45%b |
| (4S,5R,7S,13S) | (S)−15f | 6 mg | 17%a |
| (4R,5S,7R,13S) | (S)−15g | 14 mg | 73%a |
| (4R,5S,7S,13S) | (S)−15h | 7 mg | 36%a |
includes demixing, detagging and flash chromatography
repurified by chiral chromatography
contains 6% of the C7 epimer
Briefly, the preparative demixings of two trans-series ester mixtures M-(R)-14a-d and M-(S)-14a-d were straightforward (see Figures S1 and S2 in the SI). Four well-separated peaks were observed in each case, and no cross-contamination of quasiisomers occurred. The pure quasiisomers were characterized (see below), then desilylated with TBAF in THF. The crude products were purified by flash chromatography to give the eight stereoisomeric trihydroxy esters 16 shown in Table 1 in overall yields of 60–88%.
The demixings of the four lactone quasiisomer mixtures M-15 ~90 mg injections) are summarized in Table 2. These separations presented solvable problems. Demixing of the two trans-series isomers M-(R)-15a-d and M-(S)-15a-d and detagging provided the eight trans triol lactone isomers after chromatographic purification, but only five of these were pure (See Figures S3 and S4 in the SI). The two samples containing three TIPS groups and therefore zero fluorine atoms ((R)-15a and (S)-15a) where poorly retained by the column. They contained non-isomeric impurities that we think resulted from inadequate separation of this first peak from accumulated nonfluorinated impurities in the solvent front. With 10 fluorines, quasiisomer (S)-15c was the third-eluting component in its mixture, and it was contaminated with about 25% of the prior, second-eluting quasiisomer (S)-15b of the mixture.
Of the several ways to solve these problems, we found it most expeditious just to detag the three mixtures and purify the final products by preparative chiral chromatography. An (S,S)-Whelk-O-1 column was used for the triols derived from detagging of (R)-15a and (S)-15a and a Chiralcel OD column was used for the triol from (S)-15c.
In the demixing of the two cis-series quasiisomer mixtures (SI Figures S5, S6), we were more careful to separate the quasiisomers with zero fluorines from solvent front, and this time seven of the eight products were obtained in good quality. One of the third eluting quasiisomers had some cross contamination of the prior quasiisomer, and this was simply demixed a second time to remove the contaminant.
Recall that the eight quasiisomers in the cis-series (second group of eight) are not stereoisomerically pure because we could not separate the minor products of C7 after the allylation. However, because the minor product of one sample is the major product of another sample, it proved easy to analyze all eight samples by 1H NMR spectroscopy. The identity and amount (10– 25%) of each minor isomer was as expected from the allylboration results in Scheme 2 (see the SI for the actual ratios).
In preliminary experiments, one of the cis-series isomers was desilylated slowly with TBAF in THF, so we switched to HF/CH3CN/H2O for preparative detagging. All eight samples were reliably desilylated, and chiral HPLC analyses showed one major and one minor peak for each sample. Finally, preparative chiral HPLC purification provided the eight final samples, seven of which were diastereomerically pure. The eighth sample, (4S,5R,7S,13R)-1, had the misfortune of arising from quasiisomer (R)-15f with the lowest starting isomer ratio (75/25) and the tightest separation on chiral HPLC. This was obtained in an enriched 94/6 ratio of diastereomers and as such is the only one of the 24 final samples with a detectable isomeric impurity. Substantial amounts of all 16 of the final lactone isomers (3–17 mg) were obtained, as shown in Table 2.
Isomer characterization and structure assignment of Sch725674
The fluorous mixture synthesis and demixing produced in total 48 individual samples grouped as 24 with tags and 24 without tags. Within each grouping of 24, there are 8 open chain esters (14 or 16, all isomers in the trans series) and 16 closed lactones (15 or 1, all isomers in both trans and cis series).
The members of the tagged set of 24 (14 and 15) were characterized by the usual means with 1D 1H and 13C NMR spectra and HRMS. Characterization of the final 24 detagged compounds (16 and 1) depended on enantiomeric series. Members of one enantiomeric series were characterized by IR, HRMS, chiral HPLC analysis, optical rotation, 1D NMR spectra, and a set of full 2D NMR experiments (1H-1H-COSY, 1H-13C-HMQC, and 1H-13C-HMBC). In this way, substantially all the protons and carbon resonances were assigned in all the final isomers.23
Members of the other enantiomeric series where characterized by chiral HPLC analysis, 1H NMR spectroscopy and optical rotation. This produced a substantial data set (presented as Tables S1-S3, S5, S6 and copies of spectra in the SI), which could have multiple uses. Here we use selected data for comparison with each other and for structure assignment of Sch725674.
The values of the optical rotations of 1 are shown in the SI, Table S4 (the concentrations (c) in MeOH were all about 1 g/100 mL). The magnitudes of the optical rotations of the final isomers 1 can be loosely characterized as low to moderate, ranging from about ±3 up to ±40. The values are spread out enough to be informative in many cases. In other words, the structure of an unknown isomer could be reduced to two or three candidates (sometimes even one) based on optical rotation alone. The optical rotation of the natural sample of Sch725674 was not recorded, so comparison is not possible.
Fuller discussions of spectra comparisons can be found in the theses of Drs. X. Wang13 and J. Moretti.14 Here we focus on selected NMR spectral data that are especially informative. Comparison of the NMR spectra of the tagged isomers 14 and 15 is limited because these spectra depend on the structure of the tag. Unlike most prior work, the two tags here are not homologs. However, we can still compare spectra of ester 14 (open) and lactone 15 (closed) analogs that have the same tags.
The CF2 regions of the 19F spectra of two related pairs of lactones and esters are shown in Figure 5. The compounds have the same tags (three FTIPS groups) and the same configurations at C4, C5 and C7, differing only in the configuration at C13. In theory, there should be three triplets because there are three heterotopic CF2 groups (which couple to the adjacent CH2 groups but not the adjacent CF3 groups). In the two esters (right-side pair of spectra), the resonances look like broad quartets and are identical. In the two lactones (left-side pair), one set of resonances is three clear triplets and the other is three overlapping but easily assigned triplets. The difference in chemical shifts between the two lactones is remarkable given how remote the CF2 groups are from the stereocenter at C13 (12 atoms, 14 atoms and 16 atoms at the shortest counts).
Figure 5.
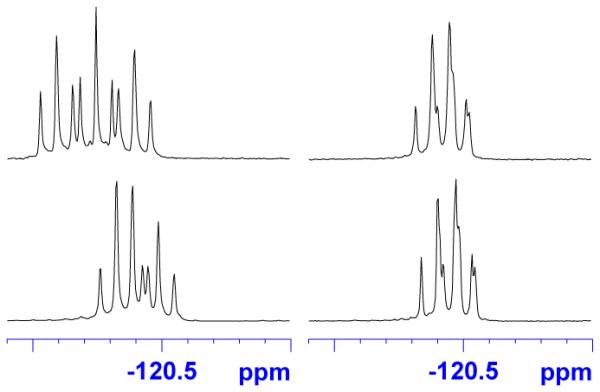
Comparison of the CF2 resonances of two lactones 15 (left) and esters 14 (right) with three fluorous silyl tags (–Si(iPr)2CH2CH2C2F5). The top spectra are (13R) isomers; the bottom spectra are (13S). The other stereocenters are each (4R,5R,7S).
This trend (ester spectra identical, lactone spectra different) held for the other three pairs of tagged C13 epimers, as summarized in Figure 6. Also, the 1H and 13C NMR spectra of the pairs of (R/S)-epimers of C13 of tagged lactones 15 were different, but the pairs of tagged esters 14 were the same. In addition, the same trend held for all of the compounds after detagging to make the free alcohols. In other words, all eight diastereomers of the lactones 1 gave clearly different 1H and 13C NMR spectra, while the four diastereomeric esters 16 (trans series only) gave two pairs of identical 1H and 13C NMR spectra. The pairs differed only in the configuration at C13.
Figure 6.
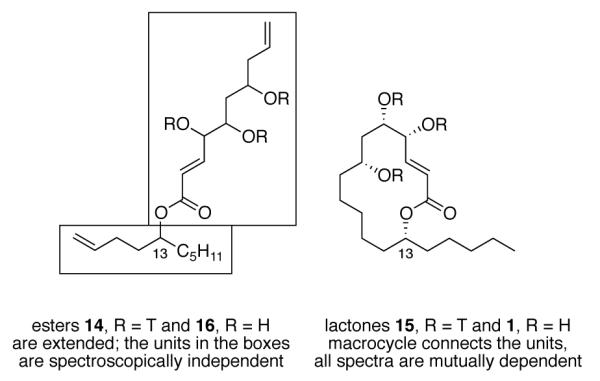
The groups of stereocenters (C13 and C4/C5/C7) behave independently in the spectra of the esters 14 and 16 but are mutually dependent in the spectra of the lactones 15 and 1.
In the open chain esters 14 and 16, the conformation of the ester C–O bond is trans, so C13 in the ester oxygen substituent is remote from the other three stereocenters in the carbonyl substituent. In no case can any spectrum (19F, 1H or 13C) sense this difference. In the lactones 15 and 1, the comparable C–O bond is probably also trans, but the two parts of the molecule are now connected though carbon ring backbone as well. A change in configuration in one part of the 14-membered ring can alter conformational populations of the ring and is therefore sensed in the spectral features of the other part of the ring.
This principle—that spectra of large rings with remote stereocenters are more sensitive to configuration changes than spectra of comparable acyclic molecules—seems intuitively sensible. But it is nonetheless impressive how the effect plays out in these libraries. In the open systems, every one of the 11 pairs of spectra compared was identical to its partner. In the closed systems, not one of the 22 pairs of spectra compared was identical.
Accordingly, each one of the final eight lactone diastereomers 1 exhibited a unique 1H and 13C NMR spectrum; no spectrum was substantially identical to any other spectrum. This makes assignment of the relative configuration of Sch725674 unambiguous; its published spectra were clearly different from seven of the diastereomers and matched only those of (4R,5S,7R,13R)-1 and its enantiomer. Since all such lactone natural products have the (13R) configuration,11 we can confidently assign the former structure to 1 as shown in Table 2.
Figure 7 illustrates some of the most obvious differences in the 1H NMR spectra of 1 with expansions of the regions of the three carbinol protons, H4, H5, and H7. Not one of these spectra has one single resonance that overlaps. This can be seen in every case by eyeball except perhaps that of H5 in spectra 4 and 5. In other words, from the chemical shift of any one resonance selected from any one of these three H’s, the stereostructure can be uniquely assigned in every case! Likewise, the alkene protons (H2, H3) exhibited large differences from isomer to isomer (see SI). In previous stereoisomer libraries, we have been surprised by the similarities in the spectra. In this library, we are surprised by the differences.
Figure 7.
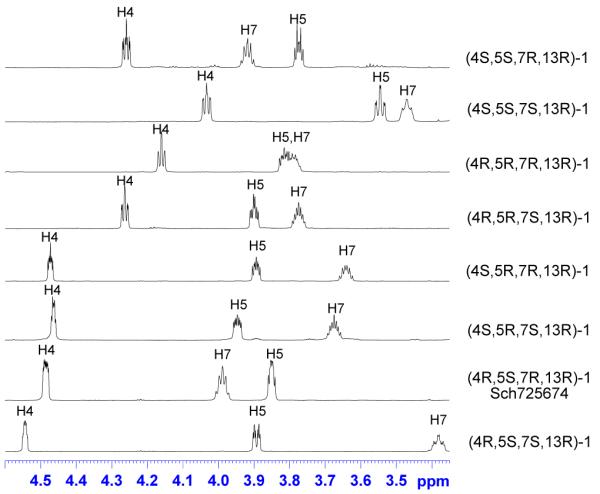
Expansions of the carbinol regions of the 1H NMR spectra (700 MHz, CD3OD) of the (13R) series of lactones 1.
There are also differences in the coupling constants from isomer to isomer, but these seem to represent primarily local effects of the group of three stereocenters. For example, H4 has vicinal protons on each side and can have a long range allylic coupling as well. When H4 and H5 are trans, these J’s play out as a broad triplet or a doublet of triplets (the first four spectra, two larger J’s and one smaller J). When H4 and H5 are cis, then a narrower peak shape results from three relatively small J’s (the last four spectra). In other words, the closure of the macrocycle changes the chemical shifts more than the coupling constants.
This conclusion is reinforced by comparing 1H NMR spectra of ester and lactone pairs that have the same configurations at C4, C5 and C7 but differ at C13. There are four sets of these groupings, one of which is shown in Figure 8. As mentioned above, the spectra of the two esters are identical to each other, and the spectra of the two lactones are different from each other.
Figure 8.
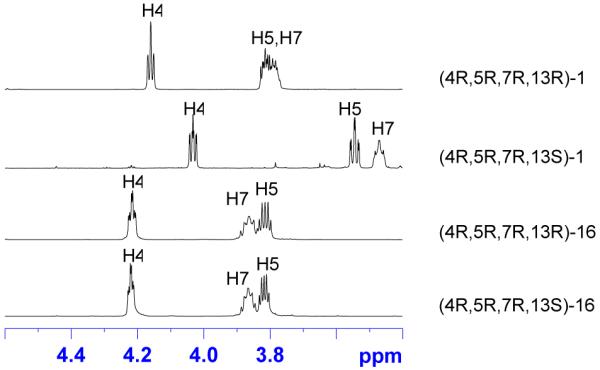
Expansions of the carbinol regions of the 1H NMR spectra of two lactones 1 (top) and two esters (bottom) 16 with the same configurations at C4, C5 and C7, differing only at C13.
In comparing the chemical shifts of these spectra of the esters to the lactones, it might be prudent to ignore H7, which is homoallylic in the esters 16 but not in the lactones 1 (the alkene is removed after RCM and reduction). No resonance of H4 or H5 of the lactone matches its ester counterpart. And while there are some differences in the lactone and ester coupling constants, it is the differences in chemical shifts that are more remarkable.
Conclusions
We have described a fluorous mixture synthesis of all sixteen stereoisomers of the macrocyclic lactone natural product Sch725674. The general approach to making four mixtures of four fluorous-tagged quasiisomers each is by now standard; however, the tagging pattern has been advanced significantly in two ways. First, only two tags are used, and only one of these is fluorous. Second, only 30 fluorine atoms are used in total to make all four quasiisomers. With the previous tag sets in comparable libraries, the smallest total number of fluorines was 46.4b The new approach is atom economical, and larger, environmentally persistent groups26 (C6F13, C8F17) are not used.
After demixing and detagging, the structure of Sch725674 was assigned as (4R,5S,7R,13R)-1 by comparison of the published spectra to the library spectra. This is the first time that a complete stereoisomer library has been made by FMS to assign a natural product of completely unknown configuration.
A substantial amount of data was produced by characterizing the 16 open esters and the 32 lactones. In each case, half of the compounds had three silyl tags and half had three free hydroxy groups. These data took significant time to collect, but provided substantial information beyond the structure of Sch725674. For example, compared to previous libraries that we have made, we were surprised by the large differences between all of the spectra of the final lactones 1. In the 28 possible comparisons of pairs of spectra, there is little overlapping at all of the 1H spectra except in the regions of the consecutive methylene groups. In contrast to the lactones, the spectra of the esters came in pairs with two isomers differing at C13 exhibiting substantially identical spectra.
It was also surprising that the differences in chemical shifts in the 1H NMR spectra of the various lactone isomers are more remarkable than the differences in coupling constants. The 13C NMR spectra presented in the SI give only chemical shift information, but again there are many differences in the lactones, and these spectra likewise suffice to assign the structure of Sch725674.
The data provide encouragement and caution for groups using various models of NMR spectra to assign configurations of macrolactones. The caution is for use of NMR spectra of model compounds to assign stereochemistry.24 The technique has great power that is increasing with the size of the databases. However, at least for 14-membered lactones, a database of acyclic stereoisomers will not be a good model set for predicting chemical shifts. For example, the database chemical shifts of esters 14 or 16 is of no use in assigning configuration of lactones 15 or 1.
The encouragement is for groups that are calculating NMR spectra of isomers, and especially the ability to calculate 13C NMR spectra has advanced in recent years.25 Here the set of 13C NMR spectra of lactones 1 are all significantly different. This provides a present challenge to NMR modelers. Pose yourself the following question: could you have confidently assigned the structure of Sch725674 if it were any one of the eight possible diastereomers? In other words, calculate the eight possible 13C NMR spectra of the diastereomers of 1 and match them to the actual spectra. If there is a unique match in each case, then you could have assigned the structure of Sch725674 only by calculation, no matter which isomer it ultimately proved to be. A structure problem like this would usually involve comparison of eight calculated spectra with one actual spectrum (the natural product). Here is the rare opportunity to compare the eight calculated spectra with all eight actual spectra.27
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health, National Institute of General Medical Sciences, for funding of this work. We thank Dr. Damodaran Krishnan and Ms. Sage Bowser for help with the NMR spectra.
ABBREVIATIONS
- FMS
fluorous mixture synthesis
- “M”
before a compound number indicates a mixture of four quasiisomers
- suffixes a-d or e-h
indicate the tagging pattern (“a” is all three TH, etc.)
- TIPS or TH is Si(iPr)3
FTIPS or TF is Si(iPr)2CH2CH2C2F5.
Footnotes
ASSOCIATED CONTENT
Supporting Information: Contains complete experimental details, tabular NMR data, supplemental Figures and copies of NMR spectra of the stereoisomer library members. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
HPLC columns were purchased from Fluorous Technologies, Inc. DPC owns an equity interest in this company. The other authors declare no competing financial interest.
REFERENCES
- 1.Luo ZY, Zhang QS, Oderaotoshi Y, Curran DP. Science. 2001;291:1766–1769. doi: 10.1126/science.1057567. [DOI] [PubMed] [Google Scholar]
- 2.Zhang QS, Curran DP. Chem. Eur. J. 2005;11:4866–4880. doi: 10.1002/chem.200500076. [DOI] [PubMed] [Google Scholar]
- 3.(a) Zhang W. Arkivoc. 2004:101–109. [PMC free article] [PubMed] [Google Scholar]; (b) Curran DP. In: The Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horváth IT, editors. Wiley-VCH; Weinheim: 2004. pp. 128–156. [Google Scholar]
- 4.(a) Wilcox CS, Gudipati V, Lu HJ, Turkyilmaz S, Curran DP. Angew. Chem. Int. Ed. 2005;44:6938–6940. doi: 10.1002/anie.200501989. [DOI] [PubMed] [Google Scholar]; (b) Curran DP, Moura-Letts G, Pohlman M. Angew. Chem. Int. Ed. 2006;45:2423–2426. doi: 10.1002/anie.200600041. [DOI] [PubMed] [Google Scholar]
- 5.(a) Zhang QS, Lu HJ, Richard C, Curran DP. J. Am. Chem. Soc. 2004;126:36–37. doi: 10.1021/ja038542e. [DOI] [PubMed] [Google Scholar]; (b) Yang F, Newsome JJ, Curran DP. J. Am. Chem. Soc. 2006;128:14200–14205. doi: 10.1021/ja064812s. [DOI] [PubMed] [Google Scholar]; (c) Jung W-H, Guyenne S, Riesco-Fagundo C, Mancuso J, Nakamura S, Curran DP. Angew. Chem. Int. Ed. 2008;47:1130–1133. doi: 10.1002/anie.200704893. [DOI] [PubMed] [Google Scholar]; (d) Curran DP, Sui B. J. Am. Chem. Soc. 2009;131:5411–5413. doi: 10.1021/ja900849f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Curran DP, Zhang QS, Lu HJ, Gudipati V. J. Am. Chem. Soc. 2006;128:9943–9956. doi: 10.1021/ja062469l. [DOI] [PubMed] [Google Scholar]; (b) Sui B, Yeh EA-H, Curran DP. J. Org. Chem. 2010;75:2942–2954. doi: 10.1021/jo100115h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wessjohann LA, Ruijter E, Garcia-Rivera D, Brandt W. Mol. Div. 2005;9:171–186. doi: 10.1007/s11030-005-1314-x. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wessjohann LA, Ruijter E. Top. Curr. Chem. 2005;243:137–184. [Google Scholar]; (b) Thomas GL, Wyatt EE, Spring DR. Curr. Opin. Drug Discov. Dev. 2006;9:700–712. [PubMed] [Google Scholar]; (c) Marsault E, Peterson ML. J. Med. Chem. 2011;54:1961–2004. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- 9.Yang S-W, Chan T-Z, Terracciano J, Loebenberg D, Patel M, Chu M. J. Antibiot. 2005;58:535–538. doi: 10.1038/ja.2005.74. [DOI] [PubMed] [Google Scholar]
- 10.(a) Adam G, Zibuck R, Seebach D. J. Am. Chem. Soc. 1987;109:6176–6177. [Google Scholar]; (b) Mortimore M, Cockerill GS, Kocienski P, Treadgold R. Tetrahedron Lett. 1987;28:3747–3750. [Google Scholar]; (c) Takano S, Shimazaki Y, Takahashi M, Ogasawara K. J. Chem. Soc., Chem. Commun. 1988:1004–1005. [Google Scholar]; (d) Seebach D, Adam G, Zibuck R, Simon W, Rouilly M, Meyer WL, Hinton JF, Privett TA, Templeton GE, et al. Liebigs Ann. Chem. 1989:1233–1240. [Google Scholar]; (e) Seebach D, Adam G, von dem Bussche-Hünnefeld C, Gisi U, Binder H. Liebigs Ann. Chem. 1990:1007–1012. [Google Scholar]; (f) Curtis NR, Holmes AB, Looney MG, Pearson ND, Slim GC. Tetrahedron Lett. 1991;32:537–540. [Google Scholar]; (g) Carling RW, Clark JS, Holmes AB, Sartor D. J. Chem. Soc., Perkin Trans. 1. 1992:95–101. [Google Scholar]; (h) Matsushita M, Yoshida M, Zhang Y, Miyashita M, Irie H, Ueno T, Tsurushima T. Chem. Pharm. Bull. 1992;40:524–527. [Google Scholar]; (i) Ley SV, Cleator E, Harter J, Hollowood C. J. Org. Biomol. Chem. 2003;1:3263–3264. doi: 10.1039/b308793j. [DOI] [PubMed] [Google Scholar]; (j) Cleator E, Harter J, Ley SV. Heterocycles. 2004;62:619–633. [Google Scholar]; (k) Sharma A, Gamre S, Chattopadhyay S. Lett. Org. Chem. 2005;2:547–549. [Google Scholar]; (l) Trenkle JD, Jamison TF. Angew. Chem., Int. Ed. 2009;48:5366–5368. doi: 10.1002/anie.200902079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Meyer WL, Schweizer WB, Beck AK, Scheifele W, Seebach D, Schreiber SL, Kelly SE. Helv. Chim. Acta. 1987;70:281–291. [Google Scholar]; (b) Carling RW, Holmes AB. Tetrahedron Lett. 1986;27:6133–6136. [Google Scholar]
- 12.Celmer WD. Pure Appl. Chem. 1971;28:413–453. doi: 10.1351/pac197128040413. [DOI] [PubMed] [Google Scholar]
- 13.Wang X. PhD Thesis of Dr. University of Pittsburgh; 2009. Chapter 2. [Google Scholar]
- 14.Moretti J. PhD Thesis of Dr. University of Pittsburgh; 2010. [Google Scholar]
- 15.Numbers with the prefix “M” are mixtures of four differentially tagged quasiisomers.
- 16.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483–2547. [Google Scholar]
- 17.Hoye TR, Jeffrey CS, Shao F. Nature Protocols. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 18.Ramachandran PV. Aldrichim. Acta. 2002;35:23–35. [Google Scholar]
- 19.We use the FTIPS abbreviation for consistency, but more appropriately this tag is an analog of an ethyldiisopropylsilyl group rather than a triisopropylsilyl group.
- 20.Denmark SE, Regens CS, Kobayashi T. J. Am. Chem. Soc. 2007;129:2774–2776. doi: 10.1021/ja070071z. [DOI] [PubMed] [Google Scholar]
- 21.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
- 22.Vougioukalakis GC, Grubbs RH. Chem. Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]
- 23.The only exception was the methylene groups at positions 10-12 in the lactones, which sometimes showed substantial overlapping in the 1H NMR spectra. Because of this, corresponding 13C peak assignments could not be made in the 1H-13C COSY experiments.
- 24.Higashibayashi S, Kishi Y. Tetrahedron. 2004;60:11977–11982. [Google Scholar]
- 25.(a) Smith SG, Goodman JM. J. Am. Chem. Soc. 2010;132:12946–12959. doi: 10.1021/ja105035r. [DOI] [PubMed] [Google Scholar]; (b) Saielli G, Bagno A. Org. Lett. 2009;11:1409–1412. doi: 10.1021/ol900164a. [DOI] [PubMed] [Google Scholar]
- 26.McCulloch A. J. Fluorine Chem. 2003;123:21–29. [Google Scholar]
- 27.The final ester and lactone samples have been deposited in the University of Pittsburgh Center for Methodology and Library Develop (CMLD) and are available for biological testing or in the event that additional spectra are needed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.