

Proteins, the working machines of the cell, operate as enzymes to catalyze chemical synthesis, as ion pumps to generate electrical voltage, and as motors to generate mechanical force. Myosin, a linear motor, jumps along an actin filament to contract striated muscle, the two-armed kinesin walks hand-over-hand on microtubuli to pull a synaptic vesicle along the nerve axon, and RNA polymerase, helicase, and the ribosome process over nucleotide strands. The flagellar drive, a rotary motor, propels bacteria though the viscous fluid. Whereas the former are powered by the hydrolysis of ATP (or other nucleotide triphosphates), the flagellar motor is powered by an electrochemical potential difference across the cytoplasmic membrane.
ATP synthase, the enzyme that produces ATP, links both types of driving forces. It produces ATP at the expense of an electrochemical potential difference or, when operating in the reverse, it generates an electrochemical difference at the expense of ATP hydrolysis, then named F-ATPase. For three decades, it had remained enigmatic how its two functions, the electrochemical and the chemical, are linked to each other. They seemed to be rather well separated in the bipartite construction of the enzyme, with an ion-conducting membrane portion, FO, and a peripheral F1-portion that, by itself, hydrolyzes ATP. Only recently, it has been fully appreciated that ion transport and the synthesis of ATP by the holoenzyme are mechanically coupled. The holoenzyme is made from two rotary motors that are mounted on a central shaft and held together by an eccentric bearing (see Fig. 1A). Depending on the demand for ATP or for ion-motive force, one motor operates in the forward direction and, by rotating the central shaft, drives the other motor backwards to operate as a generator. Of the same size as myosin and by order of magnitude smaller than the flagellar drive, ATP synthase presents a delicate blend of electrical-to-mechanical-to-chemical energy conversion (1, 2).
Figure 1.

(A) Schematic model of the H+-translocating ATP synthase adapted from ref. 2. It is based on the partial crystal structure of F1 (12), on solution structures by NMR of three subunits, namely ɛ (45), δ (46), and c (30), and on crosslinking data on the mutual arrangement of these subunits (47–52) (reviewed in ref. 53). Two of the six large subunits in the hexagon formed by subunits (αβ)3 are removed to reveal subunit γ, the central shaft. (B) Schematic model for the coupling of proton flow to torque generation (2).
The paper by Peter Dimroth et al. published in this issue of the Proceedings (3) addresses the mechanism of the electrochemical drive in the ATP synthase of the bacterium Propionigenium modestum. Its FO-portion primarily conducts Na+-cations (4), in contrast with its H+-translocating sisters in photosynthetic and/or respiratory bacteria and in the chloroplasts and mitochondria of eukarya. There is no doubt, however, that the ATP synthase of P. modestum operates according to the same general principles as its H+-translocating sister enzymes, because of the same subunit structure (see Fig. 1A) and the functionality of various chimeric constructs between P. modestum and the F1 of Escherichia coli (5, 6) and chloroplasts or cyanobacteria and E. coli (7–9). The functioning of chimeric constructs from different kingdoms of life was not quite expected for an enzyme whose genome underwent about 2 billion years of separate vertical evolution (see ref. 10).
Let us consider F1, the chemical motor of ATP synthase, first. When it is isolated and functions as ATPase, the three catalytic nucleotide binding sites on F1 rotate in their momentary role. While one site binds, the next one hydrolyzes ATP, and the third one releases the hydrolysis products. This concept by Paul Boyer (1, 11) became “visible truth” when John Walker and his collaborators obtained a crystal structure of F1 (12), which showed three catalytic binding sites on the hexagon formed by subunits (αβ)3 of F1 with subunit γ as a curved shaft in the center. These three sites were filled with ATP and ADP or were empty, depending on their respective positions relative to the concave, neutral, and convex sides of the shaft. It has been argued that torque applied to the central shaft rotates its convex surface toward the site filled with ATP and, by pushing the lower lever of the respective subunit β outwards, it opens the ATP-binding site to expel the newly synthesized but firmly bound ATP into the bulk that is the major energy-requiring step of ATP synthesis (12, 13).
The rotary mechanism is experimentally proven for the isolated F1-portion when functioning as ATPase. First evidence for a forced rotation of subunit γ relative to β was obtained by the group of R. Cross, who used cleavable crosslinks between subunit γ and β (14, 15). By polarized photochemistry with a single dye molecule as a probe on subunit γ, the group of the author resolved the rotation in time and found it compatible with the enzymatic turnover (16, 17). With a long (typically 2 μm) fluorescent actin filament as a tag on γ, the groups of M. Yoshida and K. Kinosita proved the unidirectional and ATP-driven rotation of subunit γ. The rotation of γ became “visible truth” by their stunning micromovies (18, 19). The rotation is three-stepped (17, 20), with very sudden transitions (21) between three symmetrically spaced (by 120°) angular resting positions of γ relative to (αβ)3 (18).
The FO-portion of ATP synthase probably functions as a stepper motor, too, but it is twelve-stepped rather than three-stepped for the following reasons: (i) In chloroplasts and cyanobacteria, the stoichiometry of translocated protons over synthesized ATP is 4:1 (22, 23), which amounts to 12 protons per full turn in F1; (ii) in E. coli, FO contains 12 identical copies of the proteolipid subunit, named c (24), arranged as a ring with a diameter of about 5 nm (25–27).
How such a ring, together with its partner, the large and transmembrane subunit a, might generate torque at the expense of proton flow in the H+-translocating FO of photosynthetic and respiratory organisms has been laid out by Junge et al. since 1993 (see ref. 2 and Fig. 1B). This model was quantitatively analyzed by the group of G. Oster (28). In their new article with Dimroth et al. in this issue of the Proceedings (3), the same group presents a different model for the Na+-driven rotation in FO of P. modestum.
The former model for the H+-driven FO is illustrated in Fig. 1B and, schematically, in Fig. 2A. It has been based on the then generally assumed hairpin structure of subunit c, which positioned the conserved acid residue in E. coli Asp-61 straight in the middle of the membrane (29). The expectation has been that this residue is always protonated when facing the hydrophobic lipid core of the membrane, to avoid large electrostatic penalty. The exchange of protons with either bulk phase is admitted only for those copies of c in contact with subunit a (2). This particular folding of subunit c has been supported by NMR studies of Bob Fillingame’s group on the structure of the solubilized proteolipid of E. coli in a mixed solvent (chloroform—methanol–water, 4:4:1, vol/vol/v) (30, 31) and was recently corroborated for the membrane-bound H+-ATP synthase of E. coli by using engineered disulfite bridges for zero-length crosslinks (32).
Figure 2.

A comparison of proposed models for the ionic drive of (A) the H+-ATP synthase of photosynthetic and respiring organisms (2, 28), (B) the Na+-ATP synthase of P. modestum (3), and (C) the flagellar motor (34). The “rotor” provides relay sites for ion transport between two aqueous phases and across the coupling membrane. It rotates against a “stator” (for details, see text). The diameter of the wheel in the flagellar motor (about 50 nm) is by order of magnitude larger than in ATP synthase (about 5 nm), its rotation is driven by eight motor elements instead of one, and the number of transported protons per revolution is 1,200 instead of 12. In some domains, shaded in blue, the ion-binding sites are obligatorily occupied; in others, shaded in red, they must be empty.
The NMR-structural analysis of the P. modestum proteolipid in SDS-micelles has revealed a less compact hairpin (P. Dimroth, personal communication), though it has not yet been crosschecked for the membrane. It was interpreted to position the essential acid residue Glu-65 in P. modestum, close to the cytoplasmic surface of the membrane and not in its middle. Thereby the electrostatic penalty is diminished, and the exchange of Na+ with the cytoplasmatic bulk allowed even for those copies of subunit c that are not in immediate contact with a. For the P. modestum enzyme, there is evidence for such an exchange (33).
At this point, it might be worthwhile to consider the features that are common to the proposed models for H+-FO (2), Na+-FO (3), and the flagellar drive (34) (Fig. 2 A–C): (i) A rotor ring (disk) with N cation binding sites contacts a stator, both embedded in a membrane; (ii) any rotational motion is based on Brownian fluctuations of the rotor (motor) against the stator; (iii) the fluctuations are (electrostatically) constrained to a narrow angular domain because neither can an empty (negatively charged) site pass certain domains on the rotor (blue in Fig. 2) nor an occupied (electroneutral) site others (marked in red); (iv) ion access from the two bulk phases to the binding sites is restricted to two access channels that are not colinear (Fig. 2 A and C) and one access channel plus one access domain, respectively (Fig. 2B). Thereby, the electrochemical potential difference between the two phases biases Brownian fluctuations; (v) the biased rotation generates torque and loads an elastic spring.
All three models describe the strict coupling between ion flow and rotation, which has been demonstrated for the flagellar motor (35) and the H+-ATP synthase of chloroplasts (22, 23). In the light of the basic similarity of the two alternative models for FO, it is not surprising that Dimroth et al. obtained the same dependence of the rotation rate on the load torque for both models (ref. 3, Fig. 8b ). Without a structure at atomic resolution, it may be difficult if not impossible to discriminate between different concepts for the functioning of FO based on a simulation of kinetic data alone.
The ATP synthase of P. modestum seems to differ from its sister in chloroplasts in another important property. The chloroplast enzyme can be driven either by a transmembrane voltage (Δϕ) (36–38) or, as proven under steady illumination, by the transmembrane pH-difference, ΔpH, alone. If independently varied, these two components of the electrochemical potential difference, Δμ:
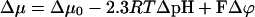 |
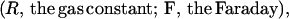 |
seemed to be equivalent even when the enzyme was operated far from equilibrium (39). The same has been found for the flagellar motor. In both cases, the equivalence has been understood in terms of Mitchell’s proton well (40). In a monospecific access channel for, say, the proton, which rapidly equilibrates with the adjacent bulk (Δμ = constant), the partial voltage drop over the channel length is converted into a pH difference of equal magnitude. For the ATP synthase of P. modestum, this equivalence been ΔpNa and Δϕ has not been observed and the voltage seems a more efficient driving force than the concentration difference of the sodium cation (41). To account for this observation, Dimroth et al. have included a tangentially oriented electrophoretic step over the “hydrophilic strip” as a new feature of their motor model (ref. 3, Fig. 2b).
In the flagellar motor, the stator is probably elastically anchored to the cell wall (34). This is why the rotor turns the flagellum relative to the large cell body (42). In ATP synthase, there is no elastic link to the outside world; instead, the enzyme carries out rapid and uniaxial Brownian rotation in the fluid membrane [rotational relaxation time, about 100 μs (17)]. Thus the stator/rotor denomination is arbitrary. To account for the load torque and for the transmission between its two rotary drives, this enzyme, too, has to contain elastic elements, which are, however, internalized.
In their article in this issue of the Proceedings, Dimroth et al. (3) neglected one very important aspect of the elastic coupling, namely the transmission between the twelve-stepped rotation in FO and the three-stepped one in F1. Strictly speaking, their description holds only for the isolated FO-portion. In the holoenzyme, FOF1, the sequential translocation of four ions causes an accumulation of elastic energy until its eventual discharge for the release of tightly bound ATP. Candidates for an elastic deformation are the intertwined helices of subunit γ as a torsional spring, the parallel helices of subunits b topped by δ and bottomed by subunit a as a parallelogram spring, and the cantilever springs of subunits β (see Fig. 1A and refs. 2 and 43). The 12-to-3 transmission implies that the first proton in a series operates against a lower torque than the fourth. We modeled such a behavior and obtained a consistent fit of the pronounced pH dependence of the rates of ATP synthesis/hydrolysis by FOF1 and of proton flow through the exposed FO (44). The pH dependence differed significantly with accumulation of torque, as in FOF1, or without, as in the bare FO.
Table 1 summarizes the properties of various motor proteins, linear and rotary. Pairwise comparison reveals the following design features: (i) Myosin vs. kinesin: the former produces high speed at the expense of processivity, and the latter runs at high processivity at the expense of speed. The slower steps in the full reaction cycle of myosin do not limit its rate of action, because they happen while the head is detached from the actin filament. (ii) RNA–polymerase and flagellar motor vs. the rest: the former can operate against large loads, because they take smaller steps than the latter. In the flagellar motor, this property is even more pronounced by the joint action of eight motor elements on the wheel. (iii) FOF1 vs. the rest: although the dual motor function is normally fully sequestered in the ATP synthase, its performance matches that of the other motors.
Table 1.
Comparison of molecular motors
| Motor | Drive | Molecular mass, 105 Da | Processivity, % | Max.
speed
|
Step size
|
Stall
load
|
|||
|---|---|---|---|---|---|---|---|---|---|
| nm/s | rev/s | nm | deg | pN | pN·nm | ||||
| Kinesin | ATP | 1 | 100 | 800 | 8 | 6 | |||
| Myosin | ATP | 5 | 1 | 8,000 | 15 | 4 | |||
| RNAP | NTP | 7 | 100 | 0.35 | 30 | ||||
| F1 | ATP | 4 | 100 | 100 | 120 | 40 | |||
| FO | PMF | 1.5 | 100 | 1,500 | 100 | (1.00) | 30 | (16) | (40) |
| Flag | PMF | ≈100 | 100 | 45,000 | 300 | (<0.4) | <1 | (300) | 4,800 |
Myosin, kinesin, RNA polymerase (RNAP), the two drives of ATP synthase (FO, F1), and the flagellar motor (Flag) are driven by nucleotide triphosphates (ATP, NTP) or by an electrochemical potential difference [here named protonmotive force, (PMF)]. The processivity is almost perfect in all motors except myosin. The data for the linear motors are taken from refs. 54–57, those for the flagellar motor from refs. 35, 42, the stall load of F1 from ref. 18, the maximum speed of F1 from ref. 39, and the data for FO were assumed to match those of F1 when operating in the holoenzyme.
The clearcut assignment of any partial function of the two motors in ATP synthase to particular subunits is of great advantage for further studies on this compact dual nanomachine.
Footnotes
A commentary on this article begins on page 4924.
References
- 1.Boyer P D. Annu Rev Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 2.Junge W, Lill H, Engelbrecht S. Trends Biochem Sci. 1997;22:123–456. doi: 10.1016/s0968-0004(97)01129-8. [DOI] [PubMed] [Google Scholar]
- 3.Dimroth P, Wang H, Grabe M, Oster G. Proc Natl Acad Sci USA. 1999;96:4924–4929. doi: 10.1073/pnas.96.9.4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laubinger W, Dimroth P. Biochemistry. 1988;27:7531–7537. doi: 10.1021/bi00419a053. [DOI] [PubMed] [Google Scholar]
- 5.Kaim G, Dimroth P. Eur J Biochem. 1995;218:937–944. doi: 10.1111/j.1432-1033.1993.tb18450.x. [DOI] [PubMed] [Google Scholar]
- 6.Laubinger W, Deckers-Hebestreit G, Altendorf K, Dimroth P. Biochemistry. 1990;29:5458–5463. doi: 10.1021/bi00475a008. [DOI] [PubMed] [Google Scholar]
- 7.Engelbrecht S, Deckers-Hebestreit G, Altendorf K, Junge W. Eur J Biochem. 1989;181:485–491. doi: 10.1111/j.1432-1033.1989.tb14750.x. [DOI] [PubMed] [Google Scholar]
- 8.Lill H, Burkovski A, Altendorf K, Junge W, Engelbrecht S. Biochim Biophys Acta. 1993;1144:278–284. doi: 10.1016/0005-2728(93)90112-s. [DOI] [PubMed] [Google Scholar]
- 9.Burkovski A, Lill H, Engelbrecht S. Biochim Biophys Acta. 1994;1186:243–246. doi: 10.1016/0005-2728(94)90184-8. [DOI] [PubMed] [Google Scholar]
- 10.Blair A, Ngo L, Park J, Paulsen I T, Saier M H. Microbiology. 1996;142:17–32. doi: 10.1099/13500872-142-1-17. [DOI] [PubMed] [Google Scholar]
- 11.Boyer P D, Kohlbrenner W E. In: Energy Coupling in Photosynthesis. Selman B R, Selman-Reimer S, editors. New York: Elsevier; 1981. pp. 231–240. [Google Scholar]
- 12.Abrahams J P, Leslie A G W, Lutter R, Walker J E. Nature (London) 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 13.Penefsky H S. J Biol Chem. 1985;260:13735–13741. [PubMed] [Google Scholar]
- 14.Zhou Y T, Duncan T M, Bulygin V V, Hutcheon M L, Cross R L. Biochim Biophys Acta. 1996;1275:96–100. doi: 10.1016/0005-2728(96)00056-4. [DOI] [PubMed] [Google Scholar]
- 15.Duncan T M, Bulygin V V, Zhou Y, Hutcheon M L, Cross R L. Proc Natl Acad Sci USA. 1995;92:10964–10968. doi: 10.1073/pnas.92.24.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabbert D, Engelbrecht S, Junge W. Nature (London) 1996;381:623–626. doi: 10.1038/381623a0. [DOI] [PubMed] [Google Scholar]
- 17.Sabbert D, Engelbrecht S, Junge W. Proc Natl Acad Sci USA. 1997;94:4401–4405. doi: 10.1073/pnas.94.9.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasuda R, Noji H, Kinosita K, Yoshida M. Cell. 1998;93:1117–1124. doi: 10.1016/s0092-8674(00)81456-7. [DOI] [PubMed] [Google Scholar]
- 19.Noji H, Yasuda R, Yoshida M, Kinosita K. Nature (London) 1997;386:299–302. doi: 10.1038/386299a0. [DOI] [PubMed] [Google Scholar]
- 20.Sabbert D, Junge W. Proc Natl Acad Sci USA. 1997;94:2312–2317. doi: 10.1073/pnas.94.6.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Häsler K, Engelbrecht S, Junge W. FEBS Lett. 1998;426:301–304. doi: 10.1016/s0014-5793(98)00358-5. [DOI] [PubMed] [Google Scholar]
- 22.Berry S, Rumberg B. Biochim Biophys Acta. 1996;1276:51–56. doi: 10.1016/s0005-2728(99)00003-1. [DOI] [PubMed] [Google Scholar]
- 23.van Walraven H S, Strotmann H, Schwarz O, Rumberg B. FEBS Lett. 1996;379:309–313. doi: 10.1016/0014-5793(95)01536-1. [DOI] [PubMed] [Google Scholar]
- 24.Jones P C, Fillingame R H. J Biol Chem. 1998;273:29701–29705. doi: 10.1074/jbc.273.45.29701. [DOI] [PubMed] [Google Scholar]
- 25.Jones P C, Jiang W P, Fillingame R H. J Biol Chem. 1998;273:17178–17185. doi: 10.1074/jbc.273.27.17178. [DOI] [PubMed] [Google Scholar]
- 26.Birkenhäger R, Deckers-Hebestreit G, Altendorf K. Eur J Biochem. 1995;230:58–67. [PubMed] [Google Scholar]
- 27.Singh S, Turina P, Bustamante C J, Keller D J, Capaldi R A. FEBS Lett. 1996;397:30–34. doi: 10.1016/s0014-5793(96)01127-1. [DOI] [PubMed] [Google Scholar]
- 28.Elston T, Wang H, Oster G. Nature (London) 1998;391:510–514. doi: 10.1038/35185. [DOI] [PubMed] [Google Scholar]
- 29.Schneider E, Altendorf K. Trends Biochem Sci. 1984;9(2):51–53. [Google Scholar]
- 30.Girvin M E, Rastogi V K, Abildgaard F, Markley J L, Fillingame R H. Biochemistry. 1998;37:8817–8824. doi: 10.1021/bi980511m. [DOI] [PubMed] [Google Scholar]
- 31.Girvin M E, Fillingame R H. Biochemistry. 1993;32:12167–12177. doi: 10.1021/bi00096a029. [DOI] [PubMed] [Google Scholar]
- 32.Fillingame R H, Jones P C, Jiang W, Valiyaveetil F I, Dmitriev O Y. Biochim Biophys Acta - Bioenergetics. 1998;1365:135–142. doi: 10.1016/s0005-2728(98)00053-x. [DOI] [PubMed] [Google Scholar]
- 33.Dimroth P, Kaim G, Matthey U. Biochim Biophys Acta. 1998;1365:87–92. doi: 10.1016/s0005-2728(98)00047-4. [DOI] [PubMed] [Google Scholar]
- 34.Meister M, Caplan S R, Berg H C. Biophys J. 1989;55:905–914. doi: 10.1016/S0006-3495(89)82889-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berry S, Berg H C. Biophys J. 1999;76:580–587. doi: 10.1016/S0006-3495(99)77226-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Junge W. Eur J Biochem. 1970;14:582–592. doi: 10.1111/j.1432-1033.1970.tb00327.x. [DOI] [PubMed] [Google Scholar]
- 37.Junge W, Rumberg B, Schroeder H. Eur J Biochem. 1970;14:575–581. doi: 10.1111/j.1432-1033.1970.tb00326.x. [DOI] [PubMed] [Google Scholar]
- 38.Schlodder E, Rögner M, Witt H T. FEBS Lett. 1982;138:13–18. [Google Scholar]
- 39.Junesch U, Gräber P. FEBS Lett. 1991;294:275–278. doi: 10.1016/0014-5793(91)81447-g. [DOI] [PubMed] [Google Scholar]
- 40.Mitchell P. Symp Soc Gen Microbiol. 1977;27:383–423. [Google Scholar]
- 41.Kaim G, Dimroth P. FEBS Lett. 1998;434:57–60. doi: 10.1016/s0014-5793(98)00969-7. [DOI] [PubMed] [Google Scholar]
- 42.Schuster S C, Khan S. Annu Rev Biophys Biomol Struct. 1994;23:509–539. doi: 10.1146/annurev.bb.23.060194.002453. [DOI] [PubMed] [Google Scholar]
- 43.Wang H Y, Oster G. Nature (London) 1998;396:279–282. doi: 10.1038/24409. [DOI] [PubMed] [Google Scholar]
- 44.Cherepanov D, Mulkidjanian A, Junge W. FEBS Lett. 1999;449:1–6. doi: 10.1016/s0014-5793(99)00386-5. [DOI] [PubMed] [Google Scholar]
- 45.Wilkens S, Dahlquist F W, Mcintosh L P, Donaldson L W, Capaldi R A. Nat Struct Biol. 1995;2:961–967. doi: 10.1038/nsb1195-961. [DOI] [PubMed] [Google Scholar]
- 46.Wilkens S, Dunn S D, Chandler J, Dahlquist F W, Capaldi R A. Nat Struct Biol. 1997;4:198–201. doi: 10.1038/nsb0397-198. [DOI] [PubMed] [Google Scholar]
- 47.Aggeler R, Ogilvie I, Capaldi R A. J Biol Chem. 1997;272:16621–16656. doi: 10.1074/jbc.272.31.19621. [DOI] [PubMed] [Google Scholar]
- 48.Ogilvie I, Aggeler R, Capaldi R A. J Biol Chem. 1997;272:16652–16656. doi: 10.1074/jbc.272.26.16652. [DOI] [PubMed] [Google Scholar]
- 49.Watts S D, Capaldi R A. J Biol Chem. 1997;272:15065–15068. doi: 10.1074/jbc.272.24.15065. [DOI] [PubMed] [Google Scholar]
- 50.Tang C L, Capaldi R A. J Biol Chem. 1996;271:3018–3024. doi: 10.1074/jbc.271.6.3018. [DOI] [PubMed] [Google Scholar]
- 51.Schulenberg B, Wellmer F, Junge W, Engelbrecht S. Eur J Biochem. 1997;249:134–141. doi: 10.1111/j.1432-1033.1997.t01-1-00134.x. [DOI] [PubMed] [Google Scholar]
- 52.Lill H, Hensel F, Junge W, Engelbrecht S. J Biol Chem. 1996;271:32737–32742. doi: 10.1074/jbc.271.51.32737. [DOI] [PubMed] [Google Scholar]
- 53.Engelbrecht S, Junge W. FEBS Lett. 1997;414:123–456. doi: 10.1016/s0014-5793(97)00997-6. [DOI] [PubMed] [Google Scholar]
- 54.Goldman Y. Cell. 1998;93:1–4. doi: 10.1016/s0092-8674(00)81137-x. [DOI] [PubMed] [Google Scholar]
- 55.Gelles J, Landick R. Cell. 1998;93:13–16. doi: 10.1016/s0092-8674(00)81140-x. [DOI] [PubMed] [Google Scholar]
- 56.Block S M. Cell. 1998;93:5–8. doi: 10.1016/s0092-8674(00)81138-1. [DOI] [PubMed] [Google Scholar]
- 57.Yanagida T, Harada Y, Ishijima A. Trends Biochem Sci. 1993;18:319–324. doi: 10.1016/0968-0004(93)90064-t. [DOI] [PubMed] [Google Scholar]