

Abstract
α-1 Antitrypsin (A1AT) is a serpin with a major protective effect against cigarette smoke–induced emphysema development, and patients with mutations of the A1AT gene display a markedly increased risk for developing emphysema. We reported that A1AT protects lung endothelial cells from apoptosis and inhibits caspase-3 activity. It is not clear if cigarette smoking or A1AT mutations alter the caspase-3 inhibitory activity of A1AT and if this serpin alters the function of other caspases. We tested the hypothesis that the caspase-3 inhibitory activity of A1AT is impaired by cigarette smoking and that the A1AT RCL, the key antiprotease domain of the serpin, is required for its interaction with the caspase. We examined the caspase-3 inhibitory activity of human A1AT purified from plasma of actively smoking and nonsmoking individuals, either affected or unaffected with chronic obstructive pulmonary disease. We also tested the caspase inhibitory activity of two mutant forms of A1AT, the recombinant human piZZ and the RCL–deleted (RCL-null) A1AT forms. A1AT purified from the blood of active smokers exhibited marked attenuation in its caspase-3 inhibitory activity, independent of disease status. In vitro exposure of the normal (MM) form of A1AT to cigarette smoke extract reduced its ability to interact with caspase-3, measured by isothermal titration calorimetry, as did the deletion of the RCL, but not the ZZ point mutation. In cell-free assays A1AT was capable of inhibiting all executioner caspases, -3, -7 and especially -6, but not the initiator or inflammatory caspases. The inhibitory effect of A1AT against caspase-6 was tested in vivo, where overexpression of both human MM and ZZ-A1AT via adeno-associated virus transduction significantly protected against apoptosis and against airspace damage induced by intratracheal instillation of caspase-6 in mice. These data indicate a specific inhibitory effect of A1AT on executioner caspases, which is profoundly attenuated by active exposure to cigarette smoking and is dependent on the protein RCL, but is not affected by the PiZZ mutation.
INTRODUCTION
α-1 antitrypsin (A1AT) deficiency is the most common genetic cause of emphysema, which is the main cause of morbidity in individuals who have A1AT deficiency and who smoke cigarettes. Emphysema along with chronic bronchitis is a major component of the chronic obstructive pulmonary disease (COPD) syndrome, a disease primarily attributed to cigarette smoke (CS) exposure that has become the third leading cause of death in the United States (1). Along with lung matrix destruction, oxidative stress and inflammation, cell death of structural components of the parenchyma is one of the key mechanisms involved in emphysema pathogenesis (2).
A1AT is a serine protease inhibitor synthesized by liver hepatocytes and secreted into the circulation, where it neutralizes neutrophil elastase. This effect accounts for inhibition of elastin degradation and excessive lung matrix proteolysis. However, A1AT’s role in emphysema extends beyond its antielastase activity. In our previous work we demonstrated that A1AT is capable of direct binding to caspase-3, thus inhibiting caspase-3–mediated apoptosis (3). Using biochemically produced A1AT conformers, we also showed that alterations of A1AT structure, such as oxidation or polymerization, may impair its antiapoptotic function. Here we report our investigation of whether A1AT inhibits the activity of other caspases and whether clinically relevant mutations of A1AT impair its anticaspase activity. In addition, we assessed whether in vivo exposure to CS is sufficient to impair the circulating A1AT anticaspase activity.
A1AT is a 52-kDa serpin with a protease-binding domain, known as the reactive center loop (RCL), which contains a critical methionine residue at position 358. Protease binding to the RCL results in an A1AT conformational change that irreversibly traps the protease within the serpin. This complex is subsequently cleared from the circulation (4). A1AT deficiency, a condition that reflects low levels of circulating A1AT, is a consequence of autosomal codominant inheritance of the Z allele, in which a single point mutation causes the substitution of glutamine 342 to lysine (5). The homozygous ZZ-A1AT is prone to polymerize intracellularly, leading to aggregates trapped in hepatocytes, which reduces the amount of circulating A1AT and decreases the antielastase capacity of plasma and lungs (6,7). The detrimental effects of CS on the lung synergize with those of A1AT deficiency, leading to severe, earlier onset of emphysema. In addition, CS oxidizes the Met358 within the RCL, which diminishes the antielastase function of the protein (8). We have shown previously that preincubation of A1AT with CS extract or hydrogen peroxide diminishes the serpin’s ability to inhibit caspase-3 activity against a fluorescently tagged substrate (3), but the CS effect on the protein–protein interaction between A1AT and caspase-3 has not been tested. Also unknown is whether the endogenous A1AT in the blood of individuals who smoke is affected in its ability to inhibit caspase-3 by the soluble components of CS absorbed in the circulation.
Caspases are cysteine proteases typically involved in the signaling cascade of apoptosis, or programmed cell death, either in the initiation or in the execution phase. The initiator caspases -2, -8, -9 and -10 cleave the prodomain of effector or executioner caspases -3, -6 and -7, leading to their activation. Caspases -1, -4 and -5 do not participate in apoptosis, but are characterized as inflammatory enzymes that regulate the innate immunity and T-cell development (9). We, along with others, identified A1AT as one of the endogenous inhibitors of caspase-3 by using cell-free assays and endothelial cell apoptosis studies, and by overexpressing A1AT via adeno-associated virus (AAV) transduction in mice instilled intratracheally with active caspase-3 (3). AAV-based augmentation strategies for A1AT have the advantage of an extended A1AT expression following a single inoculation (10). On the basis of recent studies, we chose to overexpress A1AT via the AAV serotype 8, which showed the highest levels of lung expression with the least immunogenic effects (11).
We report here that the circulating A1AT immunopurified from plasma of active smokers exhibits decreased anti–caspase-3 activity and that A1AT inhibits only executioner caspases, having the most pronounced effect on caspase-6.
MATERIALS AND METHODS
Reagents
Purified pooled human A1AT was purchased from Sigma (St. Louis, MO, USA). Recombinant active caspase-3 was from EMD Chemicals (Gibbstown, NJ, USA). Recombinant active caspase-6 was from BioVision (Mountain View, CA, USA).
Collection of blood from human subjects was performed by using a protocol approved by the Indiana University institutional review board. We recruited a group of 30 human volunteers that included COPD patients and healthy controls, age 18 years and older, who were either active smokers or ex- or nonsmokers, and provided informed consent. Active smokers without COPD (no COPD active smoking group) included volunteers with no clinical diagnosis of COPD and no symptoms of COPD such as chronic cough, dyspnea or increased sputum production and were current smokers at the time of blood collection. Healthy nonsmoking subjects (no COPD never smoking group) were defined as individuals who reported no respiratory symptoms and were either never-smokers or had a remote smoking history of less than 5 pack-years and had quit at least 2 years prior to enrollment. Patients with COPD were included on the basis of an established diagnosis by the treating physician and had pulmonary function tests (PFTs). Ex-smokers with COPD (COPD ex-smoker group) were defined as individuals with a diagnosis of COPD who had more than 20 pack-years of smoking history and quit smoking for at least 1 year before enrollment, and COPD patients who were currently smoking formed the COPD active smoking group. Exclusion criteria were pregnancy; acute illness, including fever, pneumonia, or upper respiratory infection at the time or within 4 wks prior to blood collection; α-1 antitrypsin deficiency; other pulmonary diagnoses; chronic liver disease; and inability to give informed consent. The following data were collected at the time of enrollment: age, sex, smoking history, PFT, radiological diagnosis of lung disease (if available), comorbid conditions and medications.
A1AT Purification
A1AT protein from patient plasma was purified by use of a Seize Primary Mammalian Immunoprecipitation Kit (Pierce, Rockford, IL, USA) following the manufacturer’s instructions. The antibody-coupled resin was made by using polyclonal goat anti-A1AT (1 mg/mL; 3 mL; Bethyl, Montgomery, TX, USA). For A1AT purification, 1 mL of plasma was loaded onto the column prepared as above, then, following a series of washes, the protein was eluted by using a Gentle Elution Buffer (Pierce), dialyzed in phosphate-buffered saline (PBS) and concentrated. We first assessed purified A1AT by Coomassie Blue or silver staining methods and then quantified it using bicinchoninic acid assay (Pierce), followed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotting with specific-A1AT antibodies using Western blot. Eluted fractions were tested for retained A1AT activity in cell-free elastase assays as previously described (12).
Recombinant MM-A1AT, ZZ-A1AT and RCL-Null Production
The pRC/CMV-MM and pRC/CMV-ZZ plasmids, containing either full-length human A1AT-MM or A1AT-ZZ cDNA, were kindly provided by Dr. Mark Brantly (University of Florida). hA1AT was amplified by using the following primers (containing engineered restriction sites, XhoI and NdeI, respectively): a1at-MM left: 5’-GCGCA ATACA TATGC CGTCT TCTGT CTCGT G-3’ and a1at-MM right: 5’-GCCAC TCGAG TTACC AGCTC AACCC TTCTT T-3’. The digested fragment was then sub-cloned into the NdeI and XhoI site of pET-23b (+) vector (Novagen/EMD Biosciences, Madison, WI, USA) to obtain the expression plasmid pET-23b-MM. We created the RCL-null vector using pET-23b-MM as a template with the following primers (with an engineered EcoRI restriction site): RCL left: 5’-GGCGC GAATT CCTTC ATGGG AAAAG TGGTG AAT-3’ and RCL right: 5’-GCGCC GAATT CAGCT TCAGT CCCTT TCTCG TC-3’. When ligated together, these fragments result in the deletion of the RCL (RCL-null). The resulting product was then cloned as before into a pET-23b plasmid by using NdeI and XhoI to generate the pET-23b-RCL-null plasmid. A1AT-ZZ was ligated into pET-23b following digestion of pET-23b-MM (excising the MM cDNA) and pRC/CMV-ZZ (ZZ template) with XhoI and BstEII. All subcloning was confirmed by DNA sequencing. Resulting plasmids pET-23b-MM, pET-23b-ZZ and pET-23b-RCL-null were then transformed into Escherichia coli BL21 (DE3) and used for protein production. Transformed bacteria were grown in lysogeny broth medium containing 50 μg/mL ampicillin and were induced using isopropyl β-D-1-thiogalactopyranoside (1 mmol/L; 3 h). Protein purification from bacterial pellets was performed by use of a PrepEase® Ni-IDA column (USB Corporation, Santa Clara, CA, USA) according to the manufacturer’s recommendation. Eluates were collected in 1-mL fractions and resolved on SDS-PAGE and stained with Coomassie Blue. Eluates showing a single band were concentrated further and confirmed as A1AT proteins by Western blotting with specific A1AT antibodies.
A1AT Caspase and Elastase Inhibitory Assays
Immunopurified concentrated human A1AT isolated from blood; recombinant human MM, ZZ, or RCL-null A1AT; or their respective vehicles isolated from bacterial cultures were incubated with recombinant active casaspe-3 followed by caspase enzymatic activity measurement using a fluorimetry caspase-3/7 activity kit (Promega, Madison, WI, USA) (13). The same method was used to measure the endogenous caspase-3 activity in tissues, as previously described (13). The inhibitory effect of A1AT on all caspases was screened by using fluorometric assay kits (BioVision). Serial dilutions of purified pooled human A1AT (Sigma) or vehicle were prepared and aliquoted to wells in a microtiter plate, followed by adding 0.9 U of respective active recombinant caspase (-1, -2, -3, -4, -5, -6, -7, -8, -9, -10) and caspase buffer. A fluorescent tetrapeptide substrate (25 μmol/L) specific for each respective caspase was added, then after 1 h incubation at 37°C the cleavage product was detected as fluorescence units. The slope of the fluorescence signal over time and the ratios of fluorescent signal in the presence and absence of A1AT in the reaction were calculated. As a blank, PBS was used instead of caspase. The elastase inhibitory activity of A1AT was assessed in parallel with that of caspase activity, using aliquots from the same batch of A1AT, to ensure that the dilutions of A1AT tested in the caspase inhibitory assays had preserved serpin activity. The elastase activity assays were performed by incubation of A1AT with purified active porcine pancreatic elastase (0.1 U/mL) followed by the addition of fluorescently labeled substrate (DQ-elastin, 25 μg/mL) and with the use of the Enzchek Elastase Assay Kit (Invitrogen, Carlsbad, CA, USA). The reaction was allowed to occur at room temperature, and measurements of fluorescence were performed every 5–10 min for up to 2–3 h. Dose-response inhibitory activities of A1AT against caspases were checked for a minimum of 3 independent experiments.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) was used to measure the heat released by interacting proteins using VP-ITC Microcal (noise level: 1 nanocal/s). Caspase-3 peptide (lyophilized RGTELDCGIETD, corresponding to amino acids 164–175 re-suspended in water) or purified elastase (lyophilized and then resuspended in water) was injected into a thermally controlled cell containing A1AT (lyophilized purified pooled human A1AT or recombinant A1AT resuspended in water). The injection amount and duration were adjusted according to the relaxation time of reactants and the response function of the ITC. The changes in free energy (ΔG), enthalpy (ΔH), and entropy (ΔS) upon binding were related according to the thermodynamic equation ΔG = –RT lnK = ΔH – TΔS, where R is universal gas constant, K is binding constant and T is absolute temperature.
Animal Studies
AAV constructs
All experiments were performed with a recombinant AAV vector construct, rAAV-CB-AAT. The AAV-expressing Lac Z was used as a control. The detailed structure of this construct has previously been reported (10,11). Briefly, it consisted of AAV serotype 8 inverted terminal repeats flanking an expression cassette that drives human A1AT expression from a hybrid cytomegalovirus enhancer/β-actin promoter. We pseudotyped this DNA cassette into AAV serotype 8 capsids, using a cotransfection method with purification on an iodixanol gradient (14,15). All vector preparations were titrated by using a DNA dot–blot hybridization method (14), and doses were based on vector genome equivalents. A similar method was used to insert A1AT into the AAV serotype 5 capsids used in experiments involving CS exposure.
Mouse studies involving active caspase-6 instillation were approved by the animal care and use committee of the Indiana University School of Medicine. Female C57Bl/6 mice (3 months old, 25 g) were from the Jackson’s Laboratory (Bar Harbor, ME, USA). The experiment was performed in the same shipment lot, n = 10 mice/group. Mice received 1 × 1010 particles of Lac-Z–expressing AAV (LacZ-AAV), 1 × 1010 particles of MM A1AT-expressing AAV (AAT-AAV) or 1 × 1010 particles of ZZ A1AT-expressing AAV (ZZ-AAV) intratracheally, diluted in NaCl (0.9%; 50 μL; sterile). Mice were administered either intratracheal active recombinant human caspase-6 (2.5 U, 100 μL PBS solution; BioVision) combined with transfection reagent Chariot (1 μL; Active Motif, Carlsbad, CA, USA), or Chariot reagent alone (1 μL with 100 μL PBS), following a previously used approach for the delivery of active caspase-3 (3,16). The instillation of caspase-6 was performed 4 wks following the instillation of AAV. The mouse lung was harvested 48 h following caspase-6 instillation. Trachea preparation, intratracheal delivery, and lung processing were performed as previously described (3,17).
Mouse studies involving CS exposure were approved by the animal care and use committee of the Johns Hopkins University. In vivo CS exposure was performed in C57Bl/6 mice (female, age 12 wks; n = 5–10 per group) that were exposed to CS or ambient air via nose exposure to five cigarettes/d (3R4F; Tobacco Research Institute, Lexington, KY, USA) for 3 d. AAV-GFP (used as control) or AAV-PiZZ were instilled intratracheally 2 wks prior to CS exposure (2 × 1010 particles; 50 μL).
Morphometric Analysis
The left lung was inflated under constant pressure and fixed, followed by paraffin embedding and hematoxylin–eosin staining. Standardized morphometry was performed in a blinded fashion on coded slides, as described (17).
Western Blotting and Zymograms
Samples were loaded in equal amounts of 15 μg protein (unless otherwise noted), as determined by Bradford assay (Pierce). Proteins were separated by SDS-PAGE, or native PAGE, followed by immunoblotting as previously described (18). The chemiluminescent signals were quantified by densitometry (ImageJ) and normalized by vinculin. Zymograms were performed on gelatin gels, as previously described (13). As positive controls for MMP-9 and MMP-2, human fibrosarcoma (HT-1088; Abnova Corporation, Taipei, Taiwan) whole cell lysate was used, prepared in nondenatured radioimmunoprecipitation assay cell lysis buffer.
Statistical Analysis
Statistical analysis was performed with the SigmaStat software package. We compared the differences between groups using an unpaired Student t test or one-way analysis of variance with Student–Newman–Keuls post hoc test for continuous variables and χ2 or Fisher exact test for discrete variables. All the data are expressed as mean ± standard error of the mean (SEM). Statistical difference was accepted at P <0.05.
RESULTS
Effect of Smoking Status and COPD on the A1AT–Caspase-3/7 Interaction
On the basis of our previous results in cell-free models, which showed that incubation of A1AT with CS extract altered its ability to inhibit the activity of caspase-3, we hypothesized that soluble components of CS that are absorbed in the blood of COPD patients who are smokers may affect the anti–caspase-3 function of plasma A1AT. We enrolled 29 individuals for this study (Table 1). A1AT was extracted from blood by use of an immunopurification protocol that allowed the recovery of relatively pure A1AT protein, as detected by Western blotting (Figure 1A) and silver staining (not shown) or Coomassie blue staining of column eluates (Figure 1A). We measured the caspase-3 inhibitory effects of the immunopurified A1AT in a cell-free assay, as previously described (3). As a control, commercially obtained purified A1AT from pooled human plasma was used. The results were analyzed on the basis of the smoking status (active smokers versus never- or ex-smokers) or the disease status (COPD versus no COPD) of the individuals enrolled. A1AT from plasma of actively smoking individuals, independent of disease status, showed significantly reduced ability to inhibit caspase-3/7 activity (Figure 1B). Interestingly, A1AT from plasma of COPD patients who were ex-smokers exhibited twice as much caspase inhibitory activity as did healthy never-smoking individuals.
Table 1.
Subjects’ demographic and clinical characteristics.a
| No clinical diagnosis of COPD nonsmoker | No clinical diagnosis of COPD smoker | COPD nonsmoker | COPD smoker | P | |
|---|---|---|---|---|---|
| (n = 8) | (n = 8) | (n = 5) | (n = 8) | ||
| Age, years | 53.5 ± 6.6 | 39.5 ± 7.7 | 63.8 ± 3.4 | 52.25 ± 3.6 | 0.09 |
| Race, white % | 87.5 | 62.5 | 100 | 75 | 0.37 |
| Cigarette smoking, pack-years | 7.3 ± 4.1 | 15.0 ± 5.6 | 74 ± 12.4 | 47.5 ± 15.4 | 0.0008 |
| Current smokers, % | 0 | 100 | 0 | 100 | <0.0001 |
| Cachexia, % | 0 | 0 | 20 | 37.5 | 0.0768 |
| Asthma, % | 12.5 | 12.5 | 20 | 0 | 0.68 |
| FEV1, % predicted | NA | NA | 50.6 ± 11.986 | 44.88 ± 6.337 | 0.6507 |
| FEV1/FVC, % | NA | NA | 53.2 ± 8.941 | 58.5 ± 5.039 | 0.586 |
| Emphysema diagnosed radiographically, % | NA | NA | 80 | 50 | 0.56 |
| Oral or inhaled corticosteroid use, % | 25 | 12.5 | 100 | 75 | 0.003 |
| Other medications (β-agonist, anticholinergic, methylxantine), % | 12.5 | 12.5 | 100 | 100 | <0.0001 |
Mean ± SEM. FEV1, forced expiratory volume in 1 s; NA, no data available; FVC, forced vital capacity.
Figure 1.
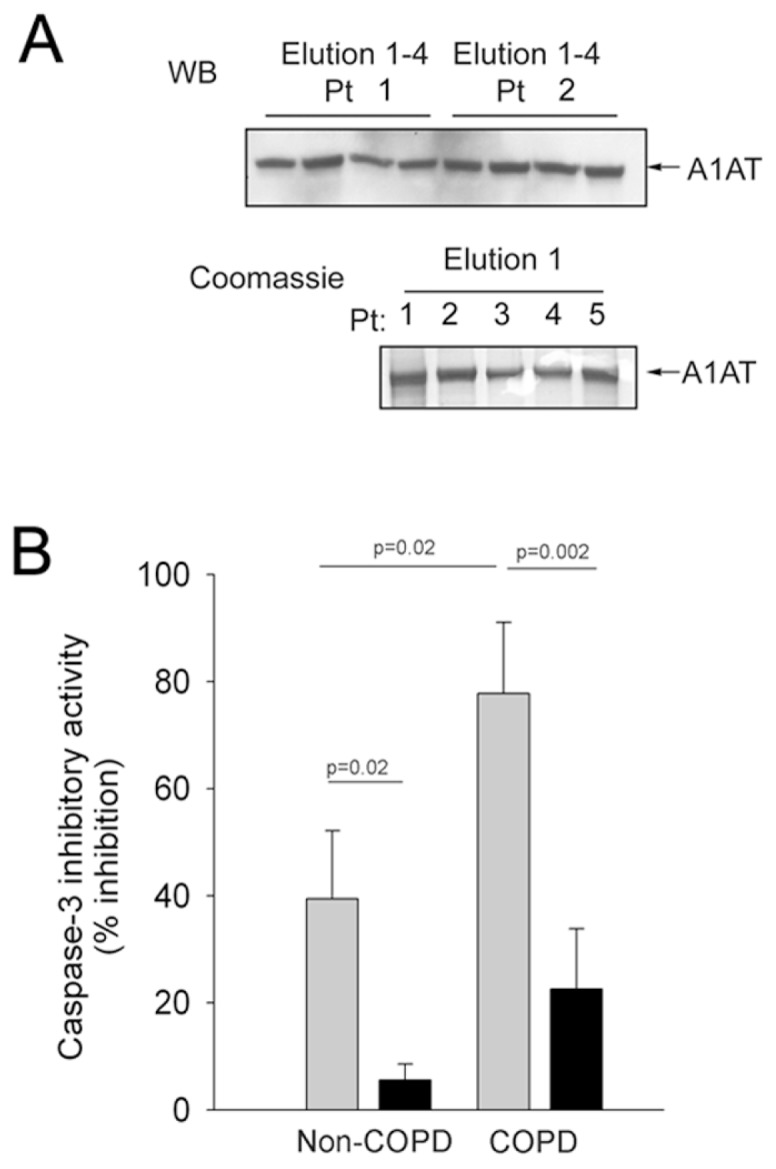
Effect of cigarette smoking and COPD on circulating A1AT’s caspase-3 inhibitory activity. (A) Purity of A1AT eluates obtained by immunopurification from plasma of volunteer subjects, tested via Western blotting (upper panel; representative blot of elutions 1–4 from two subjects) and Coomassie protein staining (lower panel; representative gel of elution 1 from five different subjects). (B) Ex vivo caspase-3 inhibitory activity (% inhibition of caspase-3 activity against a fluorescent substrate; mean + SEM) of A1AT (45.5 μg/mL) purified from the plasma of healthy no-COPD (n = 16) and COPD (n = 13) human volunteers who were either never- or ex-smokers (
 , n = 13) or actively smoking cigarettes (■, n = 16).
, n = 13) or actively smoking cigarettes (■, n = 16).
CS-Exposed A1AT Fails to Directly Interact with Caspase-3
Our previous studies demonstrated that A1AT can directly bind to and inhibit caspase-3. We also show that after exposure to CS the serpin loses its inhibitory potency compared with non–CS-exposed A1AT (3). CS is known to oxidize Met358 within the RCL of A1AT. However, it remains unclear whether exposure of A1AT to CS affects the direct protein–protein interaction between A1AT and caspase-3. To investigate this, we used ITC, a technique that allows measurement of direct protein–protein interaction. The reaction enthalpy was calculated as a function of molar ratio between the protein in the injectant and the protein in the ITC cell solution. Prior to testing of the A1AT interaction with the active caspase-3, A1AT was incubated with CS. The interaction of A1AT with elastase was used as a positive control (Figure 2A) and the interaction with the buffer was used as a negative control (Figure 2B). When native A1AT was incubated with caspase-3 an exothermic reaction was observed (Figure 2C), similar to that observed when A1AT was incubated with elastase. Preexposure of A1AT to soluble components of the CS extract caused an endothermic reaction with caspase-3 (Figure 2D), similar to the reaction of A1AT with buffer, indicating a lack of interaction between CS-exposed A1AT and caspase-3. These data are consistent with our previous caspase-3 activity assays, in which we used fluorescent substrate, which showed weaker inhibitory potency of CS-exposed A1AT. In aggregate, these data indicate that CS modifies A1AT and prevents its direct interaction with caspase-3.
Figure 2.

Interaction of CS-exposed A1AT with capsase-3. Energy (Kcal/mol) generated by the intermolecular interaction between human A1AT (100 μmol/L) added sequentially in 10-μL aliquots (for a total of 29 injections) at 540-s intervals to (A) elastase (positive control); (B) buffer (negative control); (C) caspase-3 (8.9 μmol/L); or between (D) human A1AT preexposed to CS extract (25%) and caspase-3. The energy was measured with an isothermal titration microcalorimeter at 30°C and expressed in relation to the molar ratio of the two reaction components, defined as the ratio of injected reactant to cell solution. Representative of two independent experiments.
RCL-Null, but Not ZZ-A1AT, Fails to Inhibit Caspase-3 in Cell Free Assays
Because CS is known to affect the antiprotease activity of A1AT by oxidation of Met358 in the RCL, we next investigated if the RCL was required for A1AT anti–caspase-3 activity. We produced recombinant human RCL-deleted A1AT (RCL-null), as well as MM and ZZ forms of A1AT (Figure 3A) and compared their caspase-3 inhibitory activity. We found that whereas both the MM-A1AT and ZZ-A1AT mutants had preserved caspase-3 inhibitory activity, the RCL-null A1AT mutant had significantly attenuated ability to inhibit caspase-3 activity (Figure 3B).
Figure 3.

Requirement of the RCL for the A1AT inhibition of capsase-3 activity. (A) Western blot of recombinant human MM-A1AT, ZZ-A1AT or RCL-null A1AT produced in E. coli. Representative of n = 2. (B) Kinetic activity of recombinant human caspase-3 (~1 U/μL) against a fluorescently labeled substrate in the presence of purified human recombinant MM-A1AT, ZZ-A1AT or RCL-null A1AT (45 μg/mL) (mean + SEM; n = 3–5); ns, nonsignificant statistical difference.
ZZ-A1AT Exerts Anti-Caspase-3 Effects In Vivo
To determine if the caspase-3 inhibitory effects of ZZ-A1AT also manifest in vivo, we overexpressed ZZ-A1AT using AAV transduction, as previously described (19). The CS exposure–induced caspase-3/7 activity in the lung, as determined by caspase-3 activity assay of lung tissue lysates, was not inhibited by instillation of control AAV virus (Figure 4A). In contrast, the ZZ-A1AT–expressing AAV significantly inhibited CS-induced caspase-3 activity in the lung (Figure 4A). At the same time, ZZ-A1AT did not affect either matrix metalloproteinase -9 or -2 activation induced by CS (Figure 4B) or the number of inflammatory cells present in the bronchoalveolar lavage fluid (data not shown). These results suggest that, unlike its impact on serine proteases inhibition, the ZZ mutation is not sufficient to impair the anti–caspase-3 activity of A1AT in vivo.
Figure 4.

Caspase-3 inhibitory effect of ZZ-A1AT in vivo. (A) Endogenous lung caspase-3/7 activity measured in a fluorescence activity assay at 3 d following CS exposure in mice. Mice instilled with ZZ-A1AT–expressing AAV, but not those instilled with control AAV, exhibited inhibition of CS-induced caspase-3 activity in the lung (mean + SEM; *P < 0.05; n = 3–5). (B) Zymogram of lung lysates from mice instilled with control vehicle (PBS), control AAV or ZZ-A1AT–expressing AAV exposed to air control or to CS for 3 d. hPC is a positive control (human fibrosarcoma whole cell lysate).
A1AT Inhibits Executioner Caspases -3, -6 and -7
Of the 14 mammalian caspases characterized, 7 have important roles in apoptosis, either as initiator or effector/ executioner caspases. We have previously shown that of the 3 known executioner caspases (-3, -6 and -7), A1AT inhibits caspase-3. The effect of A1AT on the other members of the caspase family is unknown. We tested this effect using a cell-free assay. Recombinant caspases were incubated with their respective fluorescently labeled substrates, and kinetics of enzymatic activity were recorded in the absence or presence of purified human A1AT. Similar to its effects on caspase-3, A1AT inhibited caspases-6 and -7 in a dose-dependent manner (Figure 5A). Interestingly, the most potent A1AT inhibitory activity was noted against caspase-6, for which even low concentrations of A1AT (50 μg/mL) had a marked inhibitory effect. In contrast, even high concentrations of the serpin (500 μg/mL) failed to inhibit other caspases such as initiator caspases or those involved in the inflammasome (Figure 5B).
Figure 5.

Effect of A1AT on the activity of different classes of caspases. Cell-free kinetic enzymatic activity was measured as follows for different classes of caspases. (A) Executioner caspases: recombinant active caspases -3 (●), -6 (▿) or -7 (
) were incubated with purified human A1AT (0–500 μg/mL). (B) Initiator caspases (recombinant active caspases -8, -9, -10, -2), executioner caspases (-3, -7, -6) or inflammatory caspases (-1, -4, -5) were incubated with purified human A1AT obtained from pooled plasma (500 μg/mL; n = 3). (C) Executioner caspase-6 incubated with purified A1AT (
) or recombinant RCL-null A1AT (■) (0–500 μg/mL; n = 3; *P < 0.05 versus caspase-6 activity incubated with vehicle and versus RCL-null A1AT).
Effects of A1AT on Caspase-6 Activity In Vivo
We previously reported that A1AT inhibits caspase-3 in vivo (3) by overexpressing A1AT via AAV transduction, followed by intratracheal instillation of active caspase-3. We used a similar approach to investigate if the effect of A1AT on caspase-6 activity observed in vitro also occurs in vivo. We overexpressed human MM-A1AT or ZZ-A1AT, or a control LacZ-A1AT vector in mouse lungs via AAV transduction, followed 4 wks later by intratracheal instillation of recombinant active caspase-6 or its vehicle control. There was evidence of human MM-A1AT and ZZ-A1AT expression in the lungs of mice at 30 d following injection of the respective AAV construct (Figure 6A). Interestingly, in an observation similar to our findings in vascular endothelial growth factor receptor (VEGFR)-inhibited mice receiving A1AT-AAV (19), we noted increased expression of human A1AT protein in the lungs of mice that received the treatment (VEGFR inhibitor, or in this case intratracheal caspase-6) compared with lungs of control mice injected with A1AT-AAV, suggesting that (proapoptotic) lung injury may facilitate the transduction of A1AT. We found that at 48 h following intratracheal instillation of recombinant active caspase-6, the lungs of these mice exhibited significantly increased apoptosis, as measured by endogenous caspase-3/7 activation (Figure 6B), compared with mice receiving the intratracheal instillation of vehicle only. Concomitantly we found rapid enlargement of airspaces, measured by a significant decrease in the surface/ volume ratio in mice instilled with caspase-6 compared with mice instilled with vehicle only (Figure 6C). Consistent with the cell-free results, mice with MM-A1AT overexpression via intratracheal instillation of MM-A1AT-AAV serotype 8, 4 wks prior to caspase-6 instillation, exhibited both significantly less apoptosis as measured by endogenous caspase-3/7 activity and less airspace enlargement (Figures 6B, C). Interestingly, overexpression of ZZ-A1AT inhibited endogenous caspase-3/7 activation, but unlike MM-A1AT, it did not significantly attenuate the airspace enlargement induced by caspase-6 instillation (Figures 6B, C).
Figure 6.

Effect of A1AT against executioner caspases in vivo. (A) Representative immunoblots of human MM-A1AT and ZZ-A1AT expression in the lungs of mice instilled with either control Lac-Z-AAV or MM-A1AT or ZZ-A1AT AAV (1 × 1010; 50 μL), followed, 4 wks later, by active caspase-6 intratracheal instillation (5 U; 50 μL; 48 h). The upper band represents A1AT immunoblots (human recombinant A1AT used as a control in the last lane), and the lower band represents vinculin immunoblots used as a loading control. (B) Endogenous lung caspase-3/7 activity and (C) lung morphometry measurement of surface/volume ratios following active caspase-6 intratracheal instillation in mice (48 h) in the presence of Lac-Z-AAV (vector) empty or overexpressing MM-A1AT, or ZZ-A1AT for 4 wks. Mean + SEM; n = 10.
DISCUSSION
Our results provide evidence that A1AT has an inhibitory activity directed only against executioner caspases. We demonstrate that this activity requires the RCL and can be profoundly inhibited by exposure of the protein to CS both in vitro and in vivo. In contrast, the single amino acid mutation in the ZZ form of A1AT did not affect its antiapoptotic activity. One of the main posttranslational modifications of A1AT in patients with COPD is that of oxidation, thought to be due to exposure to CS components (20). Reactive oxygen and nitrogen species, which are increased in smokers and COPD patients, may target and modify the A1AT methionine-358 and cysteine-232 residues of the RCL, respectively. These changes could alter the A1AT function against elastase (21–22). A similar effect may impair the ability of A1AT to directly interact with target caspases, as demonstrated by isothermal titration calorimetry assays. Interestingly, only the acute exposure to CS, but not the disease state of COPD, led to a decrease in the anti–caspase-3 activity of A1AT. This suggests that the oxidative burden that may exist in this chronic disease in the absence of smoking is not sufficient to alter the ability of A1AT to interact with the caspases in ex vivo cell-free conditions. It still remains to be determined whether the A1AT from ex-smokers with COPD has impaired ability to access intracellular caspases in vivo. We now provide additional information regarding the properties of oxidized A1AT, to include a loss of anticaspase activity, in addition to previously known loss of antielastase action and proinflammatory effects of triggering chemokine release and recruitment of inflammatory cells (23).
The fact that the RCL-null A1AT construct failed to inhibit caspase-3 or -6 activities confirmed a critical role of this domain in multiple serpin functions, because the RCL domain is essential for the antielastase activity of A1AT. Even more drastically than the oxidation of its residues, the deletion of the RCL significantly alters the structural properties of the A1AT protein. Because the conformational plasticity of the A1AT is highly vulnerable to structural changes in the protein and is required for its function as an antiprotease, it is not surprising that the RCL-null mutant failed to inhibit caspase-3 activity. However, it was somewhat surprising that the single point mutation causing the ZZ phenotype, which would be expected to affect A1AT plasticity by making it prone to polymerization, did not affect the antiapoptotic function of the protein in vitro and in vivo. A similar lack of effect of ZZ-A1AT on the antiapoptotic activity of A1AT was recently reported by Greene et al. (24). It is possible that unlike the effect on elastase, certain levels of protein polymerization do not inhibit the ability of the RCL to interact with caspases. Alam et. al. recently demonstrated that the oxidation of ZZ-A1AT precedes and promotes its polymerization (25). Because the RCL-null A1AT does not form polymers but failed to inhibit caspase-3, it is likely that the oxidation of the RCL, rather than the oxidation-induced polymerization, promotes loss of A1AT anti-apoptotic function. This possibility is supported by our previously reported finding that polymerization of A1AT obtained by heating the protein caused a significant loss of anti–capsase-3 activity, not seen with the recombinant ZZ protein. Of note, although the bacterial expression of ZZ-A1AT may generate an incompletely functional mutant due to lack of glycosylation, the ZZ-A1AT production in vivo via AAV should not have this limitation.
This report builds on our previous studies that identified a novel protective mechanism of A1AT in the lung, that of inhibition of the apoptotic effector caspase-3 (3,19). Here we identified the other executioner caspases -7 and -6, but not the initiator caspases, as unique targets for A1AT. Executioner caspases can be activated by the extrinsic and intrinsic apoptosis pathways, which in turn are triggered by stimuli such as proinflammatory cytokines or stimuli causing mitochondrial stress, respectively (26,27). Although there is some functional redundancy among executioner caspases, it has been reported that the effector caspase-6 can activate caspase-3 (28). This could explain why we observed increases in endogenous caspase-3/7 activity following intratracheal caspase-6 instillation. It is possible that the effect of the MM- or ZZ-A1AT in vivo may be a consequence of inhibition of either caspase-6 or the downstream caspase-3/7. While MM-and ZZ-A1AT comparably inhibited the casapase-6–induced rise in caspase-3/7 activity, ZZ-A1AT decreased the baseline caspase activity. A similar observation of a potent inhibition of caspase-3 by ZZ-A1AT was recently made in human bronchial epithelial cells (24), and the difference between the two proteins was attributed to ZZ-A1AT leading to trans-activation of nuclear factor–κB and up-regulation of the cellular inhibitor of apoptosis. Hence, MM- and ZZ-A1AT may inhibit apoptosis via different mechanisms, which could explain the differences we observed in the regulation of baseline caspase-3/7 activity.
Considering the vital role of apoptosis regulation in COPD, we performed a comprehensive analysis of A1AT inhibitory ability on all caspases known to be involved in apoptosis (caspase-2, -3, -6, -7, -8, -9 and-10) and also on the inflammatory caspases (-1, -4 and -5). A1AT inhibited all effector caspases (-3, -6 and -7), but none of the initiator caspases, and had only a mild but non-significant inhibitory effect on caspase-1 of the inflammatory caspases. The marked inhibitory effect on caspase-6 activity in vitro was recapitulated in vivo. We cannot rule out that other caspase activation could be inhibited in complex in vivo models in which A1AT may act on other upstream targets that lead to the activation of such caspases. This, or more favorable in vivo conditions, may explain the recently reported inhibitory effect of A1AT on caspase-1 activation in a model of myocardial infarction (29).
The clinical significance of our work is related to the finding of increased apoptosis of alveolar epithelial and endothelial cells in the lungs of COPD patients who smoke cigarettes and in animal models of emphysema (26,30–32). Our data suggest that the effect of cigarette smoking on the A1AT antiapoptotic function is reversible and may be linked to the turnover of the protein as it is synthesized from the liver or to the restoration of native A1AT by reduction of the oxidized protein. Limitations of our clinical study include the small number of patients and the fact that no PFTs were performed in the control, no-clinical-diagnosis-of-COPD-smoker group, leaving the possibility that individuals in this group may have had asymptomatic COPD. Future studies will need to address the possibility that the A1AT function at a given time may vary depending on the time since CS exposure and to also address the functional impact of normalizing the function of A1AT in lung endothelial cells on the progression of emphysema in vivo.
Finally, in addition to lung destruction there is evidence that cardiovascular morbidities are increased in COPD. Smoking cessation has been noted to result in a sharp reduction in the risk of stroke, myocardial infarction, and mortality from cardiovascular diseases in patients with or without COPD (33–35). Extrapolation of our work on pulmonary endothelium to include the antiapoptotic effect of A1AT on other vascular beds might help explain some of the rapid beneficial effects on the systemic or coronary circulation that have been noted following smoking cessation.
CONCLUSION
In conclusion, understanding and manipulating the newly discovered functions of A1AT in pulmonary and vascular biology may help the design of future therapeutic interventions for conditions in which endothelial apoptosis and dysfunction play a central pathogenic role.
ACKNOWLEDGMENTS
VA Merit Award (I Petrache); NIH-NHLBI 1P50 HL084945 (I Petrache, RM Tuder, RA Wise); NIH-NIAID T32 AI060519-05 (AD Lockett); and the Dorney-Koppel Family Foundation (RA Wise).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Kochanek KD, Xu J, Murphy SL, Minino AM, Kung HC. Deaths: preliminary data for 2009. Natl Vital Stat Rep. 2011;59:1–51. [PubMed] [Google Scholar]
- 2.Yokohori N, Aoshiba K, Nagai A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest. 2004;125:626–32. doi: 10.1378/chest.125.2.626. [DOI] [PubMed] [Google Scholar]
- 3.Petrache I, et al. alpha-1 Antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol. 2006;169:1155–66. doi: 10.2353/ajpath.2006.060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poller W, Willnow TE, Hilpert J, Herz J. Differential recognition of -antitrypsin-elastase and -antichymotrypsin-cathepsin G complexes by the low density lipoprotein receptor-related protein. J Biol Chem. 1995;270:2841–5. doi: 10.1074/jbc.270.6.2841. [DOI] [PubMed] [Google Scholar]
- 5.Owen MC, Carrell RW. alpha-1-Antitrypsin: sequence of the Z variant tryptic peptide. FEBS Lett. 1977;79:245–7. doi: 10.1016/0014-5793(77)80796-5. [DOI] [PubMed] [Google Scholar]
- 6.Mahadeva R, et al. Heteropolymerization of S, I, and Z alpha1- antitrypsin and liver cirrhosis. J Clin Invest. 1999;103:999–1006. doi: 10.1172/JCI4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott PR, Bilton D, Lomas DA. Lung polymers in Z alpha 1-antitrypsin deficiency-related emphysema. Am J Respir Cell Mol Biol. 1998;18:670–4. doi: 10.1165/ajrcmb.18.5.3065. [DOI] [PubMed] [Google Scholar]
- 8.Taggart C, et al. Oxidation of either methionine 351 or methionine 358 in α1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275:27258–65. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- 9.Nadiri A, Wolinski MK, Saleh M. The inflammatory caspases: key players in the host response to pathogenic invasion and sepsis. J Immunol. 2006;177:4239–45. doi: 10.4049/jimmunol.177.7.4239. [DOI] [PubMed] [Google Scholar]
- 10.Song S, et al. Stable therapeutic serum levels of human alpha-1 antitrypsin (AAT) after portal vein injection of recombinant adeno- associated virus (rAAV) vectors. Gene Ther. 2001;8:1299–306. doi: 10.1038/sj.gt.3301422. [DOI] [PubMed] [Google Scholar]
- 11.Liqun Wang R, et al. Recombinant AAV serotype and capsid mutant comparison for pulmonary gene transfer of alpha-1-antitrypsin using invasive and noninvasive delivery. Mol Ther. 2009;17:81–7. doi: 10.1038/mt.2008.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, et al. Expression and purification of functional human alpha-1-antitrypsin from cultured plant cells. Biotechnol Prog. 2001;17:126–33. doi: 10.1021/bp0001516. [DOI] [PubMed] [Google Scholar]
- 13.Petrache I, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11:491–8. doi: 10.1038/nm1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zolotukhin S, et al. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–67. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 15.Sirninger J, et al. Functional characterization of a recombinant adeno-associated virus 5-pseudotyped cystic fibrosis transmembrane conductance regulator vector. Hum Gene Ther. 2004;15:832–41. doi: 10.1089/hum.2004.15.832. [DOI] [PubMed] [Google Scholar]
- 16.Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol. 2003;28:555–62. doi: 10.1165/rcmb.2002-0090OC. [DOI] [PubMed] [Google Scholar]
- 17.Tuder RM, et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol. 2003;29:88–97. doi: 10.1165/rcmb.2002-0228OC. [DOI] [PubMed] [Google Scholar]
- 18.Petrache I, et al. Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1168–78. doi: 10.1152/ajplung.2001.280.6.L1168. [DOI] [PubMed] [Google Scholar]
- 19.Petrache I, et al. A novel antiapoptotic role for alpha1-antitrypsin in the prevention of pulmonary emphysema. Am J Respir Crit Care Med. 2006;173:1222–8. doi: 10.1164/rccm.200512-1842OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carp H, Miller F, Hoidal JR, Janoff A. Potential mechanism of emphysema: alpha 1-proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity. Proc Natl Acad Sci U S A. 1982;79:2041–5. doi: 10.1073/pnas.79.6.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson D, Travis J. The oxidative inactivation of human alpha-1-proteinase inhibitor. Further evidence for methionine at the reactive center. J Biol Chem. 1979;254:4022–6. [PubMed] [Google Scholar]
- 22.Miyamoto Y, Akaike T, Maeda H. S-nitrosylated human alpha(1)-protease inhibitor. Biochim Biophys Acta. 2000;1477:90–7. doi: 10.1016/s0167-4838(99)00264-2. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, et al. Oxidized {alpha}1-antitrypsin stimulates the release of monocyte chemotactic protein-1 from lung epithelial cells: potential role in emphysema. Am J Physiol Lung Cell Mol Physiol. 2009;297:L388–400. doi: 10.1152/ajplung.90373.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greene CM, et al. Anti-apoptotic effects of Z alpha1-antitrypsin in human bronchial epithelial cells. Eur Respir J. 2010;35:1155–63. doi: 10.1183/09031936.00191908. [DOI] [PubMed] [Google Scholar]
- 25.Alam S, Li Z, Janciauskiene S, Mahadeva R. Oxidation of Z alpha1-antitrypsin by cigarette smoke induces polymerization: a novel mechanism of early-onset emphysema. Am J Respir Cell Mol Biol. 2011;45:261–9. doi: 10.1165/rcmb.2010-0328OC. [DOI] [PubMed] [Google Scholar]
- 26.Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7:53. doi: 10.1186/1465-9921-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 28.Allsopp TE, et al. Caspase 6 activity initiates caspase 3 activation in cerebellar granule cell apoptosis. Cell Death Differ. 2000;7:984–93. doi: 10.1038/sj.cdd.4400733. [DOI] [PubMed] [Google Scholar]
- 29.Toldo S, et al. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51:244–251. doi: 10.1016/j.yjmcc.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Kasahara Y, Tuder RM, Cool CD, Voelkel NF. Expression of 15-lipoxygenase and evidence for apoptosis in the lungs from patients with COPD. Chest. 2000;117:260S. doi: 10.1378/chest.117.5_suppl_1.260s. [DOI] [PubMed] [Google Scholar]
- 31.Segura-Valdez L, et al. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117:684–94. doi: 10.1378/chest.117.3.684. [DOI] [PubMed] [Google Scholar]
- 32.Bartalesi B, et al. Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. Eur Respir J. 2005;25:15–22. doi: 10.1183/09031936.04.00067204. [DOI] [PubMed] [Google Scholar]
- 33.Shah AM, et al. Risk of all-cause mortality, recurrent myocardial infarction, and heart failure hospitalization associated with smoking status following myocardial infarction with left ventricular dysfunction. Am J Cardiol. 2010;106:911–6. doi: 10.1016/j.amjcard.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 34.Song YM, Cho HJ. Risk of stroke and myocardial infarction after reduction or cessation of cigarette smoking: a cohort study in Korean men. Stroke. 2008;39:2432–8. doi: 10.1161/STROKEAHA.107.512632. [DOI] [PubMed] [Google Scholar]
- 35.Wilson K, Gibson N, Willan A, Cook D. Effect of smoking cessation on mortality after myocardial infarction: meta-analysis of cohort studies. Arch Intern Med. 2000;160:939–44. doi: 10.1001/archinte.160.7.939. [DOI] [PubMed] [Google Scholar]