

Abstract
It has been shown that bridging integrator 1 (BIN1) can interact with c-myelocytomatosis (c-Myc) oncoprotein in cancer. However, the role of BIN1 in hepatocellular carcinoma (HCC) is not clear. In the present study, we investigated the expression and prognostic role of BIN1 in primary HCC and evaluated the function of BIN1 in hepatocarcinogenesis. Using real-time polymerase chain reaction and Western blot analysis, we found significantly decreased expression of BIN1 in primary HCC tumor tissues (n = 42) compared with adjacent normal tissues and in HCC cell lines. Immunohistochemistry analysis also found decreased BIN1 expression in HCC tumor tissues (n = 117). In clinicopathological analysis, loss of BIN1 expression correlated significantly (P < 0.05) with differentiation scores and tumor size. Importantly, decreased expression of BIN1 in tumors was found to be closely associated with a poor prognosis, and we conclude that BIN1 was an independent prognostic factor in a multivariate analysis. In mechanistic studies, restoring BIN1 expression in BIN1-null HCC cells significantly inhibited cell proliferation and colony formation and induced apoptosis of HCC cells. Furthermore, we found that BIN1 overexpression could significantly suppress the motility and invasion of HCC cells in vitro. Our results indicate that BIN1 may function as a potential tumor suppressor and serve as a novel prognostic marker in HCC patients. The BIN1 molecule might play an important role in tumor growth, cell motility and invasion. Modulation of BIN1 expression may lead to clinical applications of this critical molecule in the control of hepatocellular carcinoma as well as in early and effective diagnosis of this aggressive tumor.
INTRODUCTION
Bridging integrator 1 (BIN1) maps to the long arm of human chromosome 2 (2q14) and encodes multiple tissue-specific isoforms of the Myc-interacting adaptor protein (1–4). BIN1 expression is often found attenuated or even abolished in approximately 50% of the carcinoma cell lines as well as in several primary tumors, such as malignant melanoma, breast and prostate cancers, while its ectopic expression can inhibit cell proliferation and/or promote apoptosis (1,5–14). Notably, the effects of BIN1 loss on cell growth and survival appear to be contingent on cell transformation (15–18). These studies suggest that BIN1 has tumor suppressor features that are linked to cell death and differentiation decisions, and it may therefore be involved in neoplastic pathophysiology.
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide and is a major cause of cancer-related death in many countries, especially in southern China, southeastern Asia and sub-Saharan Africa. The progression of HCC is a slow process that evolves through distinct stages associated with cumulative genomic alterations. Recent studies have shown that aberrant gene expression, including oncogene overexpression and tumor suppressor down-expression, is responsible for the development of HCC. However, the molecular pathogenesis of HCC still remains unclarified. In a previous study, a lack of BIN1 expression was reported in a human HCC cell line, and overexpression of BIN1 was shown to suppress tumor cell growth (1). These findings suggested that BIN1 may play a critical role as a tumor suppressor in HCC. Nevertheless, the clinicopathological and prognostic significance of BIN1 expression remains undefined in primary HCC. Furthermore, the functional role of BIN1 in the pathogenesis and tumorigenicity of HCC is also unexplored.
In this study, we examined BIN1 expression in primary HCCs and evaluated the relationship between BIN1 expression and clinicopathological parameters of HCCs. Meanwhile, we investigated the prognostic value of BIN1 for HCC patients. Furthermore, we evaluated the functional role of BIN1 in the tumorigenesis of HCC by examining the in vitro proliferation, clone formation, cell cycle, apoptosis, motility and invasion of the BIN1 transfected HCC cells.
MATERIALS AND METHODS
Cell Culture
Human HCC cell lines, HepG2 (well differentiated, low metastatic potential), Hep3B (well differentiated, low metastatic potential) and SK-Hep1 (poorly differentiated, high metastatic potential) were obtained from American Type Culture Collection (ATCC). Huh7 (well differentiated, low metastatic potential) was obtained from the RIKEN cell bank (Ibaraki, Japan). BEL-7402 (moderately differentiated, low metastatic potential) cells were obtained from the Committee of Type Culture Collection of the Chinese Academy of Sciences (CTCCCAS, Shanghai, China). Normal liver cell line (LO2) was also obtained from CTCCCAS. All cells were cultured in 5% CO2 at 37°C in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (FBS). The mRNA and protein were extracted from all cell lines in the same culture passage (subculture to three passages) for BIN1 expression analysis.
Tissue Samples
Tissue samples, including HCC tumor tissues and adjacent noncancerous tissues (n = 42), were obtained from patients who had primary HCC surgical resection in the Sun Yat-sen University Cancer Center between 2007 and 2009. These patients did not receive any preoperative treatment, such as chemotherapy and radiotherapy. After surgical resection, the fresh tissues were frozen at −80°C and used for RNA extraction and real-time quantitative polymerase chain reaction (PCR) detection. Additional paraffin-embedded HCC samples (n = 117) were selected randomly from the patients who had primary HCC surgical resection in the Sun Yat-sen University Cancer Center between 1999 and 2001. These patients did not receive any pre-treatment either. The serial 4-μm sections were cut and used for immunohistochemical detection. Cell histological types of tumor tissues were evaluated according to World Health Organization classification criteria. Postoperative follow-up visits were conducted by our outpatient department. Follow-up included clinical and laboratory examinations every 3 months for the first 2 years, every 6 months during the third to fifth years, and then annually for an additional 5 years or until patient death, whichever occurred first. Overall survival, which was defined as the time from the operation to the patient’s death or the last follow-up, was used as a measure of prognosis. Before the study, written informed consent was obtained from each patient.
RNA Preparation and Protein Extraction
Total RNA from HCC tissues or cell lines was extracted using Trizol solution (Invitrogen, Shanghai, China) according to the manufacturer’s instructions. Total protein from HCC tissues or cell lines was extracted using radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China) according to the manufacturer’s instructions. The RNA and protein samples were stored at −80°C until use.
Real-Time Quantitative Reverse Transcription–PCR
Total RNA (2 μg) was reverse transcribed, and first-strand cDNA was synthesized using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega, Beijing, China) according to the manufacturer’s instructions. The resulting cDNA was used for real-time quantitative reverse transcription (RT)-PCR detection of BIN1. Glyceraldehyde-3-phosphate de-hydrogenase (GAPDH) or β-actin was used as an internal control. For BIN1, the 5′ primer was 5′-ACAAC GACCT GCTGT GGATG G-3′, and the 3′ primer was 5′-CGTGA CTTGA TGTCG GGGAA CT-3′. For GAPDH, the 5′ primer was 5′-CTCCT CCTGT TCGAC AGTCA GC-3′, and the 3′ primer was 5′-CCCAA TACGA CCAAA TCCGT T-3′. For β-actin, the 5′ primer was 5′-TGACC CAGAT CATGT TTGAG-3′, and the 3′ primer was 5′-CGTAC AGGGA TAGCA CAG-3′. RT-PCR was performed as follows: 95°C for 10 min, one cycle, followed by 95°C for 30 s and 60°C for 60 s (45 cycles). After RT-PCR was finished, dissociation curves were analyzed, and the relative amount of product was calculated from the threshold cycles with the instrument’s software (SDS 2.0). The experiments were performed for four times, and the statistical analysis was performed.
Western Blot
Western blot was performed to detect the protein levels of BIN1 in paired clinical specimens from HCC patients and cell lines. Total protein (50 μg) was resolved using 12% sodium dodecyl–sulfate polyacrylamide gel (SDS-PAGE). After electrophoresis, the separated proteins were transferred to PVDF membranes. The membranes were blocked with 5% nonfat milk and incubated with primary polyclonal antibody against BIN1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; at 1:250 dilution), GAPDH (Abcam, San Francisco, CA, USA; at 1:10,000 dilution) or β-actin (Proteintech Group, Chicago, IL, USA; at 1:10,000 dilution) overnight at 4°C. After washing, the membranes were probed with the horseradish peroxidase–conjugated secondary antibody and visualized with chemiluminescent system (Cell Signaling Technology, Danvers, MA, USA). The band intensity was measured using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA). GAPDH was used as a loading control.
Immunohistochemical Staining
The HCC tissue sections were deparaffinized and rehydrated. Then, the sections were boiled in ethylenediaminetetraacetic acid (EDTA) (1 mmol/L; pH 8.0) for antigen epitope retrieval. Endogenous peroxidase was blocked with 0.3% hydrogen peroxide. After rinsing, slides were incubated with a mouse polyclonal antibody against BIN1 (Santa Cruz Biotechnology; at 1:100 dilution) overnight at 4°C. After washing, the slides were incubated for 30 min with horseradish peroxidase–conjugated secondary antibody (Envision™ Detection Kit, GK500705; Gene Tech, Shanghai, China) at room temperature. After this incubation, the slides were washed three times in PBS, and the antibody complexes were colored with diaminobenzidine (DAB) and then counterstained with hematoxylin. The total BIN1 immunostaining score was calculated as the sum of the presence of positively stained tumor cells and the staining intensity. Briefly, the percent positive staining was scored as 0 (<5%, negative), 1 (5–25%, sporadic), 2 (25–50%, focal), or 3 (>50%, diffuse). The staining intensity was score as 0 (no staining), 1 (weakly stained), 2 (moderately stained) or 3 (strongly stained). The total immunostaining score was calculated with the value of percent positivity score × staining intensity score, which ranged from 0 to 9. We defined BIN1 expression levels as follows: − (score 0–1), + (score 2–3), ++ (score 4–6) and +++ (score >6). All IHC results were evaluated by two independent pathologists who were unaware of the clinical data. For inconsistent evaluations of individual slides, both observers reviewed the slide again to obtain a consensus. On the basis of their BIN1 immunostaining levels, the patients were divided into two groups: the BIN1 low group (− and +) and the BIN1 high group (++ and +++).
Expression Plasmid Construction and Transient Transfections
A eukaryotic expression plasmid of the human BIN1 gene was constructed using a pcDNA3 vector (Invitrogen, China). We constructed BIN1 cDNA expression vector on the basis of the sequence in NCBI (Accession number U68485). This BIN1 mRNA does not contain the exon 12 (−12 isoform), and the sequence has normal tumor suppressor function. The empty vector was used as negative control. Huh7 and Hep-3B cells were cultured in six-well plates until they reached 85–90% confluence, and then transient transfections were performed using Lipofectamine 2000 (Invitrogen, China) according to the manufacturer instructions. At 48 h after transfection, gene expression was confirmed via Western blot analysis.
Tumor Suppressor Function of BIN1
Cell growth rate of BIN1-expressing cells and control cells or LO2 cells were detected with the MTS cell proliferation assay. Cells were seeded in 96-well plates at a density of 4 × 103 per well. The cell growth rate was detected using a cell proliferation MTS kit (Promega, Beijing, China) according to the manufacturer’s instructions, and independent experiments were performed in triplicate. For the foci formation assay, 1 × 103 BIN1-expressing cells or control cells were plated in a six-well plate. After 10 d of culture, surviving colonies (>50 cells per colony) were counted with Giemsa staining. Colony-forming efficiency (CFE%) was defined as the ratio of the number of colonies formed in culture to the number of cells inoculated, and independent experiments were performed in triplicate.
Cell Cycle Assay
HCC cells transfected with the vector or BIN1 were collected and washed twice with ice-cold PBS and fixed with ice-cold 75% ethyl alcohol at −20ºC for 4 h. The cells were washed and resuspended in 400 μL ice-cold PBS plus RNAase and incubated in a 37ºC water bath for 30 min. Then, the cells were stained with propidium iodide at 4°C in the dark for 30–60 min. The DNA content was analyzed using a flow cytometer (Beckman Coulter, Brea, CA, USA).
Apoptosis Assay
Transfected Huh7 or Hep-3B cells were collected and washed twice with ice-cold PBS and resuspended with 1× binding buffer before incubation with annexin V-FITC (Bestbio, Shanghai, China) according to the manufacturer’s protocol. After 15 min of incubation at room temperature in the dark, propidium iodide was added, and the number of stained cells was analyzed using a flow cytometer (Beckman Coulter).
Wound-Healing Assay
Approximately 24 h after the beginning of the transfection, HCC cells or LO2 cells were seeded in a 12-well culture plate and allowed to grow to 90% confluence. A wound was then created on the cell monolayer using a pipette tip. The migration of cells toward the wound was monitored daily. Images were captured at regular time intervals. The experiment was done in triplicate.
Matrigel Invasion Assay
The matrigel invasion assay was performed using 24-well transwells consisting of polycarbonate membrane inserts with 8-μm pores (Corning, Shanghai, China), coated with a thin layer of 0.5 mg/mL Matrigel Basement Membrane Matrix (BD Biosciences, Bedford, MA, USA). HCC cells or LO2 cells (1 × 105) in 100 μL growth medium without FBS were added in the upper chamber, and 0.5 mL growth medium with 10% FBS was placed in the lower chamber. After incubation at 37°C for 48 h, the insert membranes were fixed with 75% methanol for 10 min. The cells remaining in the upper surface were removed with cotton-tipped swabs. The invaded cells on the lower surface of membranes were stained with 0.5% crystal violet. The stained cells at 10 random fields were counted under an inverted microscope. Each experiment was performed in triplicate.
Silencing BIN1 Expression in LO2 Cells
To further investigate the tumor suppressor function of BIN1, the BIN1 gene was silenced with siRNA. The siRNA sequences were as follows: BIN1-siRNA-1: 5′-GCGUC CAGAA UUUCA ACAAT T-3′; BIN1-siRNA-2: 5′-CCACU ACGAG UCCCU UCAAT T-3′; BIN1-siRNA-3: 5′-GCGUA GGUUU CUACG UCAAT T-3′; BIN1-siRNA-4: 5′-GCAAC ACCUU CACGG UCAAT T-3′; negative control: 5′-UUCUC CGAAC GUGUC ACGUT T-3′.
The siRNAs were synthesized by GenePharma Company (Shanghai, China). A total of 400 pmol siRNA was transfected into 4 × 105 LO2 cells using Lipofectamine RNAi MAX reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s protocol. After that, proliferation, migration and invasion detection were then performed.
Statistical Analysis
The correlation between BIN1 expression and clinical parameters were evaluated using χ2 tests. Survival analysis was performed using the Kaplan-Meier method, and a log-rank test was used for comparison. Univariate and multivariate analyses were performed using the Cox proportional hazards regression model. The two-tailed unpaired Student t test was used to assess differences in cell growth rate, colony formation, apoptotic frequency and invading cell numbers between control vector- and BIN1-transfected HCC cells. All calculations were performed with SPSS 16.0. The results were considered significantly different when P was <0.05.
RESULTS
Expression of BIN1 in HCC
We first examined BIN1 expression status in HCC cell lines and primary HCC tissue samples with quantitative real-time PCR and Western blot analysis. All five HCC cell lines showed significantly (P < 0.05) decreased mRNA transcript levels of BIN1, as evidenced by quantitative real-time PCR analysis when using GAPDH as a loading control, compared with the normal liver cell line LO2 (Figure 1A). To verify this finding, we repeated this experiment using a different housekeeping gene, β-actin. As shown in Figure 1B, whereas the BIN1 mRNA level of the BEL-7402 cell line was comparatively high, all the other 4 HCC cell lines revealed significantly (P < 0.05) lower BIN1 levels compared with LO2. More importantly, Western blot analysis confirmed that BIN1 protein expression levels were significantly (P < 0.05) decreased in HCC cell lines (Figures 1C, D, E) when using both GAPDH and β-actin as loading controls. In 42 paired primary HCC tissue samples, decreased mRNA transcript levels of BIN1 were found in most tumor tissues (32/42) compared with adjacent nontumor tissues, as evidenced with quantitative real-time PCR analysis (P = 0.02, Figure 1F). This tendency was also verified at the protein level with Western blot analysis (Figures 1G, H).
Figure 1.

BIN1 mRNA and protein expression were remarkably reduced in human HCC cell lines and in primary HCC surgical specimens assessed with real-time quantitative PCR and Western blot. (A, B) BIN1 mRNA expression in human HCC cell lines. The normal liver cell line LO2 was used as a positive control. The BIN1 mRNA expression level was normalized to GAPDH and LO2 or to β-actin and LO2. Experiments were performed four times (*P < 0.05 compared with LO2). (C) BIN1 protein expression in human HCC cell lines. (D, E) Density of BIN1 bands normalized to GAPDH and LO2 or to β-actin and LO2. Experiments were performed in three times (*P < 0.05 compared with LO2). (F) BIN1 mRNA expression in human primary HCC tumor tissues and adjacent nontumor liver tissues (n = 42). The BIN1 mRNA expression level was normalized to GAPDH. Experiments were performed four times. The difference in the expression levels was analyzed with the paired t test (P = 0.02). (G) BIN1 protein expression in six representative human primary HCC tumor tissues (T) and adjacent nontumor liver tissues (N). (H) Density of BIN1 bands normalized to GAPDH. Experiments were performed three times. *P < 0.05 compared T (tumor tissue) versus N (adjacent nontumor liver tissue). H: ■, N; □, T.
From previous studies, the molecular cause of BIN1 downregulation may be genetic alterations, including allelic deletions, of the BIN1 gene in cancer cells. However, loss of heterozygosity (LOH) analysis showed that the frequency of BIN1 LOH in primary cancer tumors was very low, suggesting that most losses were due to epigenetic causes (6,9,22). DNA methylation events that affect promoter activity of BIN1 offer a potential mechanism for epigenetic alteration. However, CpG methylation analyses in breast and prostate tumors have demonstrated that this event was not responsible for the majority of cases of BIN1 loss in cancer cells (22). Thus, the loss of BIN1 may be due to other upstream epigenetically regulated factors (23).
Immunohistochemical Staining of BIN1 in HCC Clinical Samples
To further investigate the relationship between BIN1 expression and various clinicopathological characteristics, as well as the prognostic role of BIN1 in HCC, 117 paraffin-embedded HCC tissue samples collected between 1999 and 2001 were used for immunohistochemical analysis. We found that BIN1 expression was positive in distant normal liver tissues and adjacent nontumor liver tissues (Figures 2A, B), but its expression was decreased in tumor tissues compared with matched nontumor samples (Figure 2B). Furthermore, BIN1 expression decreased in a stepwise fashion in well-differentiated to moderately differentiated to poorly differentiated HCC tumor tissues (Figures 2C–H). Decreased expression of BIN1 (BIN1 immunostaining levels of − and +) was found in 56 of 117 samples and correlated significantly with sex (P = 0.019), tumor size (P = 0.035) and histological differentiation (P = 0.011), but not with age, cirrhosis, serum alpha fetoprotein (AFP), hepatitis B surface antigen (HBsAg), metastasis or recurrence as seen in a χ2 analysis (Table 1). In fact, we found that low expression of BIN1 had a tendency to correlate with a higher metastasis rate of HCC despite no statistic difference (P = 0.0718, Table 1). Previous BIN1 expression detection in HCC cell lines showed the same tendency, with results of immunohistochemical analysis of tumor tissues. Among the four-well or moderately differentiated and low-metastatic HCC cell lines, BIN1 expression appeared in three cell lines (Hep3B, Huh7 and BEL-7402). Loss of BIN1 expression was found in the poorly differentiated and high-metastatic cell line SK-hep1 (Figures 1A, C).
Figure 2.

Decreased BIN1 protein expression in primary HCC surgical specimens (n = 117) detected by immunohistochemistry. (A) Distant normal liver tissues. (B) Adjacent surrounding noncancerous tissues. (C, D) Well-differentiated HCC. (E, F) Moderately differentiated HCC. (G, H) Poorly differentiated HCC. N, nontumorous tissue; T, tumor tissue (original magnification 200×). Experiments were performed two times.
Table 1.
Relationship between BIN1 expression and clinicopathological features in HCC.
| Clinicopathological variables | Number of each group | BIN1 expression | P | |
|---|---|---|---|---|
|
| ||||
| Low | High | |||
| All cases | 117 | 56 | 61 | |
| Age (years) | 0.790 | |||
| < 50 | 60 | 28 | 32 | |
| ≥50 | 57 | 28 | 29 | |
| Sex | 0.019a | |||
| Male | 101 | 44 | 57 | |
| Female | 16 | 12 | 4 | |
| Tumor size (cm in diameter) | 0.035a | |||
| <5 | 45 | 16 | 29 | |
| ≥5 | 72 | 40 | 32 | |
| Histological differentiation | 0.011a | |||
| Well | 20 | 4 | 16 | |
| Moderate | 72 | 36 | 36 | |
| Poor | 25 | 16 | 9 | |
| Liver cirrhosis | 0.255 | |||
| No | 48 | 26 | 22 | |
| Yes | 69 | 30 | 39 | |
| HBV | 0.099 | |||
| Negative | 17 | 5 | 12 | |
| Positive | 100 | 51 | 49 | |
| Serum AFP | 0.065 | |||
| <25 μg/L | 30 | 10 | 20 | |
| ≥25 μg/L | 87 | 46 | 41 | |
| Recurrence (tumor appearing in 5 years) | 0.725 | |||
| No | 86 | 42 | 44 | |
| Yes | 31 | 14 | 17 | |
| Distant metastasis | 0.0718 | |||
| No | 101 | 45 | 56 | |
| Yes | 16 | 11 | 5 | |
HBV, hepatitis B virus.
P < 0.05.
Survival Analysis
For the investigation of the prognostic value of BIN1, we analyzed the relationship between BIN1 expression and patient survival. We found that the low BIN1 expression groups showed a poorer prognosis compared to the high BIN1 expression groups, as shown by the Kaplan-Meier survival analysis (P = 0.003, log-rank test; Figure 3). The univariate analysis showed that BIN1 expression, tumor size and serum AFP were significant prognostic factors (Table 2). The multivariate analyses showed that only BIN1 expression was identified as an independent risk factor for survival (P = 0.018, Table 2). These results indicated that BIN1 could serve as prognostic marker for HCC.
Figure 3.
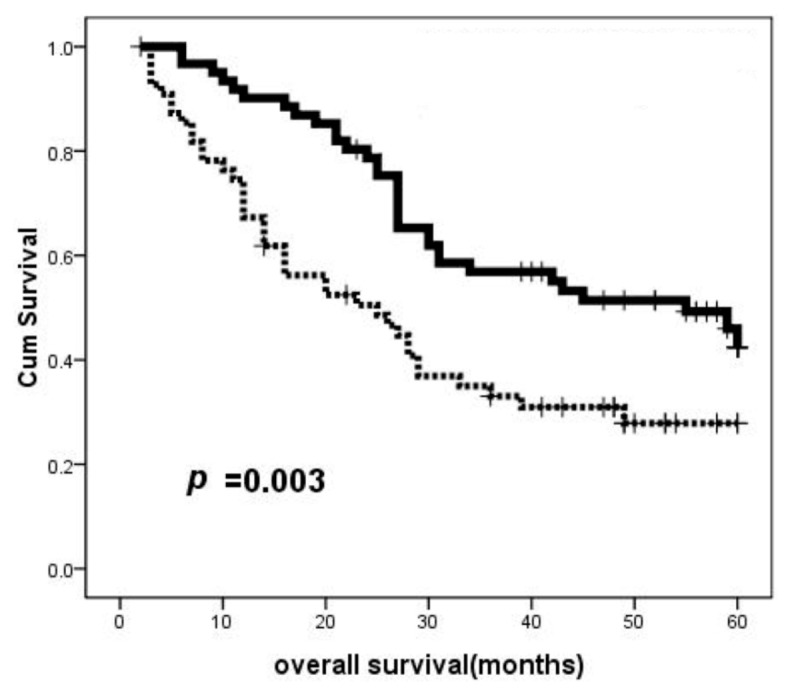
Correlation of overall survival with the expression of BIN1 in 117 HCC patients. On the basis of the results of immunohistochemical staining, log-rank test revealed that HCC patients with low BIN1 expression showed significantly poorer prognoses than patients with high BIN1 expression (P = 0.003, Kaplan-Meier method). Experiments were performed two times. Cum, cumulative. ······, low BIN1 group; ——, high BIN1 group,
Table 2.
Univariate and multivariate analysis of overall survival in HCC.
| Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| HR | 95% CI | P | HR | 95% CI | P | |
| BIN1 | 0.490 | 0.304–0.790 | 0.003a | 0.553 | 0.339–0.904 | 0.018a |
| Age | 0.900 | 0.592–1.370 | 0.624 | |||
| Sex | 0.679 | 0.341–1.353 | 0.271 | |||
| Tumor size | 1.641 | 1.051–2.562 | 0.029a | |||
| Histological grade | 1.343 | 0.955–1.887 | 0.090 | |||
| Cirrhosis | 0.782 | 0.517–1.183 | 0.244 | |||
| HBsAg status | 1.123 | 0.623–2.023 | 0.700 | |||
| Serum AFP | 1.721 | 1.036–2.861 | 0.036a | 1.406 | 0.785–2.518 | 0.252 |
| Recurrence | 1.332 | 0.860–2.061 | 0.199 | |||
| Metastasis | 1.522 | 0.859–2.696 | 0.150 | |||
CI, confidence interval; HR, hazard ratio.
P < 0.05.
Role of BIN1 in Cell Proliferation, Clone Formation, Cell Cycle and Apoptosis in Hep3B and Huh7 Cell Lines
To assess the effects of BIN1 on cell proliferation, the BIN1 expression vector and the control vector were transfected into Hep3B and Huh7 cells, respectively. Both of these cell lines had previously shown low expression of BIN1 (Figures 1A, C). The cell growth assay revealed that cell growth rates in BIN1-transfected HCC cell lines were significantly lower than those for control vector–transfected HCC cell lines (Figure 4A), but were similar to those for the normal liver cell line LO2 (Figure 6A). To further explore the tumor-suppressive effect of BIN1, clone formation was tested. The results demonstrated that the efficiency of clone formation was significantly (P = 0.013 for Hep3B, P = 0.0029 for Huh7) inhibited in BIN1-transfected HCC cells compared with control vector–transfected HCC cells (Figure 4B). To confirm the tumor suppression function of BIN1, we silenced the BIN1 expression in the LO2 cell line with siRNA. We found that silencing BIN1 expression in LO2 significantly (P < 0.05) enhanced the cell growth rate compared with mock siRNA treatment (Figure 7B).
Figure 4.

Tumor-suppressive role of BIN1 in HCC cells Hep3B and Huh7. (A) MTS assay showing the suppressive effect of BIN1 overexpression on the in vitro proliferation of HCC cell lines. Experiments were performed in triplicate. (B) Representative inhibition of colony formation in monolayer culture by BIN1. Right, quantitative analyses of foci numbers are shown as values of mean ± standard deviation. Experiments were performed in triplicate. P values were calculated using the Student t test. (C) The effect of BIN1 on cell cycle in HCC cells. The cells were transfected with BIN1 or control vector as in (A) and (B). After 48 h, the cells were collected for cell cycle distribution analysis. DNA content of BIN1 vector– or control vector–transfected cells was detected by flow cytometry. Hep-3B/Mock panel: gap 1 (G1) = 54.831, synthesis phase (S) = 23.322, gap 2 (G2) = 24.841; Hep-3B/BIN1 panel: G1 = 52.163, S = 26.885, G2 = 20.952; Huh7/Mock panel: G1 = 47.999, S = 30.405, G2 = 21.595; Huh7/BIN1 panel: G1 = 47.341, S = 29.336, G2 = 23.323. (D) Apoptosis levels of BIN1 vector– or control vector–transfected HCC cells were quantified with an annexin V and propidium iodide viability assay. The cells were transfected with BIN1 or control vector as in (A), (B) and (C). After 72 h, the cells were collected for apoptosis analysis. The percentages of annexin V– or propidium iodide–positive cells were detected by flow cytometry. Experiments were performed in triplicate. A: ●, BIN1; ■, Mock.
Figure 6.

Analysis of proliferation, migration and invasion of HCC cell lines compared with normal liver cell line. (A) Proliferation detection in HCC cell lines and normal liver cell line. The growth rate of HCC cell lines with overexpressed BIN1 was lower than the control but was similar to the normal liver cell line LO2 (*P < 0.05). (B) The migration ability of normal liver cells and BIN1 vector– or control vector–transfected HCC cells was determined by wound-healing assay. The migration ability of HCC cells with overexpressed BIN1 was lower than control and was similar to LO2. (C) Cell invasion of normal liver cells and HCC cells transfected with BIN1 or control vector was evaluated with Matrigel assay. The invasion ability of HCC cells overexpressing BIN1 was lower than control. LO2 did not show invasion. Experiments were performed in triplicate. A: ●, BIN1; ■, vector; ▲, LO2.
Figure 7.

Evaluation of proliferation, migration and invasion of LO2 transfected with control siRNA or BIN1-siRNA. (A) BIN1 mRNA detection in LO2 cells tranfected with control siRNA or BIN1-siRNA. All four BIN1-siRNAs silenced BIN1 expression effectively. *P < 0.05 compared with Mock-siRNA. We then randomly selected number 3 BIN1-siRNA for the subsequent experiments. (B) Proliferation detection of LO2 cells transfected with control siRNA or BIN1 siRNA. LO2 cells transfected with BIN1-siRNA showed a higher growth rate than control. Experiments were performed four times (*P < 0.05 compared with Mock-siRNA). (C) Migration detection of LO2 cells transfected with control siRNA or BIN1 siRNA. LO2 cells transfected with BIN1-siRNA showed a minor difference compared with the control. (D) Invasion detection of LO2 cells transfected with control siRNA or BIN1 siRNA. LO2 cells transfected with BIN1-siRNA did not alter the invasion ability. B: ●, Mock-siRNA; ■, BIN1-siRNA.
We proceeded by exploring the potential mechanism(s) underlying tumor cell growth inhibition by BIN1. We determined cell cycle distributions of control-versus BIN1 vector–transfected HCC cells with flow cytometry. As showed in Figure 4C, restored BIN1 expression did not significantly change the G1, S or G2 level of HCC cells, both in Hep3B and Huh7 cells. We then used the annexin V-FITC binding assay to explore the effects of BIN1 on apoptosis in HCC cells. Analysis of the proportion of apoptotic cells on the basis of flow cytometry revealed that apoptosis levels in HCC cells transfected with the BIN1 vector were significantly (P = 0.0094 for Hep3B, P = 0.0145 for Huh7) higher than those in cells transfected with the control vector (Figure 4D). This outcome was true for Hep3B: 29.8% (11.1% + 18.7%) versus 20.7% (10.6% + 10.1%), as well as for Huh7: 25.71% (8.71% + 17.0%) versus 17.6% (6.4% + 11.2%). These results thus suggest that BIN1 may play an important proapoptotic role in HCC development.
Previous studies demonstrated that BIN1 might affect cancer cell proliferation via multiple mechanisms. For example, BIN1 can functionally interact with Myc through its Myc binding domain (MBD) and inhibit Myc activation by recruiting the transcriptional repressor to the promoter of BIN1, resulting in inhibition of malignant cell proliferation (2). However, MBD deletion only partly relieved suppression, underscoring the importance of MBD-independent mechanisms for some types of growth inhibition by BIN1, because the N-terminal BAR-C region of BIN1 was also required to inhibit Myc transformation (2). Furthermore, other domains of BIN1, such as U1 and SH3, are required to suppress E1A or mutant p53 transformation. Thus, BIN1 could regulate malignant cell proliferation through Myc-independent as well as Myc-dependent mechanisms (2).
BIN1 mediates apoptosis of transformed primary cells also through the c-myelocytomatosis (c-Myc) pathway (15). BIN1 was not proapoptotic when overexpressed in primary cells by itself, but it instead provides a conditional linkage between c-Myc and death effector machinery (15,16). Thus, BIN1 might also regulate the HCC cell proliferation and apoptosis through the c-Myc pathway.
Role of BIN1 in Cell Migration and Invasion in HCC Cells
Finally, we assessed the role of BIN1 in HCC cell migration and invasion using wound-healing and Matrigel assays, respectively. The wound-healing assay revealed that the ability of cell migration was decreased when BIN1 was overexpressed, both in Hep3B and Huh7 cell lines (Figure 5A). In addition, BIN1 overexpression significantly inhibited cell invasion of HCC cells (P < 0.0001 and P = 0.003 for Hep3B and Huh7 cells, respectively; Figure 5B). LO2 did not show the invasion abilities in our experiment (Figure 6C). Interestingly, we found that, while the abilities of migration of HCC cell lines with overexpressed BIN1 were similar to LO2 (Figure 6B), the invasion of the BIN1-expressing HCC cells was higher than LO2, but significantly (P < 0.05) lower than HCC (see Figure 6C). However, silencing BIN1 expression in LO2 showed minimal difference, both in cell migration and invasion (Figures 7C, D).
Figure 5.

Enforced expression of BIN1 inhibited HCC cell migration and metastatic capability. (A) The migration ability of BIN1 vector– or control vector–transfected HCC cells was determined by wound-healing assay. The ability of Hep3B and Huh7 cells grown in a confluent monolayer to migrate into a linear wound created by a pipette tip was monitored for 24 h. BIN1 overexpression was confirmed by immunoblotting and the density of each band was quantitated. (B) Cell invasion of HCC cells transfected with BIN1 or control vector was evaluated with Matrigel assay. Invasive cells were stained, photographed and counted under the microscope. Right, quantitative analyses of stained cell numbers are shown as values of mean ± standard deviation. Experiments were performed in triplicate. P values were calculated using the Student t test.
DISCUSSION
Previous studies have shown that BIN1 is a potential candidate tumor suppressor involved in HCC. However, clinical implications remain undefined because of the lacking studies of human HCC tumor samples. At the same time, relative functional study of BIN1 in HCC cells is also unclear. In the present study, we evaluated the correlation of BIN1 expression level and the clinical outcome of HCC patients by using the primary HCC tissue samples (n = 117) for the first time. Furthermore, using an in vitro cell model, we also investigated the tumor suppressor role of BIN1 in HCC cells in detail. We found that the expression of BIN1 was significantly downregulated in 76% of the primary HCC tumors and in the entire HCC cell lines tested in this study, both at the mRNA level and at the protein levels. Tissue immunohistochemistry analysis also showed that decreased expression of BIN1 was found in 48% of the primary HCC samples, and downregulated expression of BIN1 significantly correlated with larger tumor size and poorly differentiated HCC. Together, these results suggest that BIN1 could be as a tumor suppressor, and alternation of this molecule may play an important role in the tumorigenesis of HCC.
In a mouse model, BIN1-null mice developed poorly differentiated tumors, characterized by reduced tubule formation, high mitotic indices and high degrees of nuclear pleomorphism (19). In human lung and lung adenocarcinoma tissues, Chang et al. found strong BIN1 expression in normal bronchial epithelia and stage I tumors. However, in case of stage II–IV lung adenocarcinoma, its expression was apparently reduced (20). This pattern was also seen in breast and prostate tumors (6,7). Our results are consistent with these studies and have suggested that the BIN1 expression might suppress the differentiation of HCC cells. As a result, the loss of BIN1 expression promotes HCC development.
Importantly, we found that patients with low BIN1 expression revealed a significantly (P = 0.003) shorter overall survival than patients with high BIN1 expression. Furthermore, we showed that BIN1 expression was an independent prognostic risk factor for HCC patients, suggesting that decreased BIN1 expression might help the characterization of HCC patients with a poor prognosis. Therefore, BIN1 may serve as a novel prognostic marker in HCC cancer patients.
Because of extremely low expression of BIN1 in Hep3B and Huh7 HCC cell lines, we transfected full-length BIN1 into these cells to further observe the functional role of this molecule in HCC. Enforced introduction of the BIN1 gene significantly suppressed cell proliferation and decreased clone formation in soft agar. In addition, silenced BIN1 expression enhanced the normal liver cell line LO2 growth rate. These studies have thus provided additional experimental evidence that BIN1 plays an important role in suppressing the development of HCCs. This result was consistent with the clinicopathological findings that decreased expression of BIN1 in HCCs was associated with larger tumor size, indicating that the loss of BIN1 may facilitate rapid HCC tumor growth.
To explore the potential mechanism for BIN1 to function as a tumor suppressor in HCC, we monitored cell cycle changes in HCC cells. Surprisingly, overexpression of BIN1 did not significantly affect the DNA content in HCC cells or cell cycle distribution compared with control cells, suggesting that BIN1 may play a minor role in the HCC cell cycle. However, we found that BIN1 is able to promote apoptosis of HCC tumor cells. This adds to the previous findings reported in melanoma, breast cancer, neuroblastoma and astrocytoma cells (5,6,13). It is known that BIN1 may interact with c-Myc and help mediate c-Myc–induced apoptosis (1–4,16). Our findings that restoring BIN1 expression induced apoptosis in HCC cells suggest that BIN1 may inhibit the proliferation of HCC cells and their colony formation through a proapoptotic pathway.
Our additional mechanistic studies showed that overexpression of BIN1 could inhibit the migration and invasion of HCC cells. These data suggest that the loss of BIN1 may favor the metastasis of HCC. These findings are consistent with our clinicopathological analysis results, which demonstrated that positive expression of BIN1 had a tendency to correlate with a lower metastasis rate of HCC. Previous studies have reported that BIN1 is linked to metastatic potential in neuroblastoma because BIN1 expression is frequently lost in metastatic neuroblastoma cell lines (21), and there was a strong trend toward lower BIN1 expression in metastatic neuroblastoma compared with localized NB (9). Similar results were also found in breast and prostate cancer studies (7,10). Thus, inhibiting the metastatic ability of tumor cells may represent another mechanism involved in the role of BIN1 as a tumor suppressor in HCC. However, for the normal liver cell line LO2, silencing BIN1 expression did not enhance the abilities of invasion. Together, our results suggest that single loss of BIN1 expression may be a prerequisite, but is not sufficient to alter the metastatic ability of cells.
CONCLUSION
In summary, in the present study, we presented evidence that BIN1 expression is significantly decreased in surgically excised HCC patient specimens as well as in HCC cell lines. We also showed that decreased BIN1 expression correlates with the degree of differentiation of HCC and predicts poor prognosis in HCC patients. Furthermore, using an in vitro model, we demonstrated that ectopic BIN1 expression could inhibit the growth, colony formation and invasion abilities of HCC cells. Of note, these results are consistent with our clinicopathological association study. Our findings thus indicate that BIN1 may be a novel prognostic marker for HCC patients, and it plays an important suppressive role in HCC development and progression.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (30973398) and Guangdong Natural Science Foundation (925100890) and was partially supported by the Gillson Longenbaugh Foundation.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Sakamuro D, Elliott K, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumor suppressor. Nature Genet. 1996;14:69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- 2.Elliott K, et al. Bin1 functionally interacts with Myc in cells and inhibits cell proliferation by multiple mechanisms. Oncogene. 1999;18:3564–73. doi: 10.1038/sj.onc.1202670. [DOI] [PubMed] [Google Scholar]
- 3.Sakamuro D, Prendergast GC. New Myc-interacting proteins: a second Myc network emerges. Oncogene. 1999;18:2942–54. doi: 10.1038/sj.onc.1202725. [DOI] [PubMed] [Google Scholar]
- 4.Prendergast GC. Mechanisms of apoptosis by c-Myc. Oncogene. 1999;18:2967–87. doi: 10.1038/sj.onc.1202727. [DOI] [PubMed] [Google Scholar]
- 5.Ge K, et al. Mechanism for elimination of a tumor suppressor: aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc Natl Acad Sci U S A. 1999;96:9689–94. doi: 10.1073/pnas.96.17.9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ge K, et al. Losses of the tumor suppressor Bin1 in breast carcinoma are frequent and reflect deficits in a programmed cell death capacity. Int J Cancer. 2000;85:376–83. [PubMed] [Google Scholar]
- 7.Ge K, et al. Loss of heterozygosity and tumor suppressor activity of Bin1 in prostate carcinoma. Int J Cancer. 2000;86:155–61. doi: 10.1002/(sici)1097-0215(20000415)86:2<155::aid-ijc2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 8.Huang H, et al. Gene expression profiling of low-grade diffuse astrocytomas by cDNA arrays. Cancer Res. 2000;60:6868–74. [PubMed] [Google Scholar]
- 9.Tajiri T, et al. Expression of a MYCN-interacting isoform of the tumor suppressor BIN1 is reduced in neuroblastomas with unfavorable biological features. Clin Cancer Res. 2003;9:3345–55. [PubMed] [Google Scholar]
- 10.Ghaneie A, et al. Bin1 attenuation in breast cancer is correlated to nodal metastasis and reduced survival. Cancer Biol Ther. 2007;6:192–4. doi: 10.4161/cbt.6.2.3587. [DOI] [PubMed] [Google Scholar]
- 11.Kennah E, et al. Identification of tyrosine kinase, HCK, and tumor suppressor, BIN1, as potential mediators of AHI-1 oncogene in primary and transformed CTCL cells. Blood. 2009;113:4646–55. doi: 10.1182/blood-2008-08-174037. [DOI] [PubMed] [Google Scholar]
- 12.Korshunov A, Sycheva R, Golanov A. Recurrent cytogenetic aberrations in central neurocytomas and their biological relevance. Acta Neuropathol. 2007;113:303–12. doi: 10.1007/s00401-006-0168-3. [DOI] [PubMed] [Google Scholar]
- 13.Galderisi U, et al. Induction of apoptosis and differentiation in neuroblastoma and astrocytoma cells by the overexpression of Bin1, a novel Myc interacting protein. J Cell Biochem. 1999;74:313–22. [PubMed] [Google Scholar]
- 14.Hogarty MD, et al. BIN1 inhibits colony formation and induces apoptosis in neuroblastoma cell lines with MYCN amplification. Med Pediatr Oncol. 2000;35:559–62. doi: 10.1002/1096-911x(20001201)35:6<559::aid-mpo14>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 15.Elliott K, Ge K, Du W, Prendergast GC. The c-Myc-interacting adapter protein Bin1 activates a caspase-independent cell death program. Oncogene. 2000;19:4669–84. doi: 10.1038/sj.onc.1203681. [DOI] [PubMed] [Google Scholar]
- 16.DuHadaway JB, Sakamuro D, Ewert DL, Prendergast GC. Bin1 mediates apoptosis by c-Myc in transformed primary cells. Cancer Res. 2001;16:3151–6. [PubMed] [Google Scholar]
- 17.DuHadaway JB, et al. Transformation selective apoptosis by farnesyltransferase inhibitors requires Bin1. Oncogene. 2003;22:3578–88. doi: 10.1038/sj.onc.1206481. [DOI] [PubMed] [Google Scholar]
- 18.Muller AJ, et al. Targeted disruption of the murine Bin1/amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol Cell Biol. 2003;23:4295–306. doi: 10.1128/MCB.23.12.4295-4306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang MY, et al. Bin1 ablation in mammary gland delays tissue remodeling and drives cancer progression. Cancer Res. 2007;67:100–7. doi: 10.1158/0008-5472.CAN-06-2742. [DOI] [PubMed] [Google Scholar]
- 20.Chang MY, et al. Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer Res. 2007;6:7605–12. doi: 10.1158/0008-5472.CAN-07-1100. [DOI] [PubMed] [Google Scholar]
- 21.Zhong X, et al. Bin1 is linked to metastatic potential and chemosensitivity in neuroblastoma. Pediatr Blood Cancer. 2009;53:332–7. doi: 10.1002/pbc.22068. [DOI] [PubMed] [Google Scholar]
- 22.Kuznetsova EB, et al. Methylation of the BIN1 gene promoter CpG island associated with breast and prostate cancer. J Carcinog. 2007;6:9. doi: 10.1186/1477-3163-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cassimere EK, Pyndiah S, Sakamuro D. The c-MYC-interacting proapoptotic tumor suppressor BIN1 is a transcriptional target for E2F1 in response to DNA damage. Cell Death Differ. 2009;16:1641–53. doi: 10.1038/cdd.2009.98. [DOI] [PubMed] [Google Scholar]