

Abstract
Tissue injury, hypoxia and significant metabolic stress activate innate immune responses driven by tumor necrosis factor (TNF)-α and other proinflammatory cytokines that typically increase damage surrounding a lesion. In a compensatory protective response, erythropoietin (EPO) is synthesized in surrounding tissues, which subsequently triggers antiinflammatory and antiapoptotic processes that delimit injury and promote repair. What we refer to as the sequelae of injury or disease are often the consequences of this intentionally discoordinated, primitive system that uses a “scorched earth” strategy to rid the invader at the expense of a serious lesion. The EPO-mediated tissue-protective system depends on receptor expression that is upregulated by inflammation and hypoxia in a distinctive temporal and spatial pattern. The tissue-protective receptor (TPR) is generally not expressed by normal tissues but becomes functional immediately after injury. In contrast to robust and early receptor expression within the immediate injury site, EPO production is delayed, transient and relatively weak. The functional EPO receptor that attenuates tissue injury is distinct from the hematopoietic receptor responsible for erythropoiesis. On the basis of current evidence, the TPR is composed of the β common receptor subunit (CD131) in combination with the same EPO receptor subunit that is involved in erythropoiesis. Additional receptors, including that for the vascular endothelial growth factor, also appear to be a component of the TPR in some tissues, for example, the endothelium. The discoordination of the EPO response system and its relative weakness provide a window of opportunity to intervene with the exogenous ligand. Recently, molecules were designed that preferentially activate only the TPR and thus avoid the potential adverse consequences of activating the hematopoietic receptor. On administration, these agents successfully substitute for a relative deficiency of EPO production in damaged tissues in multiple animal models of disease and may pave the way to effective treatment of a wide variety of insults that cause tissue injury, leading to profoundly expanded lesions and attendant, irreversible sequelae.
PATHOPHYSIOLOGY OF TISSUE INJURY
Tissue injuries caused by pathogens, trauma and hypoxia are unavoidable events in the lives of multicellular organisms. In response to these challenges, a stereotyped response has evolved that maintains a balance between inflammation and antiinflammation, characterized by six distinct stages (Figure 1). This highly orchestrated defense program is activated by the recognition of pathogen-associated molecular patterns (for example, leucine-rich repeats [1] and Toll receptor ligands [2]), as well as endogenous alarm signals (alarmins) produced by stressed cells. Alarmins are a group of molecules that include high mobility group box 1 protein (HMGB1) released by damaged cells (3) and detected by nearby immune competent cells (Figure 1, stage 1). Some form of pathogen invasion detection system is universally present in vertebrates and invertebrates (4), as well as in plants (5). These events trigger a vigorous response characterized initially by the secretion of proinflammatory cytokines (stage 2). These highly activating molecules cause the production of toxic molecules and orchestrate recruitment of local and circulating defensive cells that isolate and ultimately destroy pathogens, as well as remove damaged cells (6). Proinflammatory cytokines also isolate the site of injury by promoting vascular leakiness leading to local edema, as well as thrombosis within the surrounding vessels. Although highly effective in preventing the spread of pathogens, this innate immune response is inherently dangerous because of its self-amplifying characteristics that can result in significant or even catastrophic injury to adjacent and distant normal tissues.
Figure 1.

The innate tissue-protective response is characterized by distinct stages in which pro- and antiinflammatory cytokines and their receptors are temporally and spatially distinct. Stage 1: Pathogen-associated molecular patterns, hypoxia and molecular injury signals trigger the innate immune response driven by proinflammatory cytokines. Stage 2: Proinflammatory cytokines are produced within the injury region and diffuse into the surrounding normal tissue, priming it for self-destruction (the penumbra). Stage 3: Simultaneously, upregulation of the TPR by proinflammatory cytokines occurs within the penumbra. Macrophages recruited into the damaged area amplify injury, and the lesion progressively enlarges as the inflammatory components self-amplify and diffuse outward, causing additional damage. The high levels of proinflammatory cytokines suppress production of EPO, the ligand of the TPR. Stage 4: EPO is produced at the lesion periphery, where proinflammatory cytokine concentrations are lower, and diffuses inward, engaging the TPR, which in turn inhibits proinflammatory cytokine production and rescues cells within the penumbra from apoptosis. Stage 5: The lesion size is contained at the boundary defined by the effective inhibition of apoptosis and unrescuable cellular destruction. Stage 6: In the subacute phase, tissue receptor activation also mobilizes tissue-specific and endothelial stem cells that participate in angiogenesis and other aspects of repair. Stages 1–3 occur rapidly (minutes to hours), while stages 4–6 occur with a substantial time delay (hours to days).
To limit the uncontrolled expansion of tissue damage after activation of the innate immune system, a counterregulatory response is simultaneously triggered by inflammation or hypoxia. This protective system is characterized by a specific, spatial-temporal expression pattern of protective, antiinflammatory molecules and responses in cells surrounding the region of damage that act to stop the expansion of cellular damage from the central region of injury. A primary characteristic of this program is expression of a specific tissue-protective receptor (TPR) (stage 3) and, after a significant time delay (hours) (7), expression of its ligand, which is now known to be erythropoietin (EPO) (stage 4). EPO signals through the TPR to inhibit proinflammatory cytokine production (8), inhibit macrophage activity (9) and delimit the volume of injury by counteracting apoptosis (10) (stage 5). The TPR also acts to recruit vascular-and tissue-specific stem cells (11) and enhances tissue repair (12) (stage 6).
Because of the spatial and temporal characteristics of this system, and the relative deficiency of the endogenous TPR ligand during the early, critical phase of injury, nearby normal tissue is at high risk for destruction. This susceptibility can be obviated by administration of exogenous TPR ligands. An example of these stages of tissue injury after cerebral ischemia and the beneficial effects of exogenous EPO is illustrated in Figure 2. In extreme conditions in which systemic levels of proinflammatory cytokines are elevated (for example, sepsis caused by perforation of the gut [12] or cerebral malaria [13,14]), EPO directly suppresses proinflammatory cytokines and improves survival. It is also important to appreciate that tissue responses to inflammation and hypoxia are intimately intertwined and, as a result, activate similar responses. A key variable is oxygen availability. When oxygen is depleted or unavailable prolyl hydroxylase activity is decreased, the hypoxia inducible factor is activated. Subsequently, a large number of gene responses are activated, including the synthesis of EPO. Inflammation, acting through nuclear factor (NF)-κB, also cross-modulates the hypoxia inducible factor, and both response arms integrate into a net response that limits injury and enhances tissue recovery (15,16).
Figure 2.

Administration of EPO rescues the penumbra after cerebral ischemia. An ischemic core (IC; panel E) is caused by a 1-h occlusion of the middle cerebral artery distal to the rhinal artery in a rat, followed by reperfusion (stage 1 injury response, performed according to the methodology seen in references 8 and 81). TNFα and other proinflammatory cytokines are produced within this central volume of injury corresponding to the circulatory territory of the middle cerebral artery (MCA) and diffuse into the adjacent cerebral cortex to the right of IC (stage 2 injury response). Microglia (the macrophage equivalents of the nervous system) are recruited into the penumbra (right panels) and amplify injury. Within the penumbra, the TPR is subsequently upregulated (stage 3 injury response), but upregulation of its ligand EPO is suppressed. Therefore, EPO does not penetrate very far into the penumbra from the unsuppressed periphery, producing a large lesion when evaluated 24 h later (light gray area, panel S). However, parenteral administration of EPO (panel E) rescues the penumbra by delivering TPR ligand into this region primed for TPR activity but deficient in endogenous EPO.
ERYTHROPOIETIN
Erythropoietin (EPO) is a member of the type I cytokine superfamily that is characterized by relatively rigid globular glycoproteins with four α helices bound together by hydrophobic interactions (17). EPO-like molecules have been documented in all vertebrate lineages (18); invertebrates also have EPO-like molecules and even respond beneficially to mammalian EPO in the setting of injury (19). This 165–amino acid protein has long been appreciated for its indispensible role in the maintenance of an adequate erythrocyte population and, therefore, is required for optimal tissue oxygenation, function and survival. To accomplish this in the adult mammal, renal tubular interstitial cells detect relative hypoxia as the red cell number declines. These kidney cells then synthesize and release EPO into the circulation in a classic endocrine negative feedback control system to travel to the bone marrow and prevent programmed cell death of proerythrocytes. Low sustained concentrations of EPO are required for effective erythropoiesis because the high-affinity EPO receptor mediating red cell survival is expressed by proerythrocytes only during a brief time period during maturation (20). This interaction is facilitated by the fact that renal EPO is a highly glycosylated protein with a molecular weight of 30.4 kDa (~34 kDa on the basis of sodium dodecyl sulfate–polyacrylamide gel electrophoresis), having four sialic acid–capped oligosaccharide chains that provide a plasma half-life of 5–6 h in humans. A small pool of erythrocyte precursors (burst-forming unit erythroids and colony-forming unit erythroids) in the bone marrow continuously become responsive through the expression of the EPO receptor, but are programmed to undergo apoptosis unless EPO is present (21). In this way, the continuous loss of senescent erythrocytes (~1% each day) is precisely replaced by newly matured red cells so as to maintain equilibrium and avoid tissue hypoxia. In contrast, EPO synthesized by the brain (including the retina) is poorly sialated (22) and subsequently has a short circulating half-life, consistent with a paracrine-autocrine function. EPO synthesized locally outside of the nervous system is also presumably not highly sialated, but this hypothesis has not yet been confirmed. Removal of the oligosaccharide chains or the terminal sialic acids of EPO reduces the circulating half-life of EPO to only several minutes, which is not sufficient to support erythropoiesis (23).
EPO RECEPTOR ISOFORMS
Similar to other members of the type I cytokine superfamily, the proerythrocyte EPO receptor consists of subunits that self-assemble to form an active receptor (24). The hematopoietic receptor consists of two identical EPO receptor monomers (EPORs) that if present in the cell membrane spontaneously dimerize when within close mutual proximity (25), forming EPOR-EPOR [(EPOR)2] (Figure 3). As with other type 1 cytokine receptors, (EPOR)2 is not self-phosphorylating, but instead uses physically associated Janus kinase-2 (JAK-2) to trigger intracellular signal cascades. Signaling occurs when EPO binds in a manner that bridges the extracellular portions of each EPOR subunit and causes a small conformational change, activating JAK-2 auto-phosphorylation. This signal then cascades through several distinct molecular pathways, including signal transducers and activators of transcription (STAT) and ultimately Bcl-X (20), which activate an antiapoptotic program in proerythrocytes. Renal EPO has a high affinity (~200 pmol/L) for this receptor, and because only a few percent occupancy is needed for adequate signaling (26), the normal human serum EPO concentration is in the 1–10 pmol/L range.
Figure 3.

EPO uses receptor isoforms for signaling. A homodimer receptor (EPOR)2 is formed by the spontaneous self-assembly of EPOR monomers and mediates erythropoiesis (left). The receptor-ligand stoichiometry is 2:1 for EPOR and EPO, respectively. The hematopoietic receptor is characterized by a high affinity for EPO and requires constant, low circulating concentrations of EPO to maintain adequate erythrocyte production. Other biological activities controlled by (EPOR)2 are those associated with preservation of red cell mass in the setting of hemorrhage. These functions include induction of a prothrombotic state by activation of vascular endothelium adhesion molecules and accelerated maturation of megakaryocytes, as well as stimulation of vascular smooth muscle leading to shunting of blood from noncritical tissues. All of these activities serve to minimize blood loss. In contrast, available evidence supports the existence of a different receptor for tissue protection (middle) that involves assembly of EPOR and βCR subunits, as well as other receptor subunits (right; for example, VEGFR2) in certain tissues. On the basis of analogy to the GM-CSF receptor, the stoichiometry may be in a 2:2:2 ratio for EPO, EPOR and βCR. The TPR is characterized by a lower affinity for the endogenous ligand EPO and requires only brief receptor occupancy to initiate long-lasting biological effects. Because high doses of EPO are required for TPR activation, tissue protection by exogenous EPO is unavoidably accompanied by these potential serious adverse consequences. To circumvent these potential problems, analogs of EPO have been engineered that lack hematopoietic potency but retain tissue-protective activity. This strategy provides tissue protection while avoiding the significant potential side effects of using EPO itself.
The endothelial cell was the first non-hematopoietic cell noted to respond to EPO after it was observed that EPO caused cell mitosis and migration. However, the effective concentrations of EPO needed for this activity (1–2 nmol/L) were significantly higher than required for hematopoiesis (27). Subsequently, the nervous system (28), the kidney (29) and the heart (30) were also shown to respond to EPO, but similar to endothelial cells, required higher EPO concentrations than that needed for red cell production.
Available evidence indicates that the EPO receptor involved in tissue protection (TPR) is different from the receptor used for signaling in the hematopoietic system (Figure 3). Isolation of the EPO receptor expressed by neuronal-like cells revealed different molecular weight components, including distinct accessory proteins when compared with erythrocyte precursors (31). In contrast to low pmol/L concentrations of EPO required for erythropoiesis, tissue protection in vivo and in vitro requires nanomolar concentrations that normally are not achieved within the circulation (32). Rather, local EPO production via a typical paracrine-autocrine system in a variety of cells results in higher local concentrations within the TPR affinity range. The large differences in affinity of EPO for the erythropoietic and TPRs reduce the possibility of cross-talk between the endocrine and paracrine systems of EPO. As noted above, the 1–10 pmol/L plasma EPO concentration is well below the TPR affinity of 2–20 nmol/L (rev. in 32).
An additional line of investigation also suggested the existence of a TPR distinct from the erythropoietic receptor. Considering the well-established regions of interaction involved in binding of EPO to (EPOR)2, we and our collaborators generated a number of EPO molecules modified so as not to bind to the critical two regions of the preassembled homodimeric receptor. Therefore, these EPO derivatives were nonerythropoietic (33). For example, reacting EPO with cyanate chemically modifies positively charged lysines, including those within the receptor binding sites, to the neutral homocitrulline residue, producing carbamylated EPO (CEPO), which is not erythropoietic (34). However, testing CEPO in in vitro and in vivo models revealed that this compound is also a tissue-protective molecule with a potency equivalent to that of EPO (33). These observations clearly implicated another receptor in mediating the biological activity of CEPO. To identify this receptor, our group immobilized CEPO on a matrix and passed over it membrane fractions obtained from a variety of rat tissues known to exhibit EPO-mediated tissue protection (35). A retained protein fraction was then subjected to immunological analysis and shown to consist of the EPOR subunit and the β common receptor (βCR; also known as CD131), linked by a disulphide bond (35). The βCR is so called because it is shared by the receptors of the hematopoietic type 1 cytokines granulocyte macrophage–colony-stimulating factor (GM-CSF), interleukin (IL)-3 and IL-5 (rev. in 36). However, compared with the other known cytokine-specific receptor subunits, βCR is a much larger receptor, having a duplicated extracellular portion characteristic of type 1 cytokines, in addition to a long intracellular tail. The βCR binds to the specific cytokine α receptor subunits through disulphide bonds in a manner that confers ligand specificity (37,38), similar to what we observed for EPOR. The βCR functions as the signaling receptor subunit for GM-CSF, IL-3 and IL-5 and is known to activate a wide variety of biological responses.
As a historical note, an EPOR-βCR receptor was first hypothesized to play a role in hematopoiesis, on the basis of the observation that EPO signaling resulted in the phosphorylation of βCR in hematopoietic cells (39) and that EPOR was physically associated with the βCR in cells transfected with the EPOR and βCR subunits (40). However, a mouse model in which the βCR as well as the IL-3–-specific β receptor (a receptor isoform in the mouse, but not other mammals, that is highly homologous to βCR [41]) were knocked out showed no effects on hematopoiesis (42). Subsequently, it has become clear that the βCR-EPOR hetero-complex is used by other tissues in the tissue-protective response. A variety of tissues that have been examined for expression of βCR and EPOR, including the central (Figure 4A [35]) and peripheral (43) nervous systems, retina (44), heart (45), kidney (46), muscle (47) and the endothelium (48). Additionally, components of the immune system that are resident or migrate to regions of injury also highly express subunits for the TPR as, for example, specific subsets of macrophages (9).
Figure 4.

The TPR is characterized by expression of βCR and EPOR. (A) A variety of neurons within rat spinal cord ventral horn express both βCR and EPOR. (B) In this experiment, wild-type rat cardiomyocytes undergo apoptosis after exposure to stuarosporine in vitro (column WT). Addition of EPO protects from staurosporine (WT+EPO). In contrast, cardiomyocytes obtained from βCR KO (βc KO) animals are not protected by EPO (βc KO+EPO). (****P < 0.001 versus staurosporine alone; reproduced with adaptation from [35]).
Current evidence shows that the βCR predominantly exists on the cell surface as a dimer (49). Because the self-assembly of type 1 cytokine receptors occurs in direct proportion to the probability of interaction within the membrane (that is, on the density of receptor subunits), the probability of formation of βCR-EPOR heteromers is presumably favored by a relative surplus of βCR. This assembly is enhanced within lipid rafts, specialized regions of the cell membrane in which the receptor subunits (for example, IL-3 and βCR), among other proteins, are concentrated (50). It is also known that the α receptor subunits that provide ligand specificity for other members of the type 1 cytokine receptor family increase the probability of complexing with βCR in the presence of the ligand (51,52). CEPO, for example, has been shown to greatly increase the amount of complexed EPOR and βCR (45). Finally, in those circumstances in which EPOR is not colocalized with βCR, EPOR will self-associate and form (EPOR)2, which can also support signaling. For example, cultured neuronal-like cell lines that express only EPOR have been reported to exhibit cytoprotection by exposure to EPO (53).
As predicted by the heteroreceptor structure of βCR-EPOR, EPOR gene expression is required for brain development and full protection from tissue injury. Knockout (KO) of EPOR is embryonic lethal at 13.5 d because of ineffective erythropoiesis, and the phenotype exhibits massive cardiac developmental abnormalities (54). However, a conditional EPOR KO was constructed using a promoter that drives EPOR expression only in hematopoietic tissue. This genotype survives, but in addition to adequate red cell production, the vascular endothelium (which is derived from a hemato poietic lineage) also expresses EPOR (55), as well as βCR (56). Other tissues in the conditional KO, however, have been reported to lack EPOR expression. In tissues such as the brain and heart with parenchyma devoid of the EPOR subunit, resident vessels express both EPOR and βCR and therefore would be expected to respond to the tissue-protective effects of EPO. In this manner, preservation of vascular endothelium is in itself sufficient to salvage tissue in some injured organs (55,57). In contrast, silencing βCR function by gene KO or using neutralizing antibodies abolishes the tissue-protective properties of EPO (and nonerythropoietic derivatives). For example, in a mouse model of spinal cord injury, EPO is highly efficacious in restoring motor function after a 1-min cord compression, whereas mice in which βCR has been knocked out (35) are not protected. Similar effects were documented in vitro for cardiomyocytes (Figure 4B) and for neuropathic pain caused by sciatic nerve ligation (58). A biological role for βCR in tissue injury has been established by a number of other techniques than by gene KO to silence the βCR protein. For example, Su et al. (48) showed using the endothelium that blocking either βCR or EPOR by neutralizing antibodies or siRNA technology neutralized the ability of EPO to activate intracellular signaling cascades and ultimately the beneficial production of nitric oxide. Additionally, use of an anti-βCR antibody blocked the ability of EPO to promote endothelial cell proliferation, migration and tube formation.
DEVELOPMENT OF TISSUE-PROTECTIVE RECEPTOR–SPECIFIC LIGANDS
Although the endogenous molecule EPO signals through both the hematopoietic and TPRs as outlined above, receptor specificity is conferred by differences in receptor affinity and by the receptor concentrations within and around a region of injury. However, when EPO is administered parenterally for tissue protection, effective systemic doses unavoidably fully saturate the hematopoietic receptor. In addition to red cell production, the homodimeric receptor when activated can also mediate a number of functions associated with thrombosis and endothelial cell activation. For example, EPO acutely stimulates platelet production, increasing the number of young, highly reactive platelets within the circulation. Additionally, EPO increases E- and P-selectin expression (59,60) and therefore the likelihood of thrombosis. The existence of a receptor isoform with a lower affinity for EPO raised the possibility that EPO could be modified so as not to bind to the hematopoietic receptor, but yet still retain tissue-protective activities. A number of such compounds have been designed and validated. Modification of the EPO protein (for example, CEPO [33] or S100E EPO [61]) or peptides on the basis of the three-dimensional structure of EPO (62) results in tissue-protective molecules as potent as EPO but devoid of erythropoietic and hematopoietic activities.
EPO signaling via the TPR is similar in many respects to that used by the erythropoietic receptor. However, the temporal aspects of ligand and receptor expression lead functionally to very different results. Similar to erythropoiesis, if a cell expresses the appropriate receptor type and EPO is available, a sustained gene activation program triggers long-lasting biological effects (for example, antiapoptosis). Thus, in tissue protection, EPO acts, in effect, as a binary switch, activating sustained survival programs. In one example among many, a pioneering experiment on the tissue- protective properties of EPO demonstrated that neurons treated in vitro with neurotoxic glutamate analogs required only a 5-min exposure to EPO to provide a total and enduring protective effect (63). Similar actions occur in vivo. For example, both asialoEPO and a small peptide mimic of EPO (pyroglutamate helix B surface peptide [pHBSP] [62]) have plasma half-lives of only ~2 min. Yet both are highly effective in preventing injury in a wide range of neurological and other acute injuries after only a single parenteral dose (32,64–66).
Another example of long-lasting tissue-protective memory induced by TPR ligands was shown by the ability of EPO to antagonize wheal formation produced by intradermally administered histamine, a classic inflammatory response. After injection, histamine acts through its receptor on endothelial cells to cause a breakdown of intercellular barriers and a leakage of intravascular contents into the surrounding tissues. This localized edema can be visualized and quantified in rats by injecting Evans blue dye into the circulation (which binds to albumin) and measuring the extent of extravasation. Additionally, endothelial cells are one cell type that express the TPR constitutively (48,67) and therefore are primed to respond to tissue-protective molecules. To demonstrate the persistence of a tissue-protective effect, histamine can be injected at specific times after administration of a TPR ligand. For example, after intravenous administration of the tissue-protective peptide pHBSP (30 μg/kg), an intradermal dose of 0.12 μg histamine was injected into the skin at various time points, and the extent of dye leakage was quantified by planimetry performed on digital photographs obtained after 15 min, when wheal volume peaks (Figure 5A). The results show that a single dose of the tissue-protective molecule pHBSP (35), although having a serum half-life of only 2 min, suppresses wheal formation, in a sustained, statistically significant effect for >24 h (Figure 5B).
Figure 5.
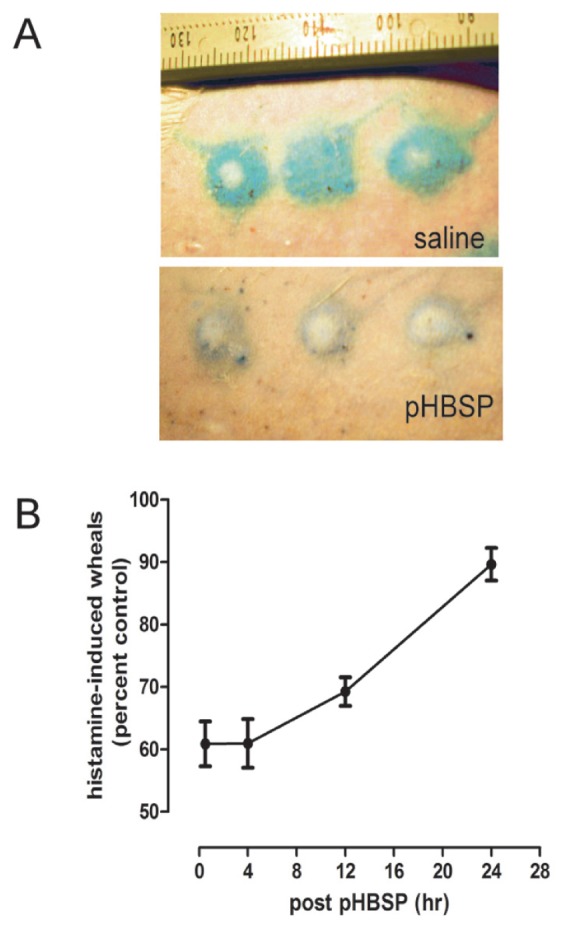
The TPR acts a molecular switch activating long-lasting tissue protection. Histamine injected intradermally activates histamine receptors that within several minutes cause intense vasoconstriction at the site of injection (wheal) that is surrounded by extravasated intravascular proteins (flare). (A) This reaction can be quantified by labeling albumin with Evans blue dye. As an example, a rat that received saline 4 h earlier exhibited a vigorous response to histamine (top). In contrast, a rat that received pHBSP 4 h earlier responded with substantially less vascular extravasation, as indicated by the smaller region less intensively labeled with Evans blue (bottom). (B) A single dose of the tissue-protective molecule pHBSP (30 μg/kg) exerts long-lasting effects, antagonizing histamine administered hours later (top; ± SEM). (Please see text for details.)
MECHANISMS OF ACTION
Multiple signaling pathways activated by the TPR have been documented in experimental studies. As noted above, a number of these pathways are similar to those used in the hematopoietic system (Figure 6). Because EPO signals through two receptor isoforms, to identify molecular pathways that are specifically associated with the TPR, availability of nonerythropoietic tissue-protective compounds (primarily CEPO and pHBSP) has been essential. It is important to note that with respect to signaling studies, asialoEPO is not useful, since it binds to (EPOR)2 with a higher affinity than EPO (it is not erythropoietic in vivo because of its very short circulating half-life).
Figure 6.

Multiple signaling pathways have been documented for the TPR. A variety of signaling pathways activated by the TPR have been determined using the nonerythropoietic tissue-protective compounds CEPO and pHBSP. Tissue-protective receptor occupancy leads to JAK-2 phosphorylation which subsequently activates a number of divergent cascades. In some tissues, these pathways overlap and are therefore redundant; but in others, each appears to serve specific functions. Both enhanced (blue boxes) and suppressed (red boxes) responses have been identified (see text for discussion).
A number of investigators have determined the TPR signaling pathways using a variety of in vivo and in vitro models. As in other type 1 cytokine signaling systems, the first step following receptor occupancy is the autophosphorylation of JAK-2. However, EPO also has been reported to signal in the kidney directly through phospholipase C and voltage- independent calcium channels (68), but this has not been examined to date using nonerythropoietic tissue-protective compounds. This effect could explain the hypertension that sometimes complicates EPO therapy of renal failure patients and, if true, likely depends on (EPOR)2. After JAK-2 phosphorylation, three dominant pathways have been identified. First, similar to erythropoiesis, the STAT pathway (involving STAT3 (heart [69,70]) and STAT5 (brain [71]) are activated, resulting in upregulation of survival signals and subsequent blockade of inflammation-induced apoptosis. Additionally, the STAT pathways activate tissue restorative functions, including neural stem cell differentiation and neurite outgrowth (71). A second major molecular pathway involves phosphatidylinositol 3-kinase (PI3K) and Akt (also known as protein kinase B), which is active in the heart (45,69,70) as well as in the liver and kidney (72). A third pathway also used by the heart, kidney and liver uses mitogen-activated protein kinases (MAPKs) (69,70,72). Both the PI3K and MAPK pathways inhibit glycogen synthase kinase 3β (GSK3β), which then directly inhibits the mitochondrial transition pore, leading to stabilization of the mitochondrion and subsequent inhibition of death signals and apoptosis (73). Additionally, effects of the MAPK and PI3K systems ultimately reduce inflammation by inhibiting NF-κB. MAPKs have also been shown to be a downstream pathway of sonic hedgehog, which is activated by TPR ligands (74). The PI3K system has also been shown by a number of groups to be critically involved in the activation of endothelial nitric oxide synthase and therefore the generation of nitric oxide by the endothelium (48,67,72). The biological consequences are vasodilation and the modulation of regional blood flow, as well as the recruitment of endothelial progenitor cells that participate in neo-angiogenesis and therefore promote wound healing.
However, within the paracrine-autocrine tissue protection system, the inflammatory and antiinflammatory arms also cross-modulate their respective ligands and their receptors, so that the final outcome depends strongly on the immediate past of the system. An initial phase of the innate immune response (which consists of proinflammatory cytokine production) strongly upregulates the TPR (Figure 1, stages 2–3). For example, tumor necrosis factor receptor 1 (TNFR1) is required for upregulation of EPOR as well as enabling the therapeutic effects of EPO in the mouse (75). Therefore, TNFR1 KO mice exhibit greater injury after brain ischemia (76), and as predicted, do not respond to EPO when exposed to a neurotoxin (75). As another example of proinflammatory cytokine production enabling tissue-protective responses, tumor necrosis factor (TNF)-α exposure primes cerebral vascular endothelial cells to respond to EPO (and CEPO) by initiating angiogenic responses (77).
In this paradigm, cells express the TPR in the immediate vicinity of injury where TNFα concentrations are high (Figure 1, stage 3). However, because TNFα also suppresses EPO expression, the cells surrounding an injury are primed to respond, but are lacking a ligand. In addition to the counter-modulatory effects of the pro- and antiinflammatory cytokine arms of the innate immune response, there are important temporal aspects that orchestrate the response. Of note, in the tissue-protective system, the receptor is expressed first, and subsequently within an hour of delay (78), EPO, its endogenous ligand, is expressed predominantly distally to the locus of injury where suppressive TNFα levels drop off (7). Furthermore, EPO expression suppresses TNFα expression. Early after injury, potentially viable cells surrounding the site express TPR, but there is little local EPO production. This result provides a window of opportunity where administration of EPO or a nonerythropoietic derivative (see below) can salvage tissue otherwise destined for self-destruction. This seemingly discoordinated program serves the purpose to allow destruction of infectious agents and damaged tissue at the injury site and prevents its spread. Yet, the swath of healthy tissue sacrificed in this process is what we recognize in the modern world as the sequelae of disease or injury.
However, in one experimental system, knockout of the βCR was reported not to abolish the tissue-protective effects of EPO. It should be noted that silencing the βCR can in itself produce an acute major reduction in injury in the mouse. For example, in a mouse model of transient coronary arterial occlusion (79), βCR KO was associated with complete resistance to a temporary occlusion of the coronary artery; permanent occlusion was required to produce significant injury. The hyperglycosylated EPO derivative darbepoietin (which has two additional oligosaccharide chains to increase its plasma half-life) was observed to have a small protective effect after permanent occlusion. This puzzling observation may be explained by the fact that the mouse has two different β receptors (42). One is common to GM-CSF, IL-3 and IL-5; the other is used only by IL-3 signaling. In βCR KO animals, IL-3 signaling is normal, whereas GM-CSF and IL-5 are completely absent. Ordinarily, a great amount of transmodulation and even transactivation exists among IL-3, IL-5 and GM-CSF (80). Animals that lack GM-CSF, IL-5 or βCR have been noted to be highly resistant to acute injury but nevertheless exhibit pulmonary pathology in the long term. It is likely that these effects can be explained by the modulation of specific leukocyte subsets such that no injury is imparted early in the KO animals, but there is an absence of an attenuation of injury in the subacute and chronic time periods.
THERAPEUTIC WINDOW OF OPPORTUNITY: IMPORTANCE OF RECEPTOR “PRIMING”
As previously discussed, in stage 3 of the injury cascade, cells surrounding the lesion are exposed to inflammatory signals that lead to the upregulation of the TPR. These cells are simultaneously programmed for apoptosis that occurs with a delay of a few hours and represents a significant volume of tissue surrounding an injury (the “penumbra”) that is potentially viable. Therefore, tissue protection can be characterized as a window of opportunity in which tissue-protective molecules can salvage cells from apoptosis. For example, in experimental stroke, EPO is effective in reducing the ultimate infarct volume if administered up to 6 h after occlusion (81). In terms of behavioral outcomes, however, the time window is much larger. EPO and its derivatives have been demonstrated to have beneficial effects when first administered as long as several days after injury. For example, in a rodent stroke model, the nonerythropoietic tissue-protective molecule CEPO has been shown to provide benefits including reduced lesion volume, attenuated neuronal losses, enhanced angiogenesis and neurogenesis when administered as late as 48 h after injury (82).
It appears to be a general characteristic of tissue injury that the TPR is not expressed in normal tissues, but only by those exposed to injury signals or metabolic stress. One exception is the hippocampus, which expresses abundant amounts of EPOR. As a central component of the neural substrate of memory, exposure to EPO (including human subjects [83]) or nonerythropoietic derivatives has been reported to markedly affect these processes. For example, CEPO was reported to enhance learning behavior as well as associated hippocampal neurogenesis at least as well as EPO in mice (84). In the central nervous system, therefore, tissue-protective molecules can affect biological function in the normal organism. Another cell type that expresses both EPOR and βCR at baseline are tissue macrophages (9). These cells are therefore “primed” for activation by tissue-protective ligands and may explain the ability of EPO to reduce the level of proinflammatory cytokines produced by macrophages, thus reducing the degree of further tissue damage. A third cell type that expresses TPR at baseline is the endothelial cell (48).
In other tissues, however, the TPR is not expressed and requires priming by injury. This observation can be illustrated by the use of tissue-protective compounds that have a very short serum half-life and, therefore, allow for temporal probing of receptor expression. For example, receptor expression, as estimated by the level of the EPOR subunit component of the TPR, is low in normal spinal cord, being expressed only in metabolically active neurons (Figure 4A) and capillary endothelial cells in relatively hypoxic white matter, accounting for enhanced learning/memory in normal animals by TPR ligands. In a rat model of spinal cord injury, after compression, there is a marked increase in EPOR immunoreactivity within the gray matter of the spinal cord (Figure 7A). In this model, administration of the short half-life EPO analog asialoEPO in advance of a compressive spinal cord injury (and documented by analysis of the serum to be absent at the time of injury) has little beneficial effect on compressive injury. In contrast, when given 1 min after injury, a significant protective affect equivalent to that of EPO was observed (Figure 7B).
Figure 7.
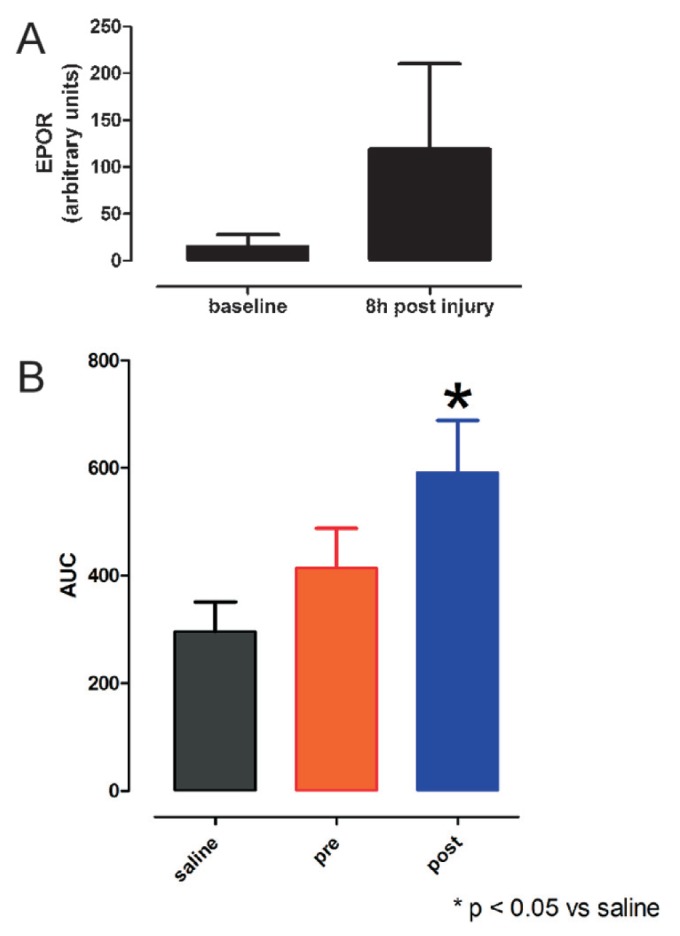
Tissue protection depends on upregulation of the TPR. (A) Rat spinal cord does not express high levels of EPOR, as estimated by immunocytochemistry. However, within a few hours after a 1-min compression of the spinal cord at level thoracic 3 (T3), EPOR immunoreactivity more than quadruples (± SEM). (B) AsialoEPO is a tissue-protective molecule derived by removing the terminal sialic acids of the oligosaccharide chains of EPO, which results in a circulating half-life of only ~2 min in vivo. However, tissue-protective activity of asialoEPO is similar to that of EPO. AsialoEPO (50 μg/kg) administered intraperitoneally 24 h before a 1-min compression at T3 in rats is associated with little protection, as assessed by a motor score derived by the area under the curve (AUC) for weekly measurements carried out to 28 d. In contrast, when asialoEPO was administered immediately after release of compression, there was a significant improvement in the motor score compared with control animals (± SEM; drawn from data presented in [87]).
βCR FORMS RECEPTOR COMPLEXES THAT INCLUDE NONCYTOKINE RECEPTORS AND OTHER PROTEINS
Recent work has shown that in addition to binding to other type 1 cytokine receptor subunits, βCR also forms complexes with noncytokine receptors as well as other proteins, including enzymes. One of the prominent effects of EPO on vascular endothelium is modulation of nitric oxide production, which has been shown to occur in large part via endothelial nitric oxide synthase. Recently, studies carried out on hypoxic endothelial cells have shown that the noncytokine receptor for vascular endothelial growth factor (VEGFR2), which is stimulated under hypoxic conditions and is highly active in neoangiogenesis, becomes an additional component of the EPOR-βCR complex. Further, this receptor subunit is essential for the production of nitric oxide (67). Another aspect of this receptor configuration has been shown by Su et al. (48) to involve endothelial nitric oxide synthase itself binding to βCR, which is required for the integration of EPO-mediated nitric oxide production. Very recently, this research group showed that AMP-activated protein kinase is also an essential component of the endothelial cell nitric oxide system (85). Further, it is of interest to note that in other cellular systems, βCR has also been described as associated with VEGFR2. For example, the βCR of the IL-3 receptor was reported to associate with VEGFR2, specifically in lipid rafts (50). The βCR-EPOR was also recently shown to directly activate NF-κB, a key proinflammatory cytokine modulator, via a direct interaction with TNF receptor–associated factor 6 (TREAF6) (86). These variations potentially provide increased complexity of function.
RECAPITULATION AND CONCLUSIONS
The innate immune response is an evolutionarily ancient and primitive self-defense mechanism that works to preserve survival after injury because discoordinated regulation of interrelated pro- and antiinflammatory cascades happen to turn on and then eventually off, sterilizing a large swath of otherwise healthy tissue surrounding the primary lesion. The dark side of this ancient survival-oriented system is that sterile and nonsterile lesions, once activated, can result in excessive tissue damage. What we know as the sequelae of injury and disease are often the consequences of this “scorched earth” strategy to destroy pathogens at the expense of a significant collateral damage to normal tissues. In recent years, an inflammation-attenuating system driven by locally produced EPO was discovered. The βCR has emerged as a central player in the reduction of inflammation and tissue injury when it is associated with EPOR. However, the relatively stereotyped defensive program is also characterized by a relative deficiency of local EPO. Development of TPR-specific, non-erythropoietic EPO mimetics that can be administered to treat the EPO deficiency offers great promise in limiting penumbral tissue losses and maximally preserving function.
ACKNOWLEDGMENTS
We thank Michael Yamin for his comments and suggestions about this manuscript.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
M Brines is an employee of Araim Pharaceuticals and is a coinventor of the tissue-protective technology described in this review. A Cerami is an inventor of the intellectual property described in part of this paper and is also the CEO of Araim Pharaceuticals that holds the patents.
REFERENCES
- 1.Kedzierski L, Montgomery J, Curtis J, Handman E. Leucine-rich repeats in host-pathogen interactions. Arch Immunol Ther Exp (Warsz) 2004;52:104–12. [PubMed] [Google Scholar]
- 2.Janssens S, Beyaert R. Role of Toll-like receptors in pathogen recognition. Clin Microbiol Rev. 2003;16:637–46. doi: 10.1128/CMR.16.4.637-646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dzik JM. The ancestry and cumulative evolution of immune reactions. Acta Biochim Pol. 2010;57:443–66. [PubMed] [Google Scholar]
- 5.Huffaker A, Pearce G, Ryan CA. An endogenous peptide signal in Arabidopsis activates components of the innate immune response. Proc Natl Acad Sci U S A. 2006;103:10098–103. doi: 10.1073/pnas.0603727103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. 2011;108:1133–45. doi: 10.1161/CIRCRESAHA.110.226936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernaudin M, et al. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:643–51. doi: 10.1097/00004647-199906000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Villa P, et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med. 2003;198:971–5. doi: 10.1084/jem.20021067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nairz M, et al. Erythropoietin contrastingly affects bacterial infection and experimental colitis by inhibiting nuclear factor-kappaB-inducible immune pathways. Immunity. 2011;34:61–74. doi: 10.1016/j.immuni.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siren AL, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A. 2001;98:4044–9. doi: 10.1073/pnas.051606598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koh SH, Noh MY, Cho GW, Kim KS, Kim SH. Erythropoietin increases the motility of human bone marrow-multipotent stromal cells (hBM-MSCs) and enhances the production of neurotrophic factors from hBM-MSCs. Stem Cells Dev. 2009;18:411–21. doi: 10.1089/scd.2008.0040. [DOI] [PubMed] [Google Scholar]
- 12.Erbayraktar Z, et al. Nonerythropoietic tissue protective compounds are highly effective facilitators of wound healing. Mol Med. 2009;15:235–41. doi: 10.2119/molmed.2009.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaiser K, et al. Recombinant human erythropoietin prevents the death of mice during cerebral malaria. J. Infect. Dis. 2006;193:987–95. doi: 10.1086/500844. [DOI] [PubMed] [Google Scholar]
- 14.Wiese L, Hempel C, Penkowa M, Kirkby N, Kurtzhals JA. Recombinant human erythropoietin increases survival and reduces neuronal apoptosis in a murine model of cerebral malaria. Malar J. 2008;7:3. doi: 10.1186/1475-2875-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rius J, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–9. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jelkmann W. Molecular biology of erythropoietin. Intern Med. 2004;43:649–59. doi: 10.2169/internalmedicine.43.649. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Ney P. EPOR signaling: 450 million years’ history. Blood. 2007;110:2225–6. [Google Scholar]
- 19.Ostrowski D, Ehrenreich H, Heinrich R. Erythropoietin promotes survival and regeneration of insect neurons in vivo and in vitro. Neuroscience. 2011;188:95–108. doi: 10.1016/j.neuroscience.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Koury MJ, Sawyer ST, Brandt SJ. New insights into erythropoiesis. Curr Opin Hematol. 2002;9:93–100. doi: 10.1097/00062752-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Lacombe C, Mayeux P. The molecular biology of erythropoietin. Nephrol. Dial. Transplant. 1999;14(Suppl 2):22–8. doi: 10.1093/ndt/14.suppl_2.22. [DOI] [PubMed] [Google Scholar]
- 22.Masuda S, et al. A novel site of erythropoietin production: oxygen-dependent production in cultured rat astrocytes. J Biol Chem. 1994;269:19488–93. [PubMed] [Google Scholar]
- 23.Tsuda E, Kawanishi G, Ueda M, Masuda S, Sasaki R. The role of carbohydrate in recombinant human erythropoietin. Eur J Biochem. 1990;188:405–11. doi: 10.1111/j.1432-1033.1990.tb15417.x. [DOI] [PubMed] [Google Scholar]
- 24.Jelkmann W, Bohlius J, Hallek M, Sytkowski AJ. The erythropoietin receptor in normal and cancer tissues. Crit Rev Oncol Hematol. 2008;67:39–61. doi: 10.1016/j.critrevonc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Ebie AZ, Fleming KG. Dimerization of the erythropoietin receptor transmembrane domain in micelles. J Mol Biol. 2007;366:517–24. doi: 10.1016/j.jmb.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 26.Krzyzanski W, Wyska E. Pharmacokinetics and pharmacodynamics of erythropoietin receptor in healthy volunteers. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:637–45. doi: 10.1007/s00210-007-0225-z. [DOI] [PubMed] [Google Scholar]
- 27.Anagnostou A, Lee ES, Kessimian N, Levinson R, Steiner M. Erythropoietin has a mitogenic and positive chemotactic effect on endothelial cells. Proc Natl Acad Sci U S A. 1990;87:5978–82. doi: 10.1073/pnas.87.15.5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konishi Y, Chui DH, Hirose H, Kunishita T, Tabira T. Trophic effect of erythropoietin and other hematopoietic factors on central cholinergic neurons in vitro and in vivo. Brain Res. 1993;609:29–35. doi: 10.1016/0006-8993(93)90850-m. [DOI] [PubMed] [Google Scholar]
- 29.Westenfelder C, Biddle DL, Baranowski RL. Human, rat, and mouse kidney cells express functional erythropoietin receptors. Kidney Int. 1999;55:808–20. doi: 10.1046/j.1523-1755.1999.055003808.x. [DOI] [PubMed] [Google Scholar]
- 30.Calvillo L, et al. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sci U S A. 2003;100:4802–6. doi: 10.1073/pnas.0630444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masuda S, et al. Functional erythropoietin receptor of the cells with neural characteristics: comparison with receptor properties of erythroid cells. J Biol Chem. 1993;268:11208–16. [PubMed] [Google Scholar]
- 32.Brines M, Cerami A. Erythropoietin- mediated tissue protection: reducing collateral damage from the primary injury response. J Intern Med. 2008;264:405–32. doi: 10.1111/j.1365-2796.2008.02024.x. [DOI] [PubMed] [Google Scholar]
- 33.Leist M, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;305:239–42. doi: 10.1126/science.1098313. [DOI] [PubMed] [Google Scholar]
- 34.Satake R, Kozutsumi H, Takeuchi M, Asano K. Chemical modification of erythropoietin: an increase in in vitro activity by guanidination. Biochim Biophys Acta. 1990;1038:125–9. doi: 10.1016/0167-4838(90)90020-g. [DOI] [PubMed] [Google Scholar]
- 35.Brines M, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci U S A. 2004;101:14907–12. doi: 10.1073/pnas.0406491101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy JM, Young IG. IL-3, IL-5, and GM-CSF signaling: crystal structure of the human beta-common receptor. Vitam Horm. 2006;74:1–30. doi: 10.1016/S0083-6729(06)74001-8. [DOI] [PubMed] [Google Scholar]
- 37.Mirza S, Chen J, Murphy JM, Young IG. The role of interchain heterodisulfide formation in activation of the human common beta and mouse betaIL-3 receptors. J Biol Chem. 2010;285:24759–68. doi: 10.1074/jbc.M109.097881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stomski FC, et al. Identification of a Cys motif in the common beta chain of the inter-leukin 3, granulocyte-macrophage colony-stimulating factor, and interleukin 5 receptors essential for disulfide-linked receptor heterodimerization and activation of all three receptors. J Biol Chem. 1998;273:1192–9. doi: 10.1074/jbc.273.2.1192. [DOI] [PubMed] [Google Scholar]
- 39.Hanazono Y, Sasaki K, Nitta H, Yazaki Y, Hirai H. Erythropoietin induces tyrosine phosphorylation of the beta chain of the GM-CSF receptor. Biochem Biophys Res Commun. 1995;208:1060–6. doi: 10.1006/bbrc.1995.1442. [DOI] [PubMed] [Google Scholar]
- 40.Jubinsky PT, Krijanovski OI, Nathan DG, Tavernier J, Sieff CA. The beta chain of the interleukin-3 receptor functionally associates with the erythropoietin receptor. Blood. 1997;90:1867–73. [PubMed] [Google Scholar]
- 41.Nicola NA, et al. Functional inactivation in mice of the gene for the interleukin-3 (IL-3)-specific receptor beta-chain: implications for IL-3 function and the mechanism of receptor transmodulation in hematopoietic cells. Blood. 1996;87:2665–74. [PubMed] [Google Scholar]
- 42.Scott CL, et al. Reassessment of interactions between hematopoietic receptors using common beta-chain and interleukin-3-specific receptor beta-chain-null cells: no evidence of functional interactions with receptors for erythropoietin, granulocyte colony-stimulating factor, or stem cell factor. Blood. 2000;96:1588–90. [PubMed] [Google Scholar]
- 43.Loesch A, Tang H, Cotter MA, Cameron NE. Sciatic nerve of diabetic rat treated with epoetin delta: effects on C-fibers and blood vessels including pericytes. Angiology. 2010;61:651–68. doi: 10.1177/0003319709360030. [DOI] [PubMed] [Google Scholar]
- 44.Colella P, et al. Non-erythropoietic erythro-poietin derivatives protect from light-induced and genetic photoreceptor degeneration. Hum Mol Genet. 2011;20:2251–62. doi: 10.1093/hmg/ddr115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu X, et al. Carbamylated erythropoietin protects the myocardium from acute ischemia/ reperfusion injury through a PI3K/Akt-dependent mechanism. Surgery. 2009;146:506–14. doi: 10.1016/j.surg.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 46.Kitamura H, et al. Nonerythropoietic derivative of erythropoietin protects against tubuloint-erstitial injury in a unilateral ureteral obstruction model. Nephrol Dial Transplant. 2008;23:1521–8. doi: 10.1093/ndt/gfm842. [DOI] [PubMed] [Google Scholar]
- 47.Joshi D, et al. Review of the role of erythropoietin in critical leg ischemia. Angiology. 2010;61:541–50. doi: 10.1177/0003319709358697. [DOI] [PubMed] [Google Scholar]
- 48.Su KH, et al. β common receptor integrates the erythropoietin signaling in activation of endothelial nitric oxide synthase. J Cell Physiol. 2011;226:3330–9. doi: 10.1002/jcp.22678. [DOI] [PubMed] [Google Scholar]
- 49.Carr PD, et al. Structure of the complete extracellular domain of the common beta subunit of the human GM-CSF, IL-3, and IL-5 receptors reveals a novel dimer configuration. Cell. 2001;104:291–300. doi: 10.1016/s0092-8674(01)00213-6. [DOI] [PubMed] [Google Scholar]
- 50.Saulle E, et al. Colocalization of the VEGF-R2 and the common IL-3/GM-CSF receptor beta chain to lipid rafts leads to enhanced p38 activation. Br J Haematol. 2009;145:399–411. doi: 10.1111/j.1365-2141.2009.07627.x. [DOI] [PubMed] [Google Scholar]
- 51.Bagley CJ, Woodcock JM, Stomski FC, Lopez AF. The structural and functional basis of cytokine receptor activation: lessons from the common beta subunit of the granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), and IL-5 receptors. Blood. 1997;89:1471–82. [PubMed] [Google Scholar]
- 52.Lopez AF, et al. Molecular basis of cytokine receptor activation. IUBMB Life. 2010;62:509–18. doi: 10.1002/iub.350. [DOI] [PubMed] [Google Scholar]
- 53.Um M, Gross AW, Lodish HF. A “classical” homodimeric erythropoietin receptor is essential for the antiapoptotic effects of erythropoietin on differentiated neuroblastoma SH-SY5Y and pheochromocytoma PC-12 cells. Cell Signal. 2007;19:634–45. doi: 10.1016/j.cellsig.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 54.Wu H, Lee SH, Gao J, Liu X, Iruela-Arispe ML. Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development. 1999;126:3597–605. doi: 10.1242/dev.126.16.3597. [DOI] [PubMed] [Google Scholar]
- 55.Teng R, et al. Acute erythropoietin cardio-protection is mediated by endothelial response. Basic Res Cardiol. 2011;106:343–54. doi: 10.1007/s00395-011-0158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colotta F, et al. Differential expression of the common beta and specific alpha chains of the receptors for GM-CSF, IL-3, and IL-5 in endothelial cells. Exp Cell Res. 1993;206:311–7. doi: 10.1006/excr.1993.1151. [DOI] [PubMed] [Google Scholar]
- 57.Xiong Y, et al. Erythropoietin improves histological and functional outcomes after traumatic brain injury in mice in the absence of the neural erythropoietin receptor. J Neurotrauma. 2010;27:205–15. doi: 10.1089/neu.2009.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swartjes M, et al. ARA290, a peptide derived from the tertiary structure of erythropoietin, produces long-term relief of neuropathic pain: an experimental study in rats and beta-common receptor knockout mice. Anesthesiology. 2011;115:1084–92. doi: 10.1097/ALN.0b013e31822fcefd. [DOI] [PubMed] [Google Scholar]
- 59.Allegra A, Buemi M, Corica F, Aloisi C, Frisina N. Erythropoietin administration induces an increase of serum levels of soluble E-selectin and soluble intercellular adhesion molecule 1. Nephron. 1999;82:361–2. doi: 10.1159/000045456. [DOI] [PubMed] [Google Scholar]
- 60.Heinisch BB, et al. The effect of erythropoietin on platelet and endothelial activation markers: a prospective trial in healthy volunteers. Platelets. 2011 Nov 18; doi: 10.3109/09537104.2011.631621. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 61.Montero M, et al. Comparison of neuro-protective effects of erythropoietin (EPO) and carbamylerythropoietin (CEPO) against ischemia-like oxygen-glucose deprivation (OGD) and NMDA excitotoxicity in mouse hippocampal slice cultures. Exp Neurol. 2007;204:106–17. doi: 10.1016/j.expneurol.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 62.Brines M, et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc Natl Acad Sci U S A. 2008;105:10925–30. doi: 10.1073/pnas.0805594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience. 1997;76:105–16. doi: 10.1016/s0306-4522(96)00306-5. [DOI] [PubMed] [Google Scholar]
- 64.Erbayraktar S, et al. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuro-protective activity in vivo. Proc Natl Acad Sci U S A. 2003;100:6741–6. doi: 10.1073/pnas.1031753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mori S, Sawada T, Okada T, Kubota K. Erythropoietin and its derivative protect the intestine from severe ischemia/reperfusion injury in the rat. Surgery. 2008;143:556–65. doi: 10.1016/j.surg.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 66.Okada T, Sawada T, Kubota K. Asialoery-thropoietin has strong renoprotective effects against ischemia-reperfusion injury in a murine model. Transplantation. 2007;84:504–10. doi: 10.1097/01.tp.0000277672.02783.33. [DOI] [PubMed] [Google Scholar]
- 67.Sautina L, et al. Induction of nitric oxide by erythropoietin is mediated by the β common receptor and requires interaction with VEGF receptor 2. Blood. 2010;115:896–905. doi: 10.1182/blood-2009-04-216432. [DOI] [PubMed] [Google Scholar]
- 68.Marrero MB, Venema RC, Ma H, Ling BN, Eaton DC. Erythropoietin receptor-operated Ca2+ channels: activation by phospholipase C-gamma 1. Kidney Int. 1998;53:1259–68. doi: 10.1046/j.1523-1755.1998.00887.x. [DOI] [PubMed] [Google Scholar]
- 69.Ahmet I, et al. A small nonerythropoietic helix B surface peptide based upon erythropoietin structure is cardioprotective against ischemic myocardial damage. Mol Med. 2011;17:194–200. doi: 10.2119/molmed.2010.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ueba H, et al. Cardioprotection by a non-erythropoietic, tissue-protective peptide mimicking the 3D structure of erythropoietin. Proc Natl Acad Sci U S A. 2010;107:14357–62. doi: 10.1073/pnas.1003019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fu ZQ, et al. Effect of carbamylated erythropoietin on major histocompatibility complex expression and neural differentiation of human neural stem cells. J Neuroimmunol. 2010;221:15–24. doi: 10.1016/j.jneuroim.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 72.Patel NS, et al. A nonerythropoietic peptide that mimicks the 3D structure of erythropoietin reduces organ injury/dysfunction and inflammation in experimental hemorrhagic shock. Mol Med. 2011;17:883–92. doi: 10.2119/molmed.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moon C, et al. Erythropoietin, modified to not stimulate red blood cell production, retains its cardioprotective properties. J Pharmacol Exp Ther. 2006;316:999–1005. doi: 10.1124/jpet.105.094854. [DOI] [PubMed] [Google Scholar]
- 74.Wang L, et al. The Sonic hedgehog pathway mediates carbamylated erythropoietin-enhanced proliferation and differentiation of adult neural progenitor cells. J Biol Chem. 2007;282:32462–70. doi: 10.1074/jbc.M706880200. [DOI] [PubMed] [Google Scholar]
- 75.Taoufik E, et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc Natl Acad Sci U S A. 2008;105:6185–90. doi: 10.1073/pnas.0801447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–7. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 77.Wang L, et al. Tumor necrosis factor alpha primes cerebral endothelial cells for erythropoietin-induced angiogenesis. J Cereb Blood Flow Metab. 2011;31:640–7. doi: 10.1038/jcbfm.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Junk AK, et al. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2002;99:10659–64. doi: 10.1073/pnas.152321399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanellakis P, et al. Darbepoetin-mediated cardioprotection after myocardial infarction involves multiple mechanisms independent of erythropoietin receptor-common beta-chain heteroreceptor. Br J Pharmacol. 2010;160:2085–96. doi: 10.1111/j.1476-5381.2010.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Woodcock JM, et al. The human granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor exists as a preformed receptor complex that can be activated by GM-CSF, interleukin-3, or interleukin-5. Blood. 1997;90:3005–17. [PubMed] [Google Scholar]
- 81.Brines ML, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci U S A. 2000;97:10526–31. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiong Y, et al. Effects of posttraumatic carbamylated erythropoietin therapy on reducing lesion volume and hippocampal cell loss, enhancing angiogenesis and neurogenesis, and improving functional outcome in rats following traumatic brain injury. J Neurosurg. 2011;114:549–59. doi: 10.3171/2010.10.JNS10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miskowiak K, O’Sullivan U, Harmer CJ. Erythropoietin enhances hippocampal response during memory retrieval in humans. J Neurosci. 2007;27:2788–92. doi: 10.1523/JNEUROSCI.5013-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leconte C, et al. Comparison of the effects of erythropoietin and its carbamylated derivative on behaviour and hippocampal neurogenesis in mice. Neuropharmacology. 2011;60:354–64. doi: 10.1016/j.neuropharm.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 85.Su KH, et al. AMP-activated protein kinase mediates erythropoietin-induced activation of endothelial nitric oxide synthase. J Cell Physiol. 2011 Oct 20; doi: 10.1002/jcp.23052. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 86.Meads MB, Li ZW, Dalton WS. A novel TNF receptor-associated factor 6 binding domain mediates NF-kappa B signaling by the common cytokine receptor beta subunit. J Immunol. 2010;185:1606–15. doi: 10.4049/jimmunol.0902026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grasso G, et al. Amelioration of spinal cord compressive injury by pharmacological preconditioning with erythropoietin and a nonerythropoietic erythropoietin derivative. J Neurosurg Spine. 2006;4:310–8. doi: 10.3171/spi.2006.4.4.310. [DOI] [PubMed] [Google Scholar]