

Abstract
Phagocytosis of apoptotic cells by macrophages, known as efferocytosis, is a critical process in the resolution of inflammation. High mobility group box 1 (HMGB1) protein was first described as a nuclear nonhistone DNA-binding protein, but is now known to be secreted by activated cells during inflammatory processes, where it participates in diminishing efferocytosis. Although HMGB1 is known to undergo modification when secreted, the effect of such modifications on the inhibitory actions of HMGB1 during efferocytosis have not been reported. In the present studies, we found that HMGB1 secreted by Toll-like receptor 4 (TLR4) stimulated cells is highly poly(ADP-ribosyl)ated (PARylated). Gene deletion of poly(ADP)-ribose polymerase (PARP)-1 or pharmacological inhibition of PARP-1 decreased the release of HMGB1 from the nucleus to the extracellular milieu after TLR4 engagement. Preincubation of macrophages or apoptotic cells with HMGB1 diminished efferocytosis through mechanisms involving binding of HMGB1 to phosphatidylserine on apoptotic cells and to the receptor for advanced glycation end products (RAGE) on macrophages. Preincubation of either macrophages or apoptotic cells with PARylated HMGB1 inhibited efferocytosis to a greater degree than exposure to unmodified HMGB1, and PARylated HMGB1 demonstrated higher affinity for phosphatidylserine and RAGE than unmodified HMGB1. PARylated HMGB1 had a greater inhibitory effect on Ras-related C3 botulinum toxin substrate 1 (Rac-1) activation in macrophages during the uptake of apoptotic cells than unmodified HMGB1. The present results, showing that PARylation of HMGB1 enhances its ability to inhibit efferocytosis, provide a novel mechanism by which PARP-1 may promote inflammation.
INTRODUCTION
The uptake and clearance of apoptotic cells by macrophages and other phagocytic cells, a process known as efferocytosis, plays a central role in the resolution of inflammation. Efferocytosis participates in decreasing the severity of inflammation by diminishing the number of apoptotic cells progressing to necrosis, thereby preventing the release of proinflammatory intracellular contents into the extracellular milieu (1). Phagocytosis of apoptotic cells also diminishes inflammation through inducing release of anti-inflammatory mediators, such as TGF-β1, by the engulfing phagocytic cell and by suppressing macrophage production of proinflammatory cytokines (2,3). Impaired clearance of apoptotic cells is associated with autoimmune and inflammatory diseases including rheumatoid arthritis (4,5), chronic obstructive pulmonary disease (6,7), acute lung injury (8,9) and cystic fibrosis (7).
One of the early characteristics of apoptosis is the redistribution of phosphatidylserine (PS) from the inner to the outer membrane leaflet (10). PS serves as an “eat-me” signal, allowing the recognition of the target cell by a broad set of receptors localized on the surface of the phagocyte, including integrins, brain-specific angiogenesis inhibitor 1 (BAI-1), T-cell immunoglobulin mucin-4 (TIM-4) and stabilin-2 (11–13). In recent studies, we and others have shown that the receptor for advanced glycation end products (RAGE) is directly involved in enhancing the ingestion of apoptotic cells by macrophages through binding to PS (14,15).
High mobility group box 1 protein (HMGB1) was originally described as a nuclear nonhistone protein with DNA-binding and transcriptional activities (16–19). It is now known that HMGB1 can also be released into the extracellular milieu and acts as a proinflammatory mediator. Several mechanisms by which HMGB1 participates in enhancing inflammatory reactions have been described, including inducing the release of proinflammatory mediators (17,20,21), increasing chemotaxis and accumulation of neutrophils in inflammatory sites and diminishing the phagocytosis and clearance of apoptotic cells (22–24). Both TLR4 and RAGE were identified as receptors for HMGB1 (25,26). We recently showed that the C-terminal domain of HMGB1, a motif required for binding to RAGE, is required to inhibit ingestion of apoptotic cells by macrophages (27). However, there is no evidence at present that binding between HMGB1 and RAGE is involved in modulating efferocytosis.
Poly(ADP)-ribose polymerase (PARP) is a nuclear enzyme that catalyzes the transfer of ADP-ribose moieties from NAD+ to itself and other acceptor proteins in response to DNA damage (28). A role for PARP-1 in contributing to inflammation is well described (29). It has been shown that HMGB1 is PARylated by PARP-1 and also that activation of PARP-1 induces release of HMGB1 from the nucleus (30). However, the previous studies did not identify if posttranslational modifications of extracellular HMGB1, such as PARylation, could affect interactions between HMGB1 and its receptors.
In the present experiments, we examined the possibility that HMGB1 could be PARylated in activated cells, and, if so, whether PARylation may play a role in the ability of HMGB1 to diminish the ingestion of apoptotic cells by macrophages. We found that PARylated HMGB1 is secreted by cells stimulated through TLR4 engagement and also that PARylated HMGB1 was more effective in diminishing efferocytosis than unmodified HMGB1.
MATERIALS AND METHODS
Mice
Male C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD, USA). Transgenic RAGE−/−mice were a gift from A. Bierhaus (University of Heidelberg, Heidelberg, Germany). Mice were housed and studied at the University of Alabama at Birmingham using protocols approved by the Institutional Animal Care and Use Committee. Experiments were performed using 8- to 10-wk-old mice.
Reagents
Roswell Park Memorial Institute (RPMI) media, penicillin-streptomycin, Brewer thioglycollate and PKH26 were from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (FBS) (heat-inactivated) was from Gibco-BRL (Grand Island, NY, USA). The HMGB1 enzyme-linked immunosorbent assay (ELISA) kit was from IBL International (Hamburg, Germany). The Rac activation assay kit was from Cytoskeleton (Denver, CO, USA). Mouse RAGE chimeric protein was from R&D Systems (Minneapolis, MN, USA). PS was from Avanti Polar Lipids (Alabaster, AL, USA). Recombinant mouse MFG-E8 and mouse Gas6 as well as recombinant human αVβ3-integrin were from R&D Systems. Recombinant mouse αVβ5-integrin was from Millipore (Billerica, MA, USA). PARP-1 and poly(ADP-ribose) glycohydrolase (PARG) proteins were from Enzo Life Sciences (Exeter, UK). Rabbit anti-HMGB1 polyclonal antibody was from Abcam (Cambridge, MA, USA). Goat anti-RAGE (sc-8230) and rabbit anti-His (sc-803) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Fluorescein isothiocyanate (FITC)-conjugated anti-CD11b and allophycocyanin-conjugated anti-CD 90.2 antibodies were from BD (Sparks, MD, USA). Poly(ADP-ribose) (PAR) antibody was from AXXORA (San Diego, CA, USA).
Construction of Expression Plasmids and Adenoviral Vectors Expressing Wild-Type PARP-1-EYFP or E988K PARP-1-EYFP
Full-length HMGB1 (wild-type [WT], 1–215 amino acids), C-tail deletion mutant (ΔC, 1–185 amino acids), A-box (1–81 amino acids) and B-Box (95–185) fragments were generated by polymerase chain reaction amplification and cloned into pTriEx-4 Neo vector (Novagen, Rockland, MA, USA) in frame with N-terminal 6XHis tag for bacterial expression and purification of recombinant protein. HMGB1-His, ΔC-His, A-box-His and B-box-His recombinant proteins were purified using nickel-nitrilotriacetic acid-agarose (Qiagen, Valencia, CA, USA). The amounts and purity of recombinant proteins were determined using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining with bovine serum albumin (BSA) as a standard. All construct sequences were confirmed by DNA sequencing.
To generate a stable cell line that over-expressed Flag-tagged HMGB1, J447-mouse macrophage cells were transfected with the HMGB1-Flag construct. A clone that stably expressed HMGB1-Flag was selected and expanded for purification of HMGB1-Flag after lipopolysaccharide (LPS) treatment. Cells (grown to confluence in a 10 × 150 mm dish) were lysed under nondenaturing conditions and incubated with anti-Flag agarose beads (M2-agarose; Sigma-Aldrich) for 2 h, with rotation at 4°C after, which the beads were washed three times with 50 mmol/L Tris buffer (pH 7.5). Beads binding Flag-tagged proteins were eluted using the 3XFlag peptide (250 μg/mL) in 50 mmol/L Tris buffer. Proteins were concentrated using Amicon Ultra 0.5 filters (Ultracell 10K; Millipore). For denaturing conditions, the beads were resuspended in protein loading buffer with 4% SDS, boiled for 5 min and resolved by 2–12% gradient SDS-PAGE and transferred to a nitrocellulose membrane for subsequent probing with monoclonal antibodies against HMGB1 and PAR. For the construction of the WT PARP-1-EYFP expression vector, PARP-1 cDNA was cloned into the Gateway entry vector pENTR/SD/D-TOPO (Invitrogen). The expression clone was generated by performing a left and right (LR) recombination reaction between the entry clone and the destination vector pAd/CMV/V5-DEST (Invitrogen).
Knockdown of PARP-1 or HMGB1 in J447 mouse cells was achieved using lentiviral vectors (sc-29438-V and sc-37983-V, respectively; Santa Cruz Biotechnology) encoding targeting shRNA, according to the manufacturer’s specifications and instructions, before treatment with LPS. At 48 h after infection, the expression of PARP-1, HMBG1 and actin was assessed by immunoblot analysis and compared with that in uninfected cells. Cells transduced with either virus were treated with LPS for 24 h, after which Flag-HMGB1 pull-down was conducted and levels of PARylated HMGB1 were determined by immunoblot.
Isolation and Treatment of Mouse Peritoneal Macrophages
Peritoneal macrophages were elicited from 8- to 10-wk-old mice by intraperitoneal injection of 1 mL 3% thioglycollate, and cells were harvested 4 d later by peritoneal lavage. Macrophages (0.5 × 106) were plated in 24-well plates in RPMI media containing 5% FBS. After 1 h at 37°C, nonadherent cells were removed by washing with medium. One hour before performance of phagocytosis assays, the macrophages were washed with fresh serum-free medium. WT and PARP-1−/− mouse embryonic fibroblasts (MEFs) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, penicillin and streptomycin. Cells were treated with 100 ng/mL LPS (AXXORA) for 24 h in the absence or presence of 50 μmol/L of the PARP-1 inhibitor NU1025 (Santa Cruz Biotechnology). PARP-1−/− MEFs were transduced with an adenoviral vector expressing an EYFP-WT PARP-1 (Ad-EYFP-PARP-1) or enzymatically inactive EYFP-E988K PARP-1 (Ad-EYFP-E988K PARP-1) or left untransduced.
J447-mouse cells transfected with HMGB1-Flag were treated or not with 100 ng/mL LPS for 24 h. Intracellular and extracellular (from medium) HMGB1-Flag was purified using 3× Flag peptide.
Induction of Apoptosis in Thymocytes
To induce apoptosis, murine thymocytes were resuspended in RPMI 1640 medium containing 5% FBS and 1 mmol/L dexamethasone and incubated at 37°C in 5% CO2 for 12 h. At this time point, 90% of the thymocytes were apoptotic, as assessed by flow cytometry with annexin V and propidium iodide staining.
In Vitro and In Vivo Phagocytosis Assays
Phagocytosis of apoptotic thymocytes by macrophages was assayed by adding106 apoptotic thymocytes at 37°C for 60 min to each well of a 24-well plate containing adherent macrophage monolayers. For studies investigating inhibition of phagocytosis, apoptotic thymocytes were preincubated with 1 μg/mL recombinant HMGB1 or PARylated HMGB1 and compared with control BSA-treated thymocytes, as described in the figure legends. Noningested cells were removed after incubation of apoptotic thymocytes with macrophages by washing three times with phosphate-buffered saline (PBS).
In vivo phagocytosis was determined by labeling apoptotic thymocytes with PKH26 and incubating 106 apoptotic thymocytes with 2 μg non-PARylated or PARylated HMGB1 or mouse albumin in 100 μL PBS. The thymocytes were then intratracheally injected into isofluorane-anesthetized mice. After 90 min, the mice were sacrificed and bronchoalveolar lavage (BAL) was performed.
For in vivo and in vitro phagocytosis assays, the samples were resuspended in 1% PBS/BSA, FITC-CD11b antibody (Ab) (macrophage marker) and allophycocyanin-conjugated anti-CD (APC-CD) 90.2 Ab (thymocyte marker). Flow cytometry was performed, and the phagocytosis index was calculated as the ratio of FITC+PKH26+APC− cells to all cells gated.
Poly(ADP-ribosyl)ation In Vitro
Purified recombinant His-tagged HMGB1, ΔC-HMGB1, A box or B box were incubated with purified PARP-1 in a mixture (25 μL) containing reaction buffer (100 mmol/L Tris-HCl, 1 mmol/L DTT, 10 mmol/L MgCl2), sonicated salmon sperm DNA and 2 mmol/L NAD (Sigma) for 30 min at 37°C. The reaction was either terminated by the addition of an equal volume of SDS sample buffer and heating at 95°C for 5 min or subjected to PARG treatment. Samples were then subjected to immunoblot analysis with antibodies to PAR or anti-His antibodies.
Rac-1 Activation Assays
Apoptotic thymocytes were treated with 1 μg/mL BSA, HMGB1 or PARylated HMGB1 for 2 h followed by washing three times with PBS. Apoptotic thymocytes were added to macrophages for the indicated time periods (5 and 30 min). The macrophages were then washed three times with PBS and lysed. Macrophages preincubated with media only were used as a negative control. To determine Ras-related C3 botulinum toxin substrate 1 (Rac-1) activation, a Rac-1 activation assay kit was used (Cytoskeleton) according to the manufacturer’s instructions.
Indirect Immunofluorescence
WT and RAGE−/− peritoneal macrophages grown on chamber slides or thymocytes (apoptotic or viable) were preincubated with 1 μg/mL HMGB1 or PARylated HMGB1 for 2 h. Cells were then fixed with 4% formaldehyde in PBS for 5 min. After being washed with PBS and blocked with 1% BSA in PBS, the cells were incubated with anti-HMGB1 antibodies at 4°C overnight. After being washed three times with PBS, the cells were incubated with Alexa Fluor 488–conjugated secondary antibodies for 1 h at room temperature. After counterstaining with DAPI, antibody–antigen complexes were detected by a fluorescence microscope (Nikon) using a 40× objective lens.
Solid-Phase ELISA
The solid-phase ELISA for HMGB1 binding to PS or RAGE was carried out as follows: recombinant chimeric RAGE or a solution of PS at a concentration of 3 mg/mL was added to 96-well plates for 2 h at room temperature, followed by washing with 1% BSA in PBS. HMGB1 or PARylated HMGB1 was then added to the wells and incubated at room temperature for 2 h. HMGB1 that bound to the wells was quantified by ELISA with anti-HMGB1 and peroxidase-conjugated secondary Abs. Washing with PBS containing 0.05% Tween 20 was performed between each step. To assess competition between non-PARylated HMGB1 or PARylated HMGB1 and RAGE for binding to PS, the assay was modified by adding RAGE after HMGB1 treatment.
HMGB1 levels in culture supernatants were determined using commercial ELISA kits from IBL International (Hamburg, Germany) according to the manufacturer’s protocols. Briefly, 100 μL sample diluent was added to each well and then 10 μL standard, sample or control was added to the well. The plate was incubated for 24 h at 37°C. After washing, 100 μL/well of anti-HMGB1 antibody conjugated to peroxidase was added and the plate was incubated at room temperature for 2 h. After washing, substrate was added to each well. The enzyme reaction was incubated for 30 min at room temperature. The chromogenic substrate reaction was stopped by the addition of stop solution. Absorbance was determined using an automated plate reader at an excitation wavelength of 450 nm.
Statistical Analysis
Data are presented as the means ± standard deviation (SD) for in vitro experiments or means ± standard error of the mean (SEM) for in vivo experiments for each condition. One-way analysis of variance followed by Tukey-Kramer analysis was performed for comparisons among multiple groups, and a Student t test was used for comparisons between two groups. A value of P < 0.05 was considered significant.
All supplementary materials are available online at www.molmed.org.
RESULTS
HMGB1 Secreted by TLR4-Stimulated Cells Is Highly PARylated
A role for PARP-1 in the nuclear release of HMGB1 from DNA-damaged necrotic cells was previously demonstrated (30). PARP-1 is capable of PARylating both full-length HMGB1 as well as HMGB1 lacking the C-terminal tail (ΔC HMGB1) (30). However, it is not known whether HMGB1 that is secreted by activated cells is PARylated. To determine if PARP-1 is required for HMGB1 release by TLR4-stimulated cells, WT and PARP-1−/− MEFs were treated with LPS for 24 h. As shown in Figure 1A, PARP-1−/− fibro-blasts secrete significantly less HMGB1 than WT fibroblasts after incubation with LPS. To further delineate if PARP-1 is necessary for HMGB1 release after LPS exposure, WT fibroblasts were treated with NU1025, a noncompetitive PARP-1 inhibitor (31), before incubation with LPS. The inclusion of NU1025 in the cultures decreased HMGB1 release compared with that found with vehicle-treated cells (Figure 1B).
Figure 1.

Extracellular HMGB1 is highly PARylated in cells stimulated with LPS. (A) PARP-1 gene deletion inhibits LPS-induced HMGB1 release. WT or PARP-1−/− MEFs were treated with 100 ng/mL LPS for 24 h at 37°C and then extracellular HMGB1 was secreted into the culture supernatant quantified by ELISA. **P < 0.01 compared with nontreated cells. Assay was done in triplicate. (B) Inhibition of PARP-1 reduces LPS-induced HMGB1 release. WT MEF cells were treated with LPS in the presence or absence of the noncompetitive PARP-1 inhibitor NU1025 for 24 h, after which extracellular HMGB1 was quantified by ELISA. **P < 0.01 compared with nontreated cells. (C) The decrease in HMGB1 release by LPS-stimulated PARP-1−/− MEFs is reversed by reconstitution of expression of WT PARP-1 but not by enzymatically inactive PARP-1 (E988K). PARP-1−/−MEFs were infected with an adenoviral vector expressing EYFP-WT PARP-1 (Ad-WT-PARP-1) or EYFP-E988K PARP-1 (Ad-E988K-PARP-1) or left uninfected. Forty-eight hours later, the cells were cultured for 24 h with 100 ng/mL LPS or left untreated (Con). Extracellular HMGB1 was quantified by ELISA. *P < 0.05 compared with uninfected PARP-1−/− MEFs. **P < 0.01 compared with untreated WT MEFs. (D) HMGB1 secreted after TLR4 stimulation is highly PARylated. J447 mouse cells expressing Flag-HMGB1 were treated with 100 ng/mL LPS for 24 h or left untreated (Con). The culture supernatants and protein extracts prepared from the cells were then subjected to pull-down with anti-Flag agarose beads. The resulting precipitates were subjected to immunoblot analysis with antibodies to PAR and HMGB1. A representative gel is shown. Two additional independent experiments provided similar results. (E) Flag-HMGB1 pull-down under nondenaturing conditions and after PARP-1 and HMGB1 knockdown. J447 mouse cells expressing Flag-HMGB1 were treated with 100 ng/mL LPS for 24 h or left untreated (Con). The protein extracts were then subjected to pull-down with anti-Flag agarose beads under nondenaturing conditions. The resulting precipitates were subjected to immunoblot analysis with antibodies to PAR or HMGB1. (F) Flag-HMGB1 pull-down under denaturing conditions after PARP-1 and HMGB1 knockdown. The same experiment was conducted with J447-mouse cells expressing Flag-HMGB1 after PARP-1 or HMGB1 knockdown. The cells were transduced with lentiviruses encoding control shRNA (shRNA-Con), shRNA targeting HMGB1 (shRNA-HMGB1) or shRNA targeting PARP-1 (shRNA-PARP-1); expression of PARP-1, HMGB1 and actin was assessed by immunoblot analysis and compared with that in uninfected cells. (G) These infected or uninfected cells were treated with 100 ng/mL LPS for 24 h or left untreated. The protein extracts were then subjected to pull-down with anti-Flag agarose beads. The resulting precipitates were subjected to immunoblot analysis with antibodies to PAR or Flag. Results from representative experiments are shown. A second set of independent experiments provided similar findings.
To characterize if the ability of PARP-1 to enhance HMGB1 release is due to the presence of PARP-1 protein alone or whether PARP-1 enzymatic activity also is necessary, PARP-1−/− fibroblasts were transduced using adenovirus gene transfer with Ad-WT PARP-1 or modified, enzymatically inactive Ad-E988K-PARP-1. As shown in Figure 1C, the reestablishment of enzymatically active PARP-1 in PARP-1−/− fibroblasts fully restored release of HMGB1, whereas the addition of enzymatically inactive PARP-1 did not. These results demonstrate that the enzymatic activity of PARP-1 is required for HMGB1 release from the nucleus to the extracellular milieu in TLR4-stimulated cells.
Because of the involvement of PARP-1 in HMGB1 secretion, we examined whether HMGB1 secreted after TLR4 engagement is PARylated. Treatment of J447 cells expressing Flag-HMGB1 with LPS resulted in a marked increase in the PARylation of both intracellular and extracellular HMGB1 as assessed by Western blots using anti-PAR antibodies (Figure 1D). To rule out the possibility that the PARylation was from proteins interacting with HMGB1 rather than from HMGB1 itself, we repeated the experiment under nondenaturing conditions (Figure 1E). Because the PAR pattern was similar under denaturing or nondenaturing conditions, we concluded that the PARylation was from HMGB1 itself. PARP-1 or HMGB1 knockdown with lentiviral vectors encoding shRNA targeting PARP-1 or HMGB1 (see Figure 1E) significantly reduced HMGB1 PARylation upon LPS treatment of J447 cells (Figure 1F), confirming the specificity and validity of our observations.
PARylated HMGB1 Inhibits Efferocytosis More Effectively than Non-PARylated HMGB1
In previous studies, we demonstrated that HMGB1 inhibits efferocytosis through binding to PS on apoptotic cells and to αvβ3-integrin as well as to RAGE on macrophages (23,32). However, it is not known if PARylation of HMGB1 has an effect on the uptake of apoptotic cells by macrophages. To investigate this issue, PARylated HMGB1 or non-PARylated HMGB1 was incubated with apoptotic thymocytes for 2 h before addition to macrophages. As shown in Figure 2A, PARylated HMGB1 was more effective in inhibiting the phagocytosis of apoptotic cells than non-PARylated HMGB1.
Figure 2.

PARylated HMGB1 inhibits phagocytosis of apoptotic cells by macrophages more effectively than non-PARylated HMGB1. (A) Phagocytosis assays were performed after preincubation of apoptotic thymocytes for 2 h with 1 μg/mL recombinant HMGB1 or PARylated HMGB1. *P < 0.05 and **P < 0.01 compared with control BSA-treated thymocytes. #P < 0.05 compared with HMGB1-treated thymocytes. (B) After preincubation of thymocytes for 2 h with PARylated or unmodified HMGB1, phagocytosis assays were performed for 5 and 30 min. The macrophages were then washed three times, and cell lysates were prepared to determine Rac-1 activation. As a negative control, macrophages were preincubated with media in the absence of thymocytes. White bars represent unmodified HMGB1, and gray bars represent PARylated HMGB1; *P < 0.05 versus PARylated HMGB1. (C) Phagocytosis assays were performed after preincubation of macrophages for 2 h with 1 μg/mL recombinant HMGB1 or PARylated HMGB1. *P < 0.05 compared with control BSA-treated macrophages. #P < 0.05 compared with HMGB1-treated macrophages. (D.1) Phagocytosis assays were performed after preincubation of macrophages for 2 h with 1 μg/mL PARylated HMGB1 or free PAR and compared with control BSA-treated macrophages. (D.2) PARP-1 protein was incubated in a poly(ADP-ribosyl)ation reaction containing NAD and activated DNA for 30 min. PARG was then added to the reaction for 30 min. Separation of free PAR from PARP-1 was performed using Amicon Ultra 0.5 filters. A portion (10%) of the reaction mixture was used for immunoblot analysis with antibodies to PAR. A representative gel is shown. A second independent experiment provided similar results. All phagocytosis experiments were performed at least three independent times with similar results. (E) Apoptotic thymocytes (107) preincubated for 2 h with 2 μg HMGB1, PARylated HMGB1 or BSA were administered intratracheally in 50 μL PBS into anesthesized mice. BAL fluid was collected 90 min later. The samples were resuspended in PBS with 1% BSA and stained with FITC-CD11b Ab (macrophage marker) and APC-CD 90.2 Ab (thymocyte marker). Flow cytometry was performed, and the phagocytosis index was calculated as the ratio of FITC+PKH26+APC− cells to all cells gated. n = 3 mice in each group. *P < 0.05 compared with control. #P < 0.05 compared with HMGB1-treated thymocytes. A second independent experiment provided similar results.
Activation of Rac-1 normally occurs during efferocytosis (33,34). To determine if PARylated HMGB1 affects efferocytosis-associated Rac-1 activation differently than unmodified HMGB1, thymocytes were incubated with unmodified or PARylated HMGB1 followed by incubation with macrophages. Macrophages preincubated with media alone were used as a negative control. Activation of Rac-1 was then determined 5 and 30 min later. As shown in Figure 2B, incubation of thymocytes with PARylated HMGB1 diminished efferocytosis-associated Rac-1 activation to a greater extent than unmodified HMGB1.
To determine if the inhibitory effect of PARylated HMGB1 on phagocytosis specifically occurred through binding to apoptotic cells or whether interaction of PARylated HMGB1 with macrophages also resulted in diminished efferocytosis, peritoneal macrophages were incubated with either PARylated or non-PARylated HMGB1 for 2 h and then washed before incubation with apoptotic thymocytes. As shown in Figure 2C, PARylated HMGB1, after being incubated with macrophages, inhibited efferocytosis to a greater extent than non-PARylated HMGB1. To eliminate the possibility that the presence of PAR is responsible for the decrease in efferocytosis induced by PARylated HMGB1, free PAR or PARylated HMGB1 was incubated with peritoneal macrophages for 2 h before the addition of apoptotic thymocytes. The presence of free PAR alone had no effect on macro phage phagocytosis of apoptotic cells (Figure 2D).
To determine if PARylation of HMGB1 enhanced its ability to inhibit efferocytosis in vivo, apoptotic thymocytes were incubated with PARylated or non-PARylated HMGB1 for 2 h and then administered intratracheally to mice. After 90 min, the mice were euthanized and bronchoalveolar lavage was performed to assess the phagocytic index of alveolar macrophages. As shown in Figure 2E, PARylated HMGB1 inhibited the phagocytosis of apoptotic cells by alveolar macrophages to a greater extent than non-PARylated HMGB1.
PARylated HMGB1 Binds PS and RAGE with Higher Affinity than Non-PARylated HMGB1
To determine if PARylated HMGB1 binds to a receptor or ligand involved in efferocytosis with higher affinity than non-PARylated HMGB1, we incubated PARylated or non-PARylated HMGB1 with receptors or opsonins that are known to interact directly or indirectly with HMGB1 during efferocytosis, specifically αvβ3, αvβ5, Gas6, MFG-E8, RAGE and PS. As shown in Figures 3A and B, PARylated HMGB1 did not bind to αvβ3, αvβ5, Gas6 or MFG-E8 with higher affinity than did non-PARylated HMGB1. In fact, PARylated HMGB1 was found to bind to αvβ3, Gas6, and MFG-E8 with much lower affinity than non-PARylated HMGB1. No significant difference in binding to αvβ5 was found between non-PARylated and PARylated HMGB1 (Figure 3A, right panel). However, as shown in Figure 3C, PARylated HMGB1 binds to PS and RAGE with higher affinity than does non-PARylated HMGB1. These results suggest that inhibition of phagocytosis of apoptotic thymocytes by macrophages may be mediated through enhanced binding of PARylated HMGB1 to PS and RAGE.
Figure 3.

PARylated HMGB1 binds to PS and RAGE with higher affinity than non-PARylated HMGB1. (A) After being precoated with the integrins αVβ3 or αVβ5, 96-well plates were incubated with 1 μg/mL HMGB1 or PARylated HMGB1 for 2 h. The plates were then washed and bound HMGB1 was quantified by ELISA, as described in Materials and Methods. **P < 0.01 compared with HMGB1. (B) Gas6 and MFG-E8–precoated 96-well plates were incubated with 1 μg/mL HMGB1 or PARylated HMGB1 for 2 h. The plates were then washed and bound HMGB1 was quantified by ELISA. *P < 0.05 and **P < 0.01 compared with HMGB1. (C) Recombinant RAGE dissolved in PBS or PS dissolved in methanol was incubated in 96-well plates. The plates were then incubated with 1 μg/mL HMGB1 or PARylated HMGB1 for 2 h. Bound HMGB1 was quantified by ELISA. *P < 0.05 compared with HMGB1. Assays were done in triplicate.
PARylated HMGB1 Binds to PS on Apoptotic Cells and to RAGE on Macrophages with Higher Affinity than Non-PARylated HMGB1
We used immunofluorescence microscopy to directly determine the binding of PARylated and non-PARylated HMGB1 to PS on apoptotic thymocytes and to RAGE on macrophages. As shown in Figures 4A and B, PARylated HMGB1 bound to the surface of both apoptotic thymocytes and macrophages with increased affinity compared with that found with non-PARylated HMGB1. To determine if the increased binding of PARylated HMGB1 to apoptotic thymocytes is due to interaction with PS, PARylated HMGB1 was added to viable thymocytes, which do not have PS exposed on their outer membranes. As shown in Figure 4C, there was no binding of PARylated HMGB1 to viable thymocytes. Similarly, using RAGE−/− macrophages, we found that the absence of RAGE from the macrophage surface was associated with significantly decreased binding of PARylated HMGB1 (Figure 4D). However, unlike our findings showing no binding of HMGB1 to viable thymocytes, which do not express PS, there still was residual binding of PARylated HMGB1 to RAGE−/− macrophages. These data suggest that macrophage receptors in addition to RAGE interact with PARylated HMGB1.
Figure 4.

PARylated HMGB1 binds with increased affinity to PS on apoptotic cells and to RAGE on macrophages compared with unmodified HMGB1. (A) Apoptotic thymocytes or WT macrophages were incubated with 1 μg/mL HMGB1 or PARylated HMGB1 for 2 h at 37°C or left untreated (Control), and then HMGB1 binding was examined by immunofluorescence. Representative images are shown. Two additional independent experiments provided similar results. (B) Quantification of HMGB1 binding to apoptotic cells or macrophages using ImageJ software (n = 10 cells from three independent experiments). Means ± SEM are shown. *P < 0.05 and **P < 0.01 compared with control. (C, D) PARylated HMGB1 does not bind to viable thymocytes and binds with diminished affinity to RAGE−/− macrophages. Viable thymocytes (C) or RAGE−/− macrophages (D) were incubated with 1 μg/mL PARylated HMGB1 for 2 h at 37°C, and then HMGB1 binding was examined by immunofluorescence. Representative images are shown. Two additional independent experiments provided similar results.
PARylated HMGB1 Inhibits RAGE Binding to PS More Effectively than Non-PARylated HMGB1
We next investigated whether extra-cellular HMGB1 can modulate RAGE binding to PS by using a solid-phase ELISA in which wells precoated with PS were exposed to increasing concentrations of nonmodified or PARylated HMGB1, and then interaction between PS and RAGE was determined. As shown in Figure 5A, unmodified HMGB1 bound to PS and also inhibited RAGE binding to PS in a dose-dependent manner. PARylated HMGB1 was more effective in diminishing the binding of RAGE to PS than non-PARylated HMGB1 (Figure 5B).
Figure 5.
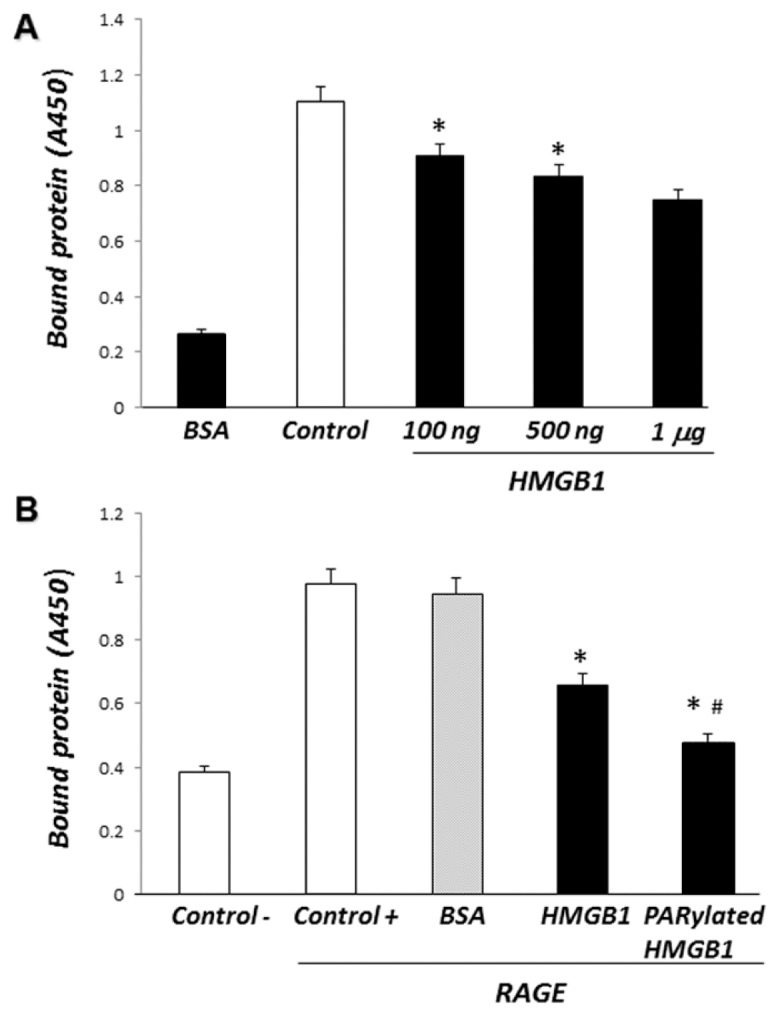
PARylated HMGB1 inhibits the binding of RAGE to PS more effectively than non-PARylated HMGB1. (A) PS-precoated 96-well plates were incubated with increasing concentrations of recombinant HMGB1 (100 ng/mL, 500 ng/mL or 1 μg/mL) for 2 h at room temperature. Recombinant mouse chimeric RAGE was then added to the wells for 2 h. The plates were washed and bound RAGE was quantified by ELISA. All assays were performed in triplicate. *P < 0.05 compared with control. (B) PS-precoated 96-well plates were incubated with recombinant HMGB1, PARylated HMGB1 or BSA (1 μg/mL) for 2 h at room temperature. Recombinant mouse chimeric RAGE was then added to the wells for 2 h. The plates were washed and bound RAGE was quantified by ELISA. All assays were done in triplicate. *P < 0.05 compared with control. #P < 0.05 compared with HMGB1.
Poly(ADP-ribosyl)ation of the HMGB1 C-Terminal Domain and A-Box by PARP-1 Increases Their Ability to Inhibit Efferocytosis
Previous studies have shown that HMGB1 and HMGB1 lacking the C-terminal tail can be PARylated by PARP-1 (30). However, it is not known whether the A and/or B box of HMGB1 can be PARylated. Using an in vitro poly(ADP-ribosyl)ation system, we found that His-tagged full-length HMGB1 and truncated C-terminal HMGB1 (ΔC-HMGB1) as well as the HMGB1 A box, but not the HMGB1 B box, can be modified by PARP-1 (Figure 6A).
Figure 6.

Poly(ADP-ribosyl)ation by PARP-1 of HMGB1 lacking the C-terminal tail and of HMGB1 A-Box, but not HMGB1 B-Box, potentiates the inhibitory effects of HMGB1 on efferocytosis. (A) PARylation of full-length HMGB1 and HMGB1 domains by PARP-1. Full-length HMGB1-His, ΔC-HMGB1-His, A Box-His or B Box-His (500 ng/mL) were incubated with recombinant PARP-1 in a poly(ADP-ribosyl)ation reaction containing NAD and activated DNA for 30 min. The reactions were terminated with sample buffer and subjected to immunoblot analysis with antibodies to PAR or His. Representative gels are shown. Two additional independent experiments provided similar results. (B) Phagocytosis assays were performed after preincubation of apoptotic thymocytes with 1 μg/mL unmodified or PARylated HMGB1-His, ΔC-HMGB1-His, A Box-His or B Box-His for 2 h. The thymocytes were then washed before being added to macrophages. *P < 0.05 compared with control. #P < 0.05 compared with thymocytes treated with non-PARylated HMGB1 or HMGB1 domains.
We next examined whether PARylation of ΔC-HMGB1 and the HMGB1 A box modifies their ability to affect efferocyto-sis. In these experiments, macrophages were preincubated with full-length HMGB1, ΔC-HMGB1, A box or B box that were either unmodified or had been subjected to in vitro poly(ADP-ribosyl)ation. After a 2-h incubation with HMGB1 or HMGB1 truncation mutants, apoptotic thymocytes were added to the macro phages and phagocytosis assays were performed. As shown in Figure 6B, PARylated ΔC-HMGB1 and A box inhibited efferocytosis, whereas their equivalent unmodified proteins failed to do so.
DISCUSSION
The release of the nuclear protein HMGB1 to the extracellular milieu has proinflammatory effects (35,36). High levels of circulating HMGB1 are found in acute and chronic inflammatory conditions, including severe infection, arthritis and ischemia/reperfusion injury (4,5). One mechanism by which HMGB1 can enhance inflammatory reactions is by inhibiting the recognition and clearance of apoptotic cells by macrophages and other phagocytes, thereby allowing apoptotic cells to progress to necrosis, with release of proinflammatory intracellular contents into the interstitial space (23). Because previous studies (17,20, 24,32) showed that highly purified HMGB1 has diminished effects on cell activation, such as cytokine production, the question of how HMGB1 contributes to inflammation remains an important question. A potential mechanism by which HMGB1 may acquire proinflammatory activity is through posttranslational modifications. Modifications of HMGB1 described in previous studies include phosphorylation (37,38) and acetylation (39–42). Monocytes and macrophages acetylate HMGB1 extensively after TLR4 engagement (42). Other studies reported that HMGB1 is phosphorylated in RAW 264.7 cells after TNF stimulation (38). However, none of these reports examined whether PARylation of HMGB1 occurred after cell activation or if PARylation of HMGB1 could affect its activity in relevant processes that contribute to inflammatory tissue injury.
In previous studies, the release of HMGB1 from the nucleus of DNA-damaged cells was shown to depend on PARP-1 activity (30). The present experiments confirm crosstalk between HMGB1 and PARP-1 and demonstrate that PARP-1 enzymatic activity is crucial for the efficient release of HMGB1 to the extracellular milieu after cellular activation through TLR4 engagement. In particular, we found that after LPS stimulation, PARP-1−/− MEFs as well as WT MEFs treated with a PARP inhibitor released significantly less HMGB1 than WT fibroblasts. Restoration of PARP-1 expression in PARP-1−/− fibroblasts reestablished secretion of HMGB1, whereas the addition of the enzymatically inactive PARP-1 mutant (E988K) did not. Thus, these experiments show that release of HMGB1 to the extracellular milieu requires not only direct protein–protein interaction with PARP-1, but also PARP-1 enzymatic activity, which results in HMGB1 poly(ADP-ribosyl)ation. These observations are supported by the fact that HMGB1, which is secreted after LPS treatment, is highly PARylated. Of note, in addition to extracellular HMGB1 being highly PARylated after release from TLR4-stimulated cells, intracellular HMGB1 that was isolated 24 h after LPS exposure also demonstrated PARylation. Therefore, not only do these studies suggest that poly(ADP-ribosyl)ation contributes to the release of HMGB1 into the extracellular milieu, but also that PARylation may contribute to the translocation of HMGB1 from the nucleus to cytoplasm. However, it is presently unclear if the structural type of PAR of intracellular and extracellular HMGB1 is the same; additional investigations are necessary to resolve this question.
CONCLUSION
Our data demonstrate that PARylation of HMGB1 by PARP-1 increases inhibition of efferocytosis and suggest a novel mechanism by which PARP-1 may contribute to inflammation (Figure 7). In particular, PARP-1 through PARylating HMGB1 may enhance the inhibitory effects of HMGB1 to diminish the clearance of apoptotic neutrophils and other apoptotic cells from inflammatory foci. As previously demonstrated (23,32), the present experiments confirmed that incubation of either apoptotic cells or macrophages with unmodified HMGB1 resulted in diminished efferocytosis. However, preincubation of macrophages or apoptotic cells with PARylated HMGB1 inhibited efferocytosis to a greater degree than exposure to unmodified HMGB1. The mechanism for such inhibitory actions of PARylated HMGB1 appears to be through binding more effectively to PS on the apoptotic cell membrane and to RAGE on the macrophage than non-PARylated HMGB1. The in vivo relevance of HMGB1 PARylation was shown in the present experiments by the enhanced ability of PARylated HMGB1 to diminish the uptake of intratracheally administered apoptotic thymocytes by alveolar macrophages.
Figure 7.

Proposed mechanism by which PARylation enhances HMGB1-associated inhibition of efferocytosis. PARylation increases the ability of HMGB1 to inhibit efferocytosis by binding more efficiently to PS on apoptotic cells and to RAGE on macrophages, thereby diminishing association of the apoptotic cell with the macrophage by interfering with binding between RAGE and PS.
In previous studies, we found that HMGB1 inhibits efferocytosis through binding to PS on apoptotic cells (23) and to αvβ3 and RAGE on macrophages (32,43). In addition, we and others have shown that RAGE has the ability to bind directly to PS (14,15). In the present experiments, we demonstrated that HMGB1 inhibits efferocytosis by blocking interactions between RAGE and PS and also that PARylated HMGB1 binds with greater affinity to both PS and RAGE than unmodified HMGB1. It is noteworthy that preincubation of PARylated HMGB1 with RAGE−/− macrophages did not completely prevent binding of PARylated HMGB1, suggesting that PARylated HMGB1 can interact with macrophage receptors other than RAGE.
Full-length and HMGB1 lacking the C-terminal tail have been shown to be PARylated by PARP-1 (30). In the present experiments, we confirmed such PARylation by PARP-1 and also demonstrated that the HMGB1 A box is PARylated by PARP-1. In contrast, HMGB1 B box failed to be modified by PARP-1. These results suggest that poly(ADP-ribosyl)ation sites reside within amino acids 1–81 and 186–215 of HMGB1.
Activation of PARP-1 has been shown to trigger relocalization of HMGB1 away from chromatin, allowing HMGB1 to be released from necrotic cells (30). However, there was no information available concerning a potential effect of PARylation on the proinflammatory activity of released HMGB1. In a previous study, we demonstrated that full-length HMGB1 diminished the uptake of apoptotic neutrophils, and this action was lost when the C-terminal tail was absent (27). In the present experiments, we found that PARylated HMGB1 lacking the C-terminal tail does not completely lose its inhibitory effect on efferocytosis, indicating that the presence of the C-terminal tail is not required for ability of HMGB1 to diminish efferocytosis as long as PARylation occurs. Furthermore, we found that PARylated HMGB1 A box inhibits the phagocytosis of apoptotic cells, whereas unmodified A box does not have any effect. Further work is necessary to determine the minimum number and location of PARylation sites in HMGB1 that need to be modified by PARP-1 to inhibit efferocytosis.
Supplemental Data
ACKNOWLEDGMENTS
This work was supported by grant 11SDG5330014 from the American Heart Association to M Zerfaoui.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med. 1999;160:S5–11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 2.Voll RE, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 3.Fadok VA, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ulloa L, Batliwalla FM, Andersson U, Gregersen PK, Tracey KJ. High mobility group box chromosomal protein 1 as a nuclear protein, cytokine, and potential therapeutic target in arthritis. Arthritis Rheum. 2003;48:876–81. doi: 10.1002/art.10854. [DOI] [PubMed] [Google Scholar]
- 5.Czura CJ, Yang H, Amella CA, Tracey KJ. HMGB1 in the immunology of sepsis (not septic shock) and arthritis. Adv Immunol. 2004;84:181–200. doi: 10.1016/S0065-2776(04)84005-7. [DOI] [PubMed] [Google Scholar]
- 6.Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–82. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- 7.Vandivier RW, et al. Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. Am J Physiol Lung Cell Mol Physiol. 2009;297:L677–86. doi: 10.1152/ajplung.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishii Y, et al. Elimination of neutrophils by apoptosis during the resolution of acute pulmonary inflammation in rats. Lung. 1998;176:89–98. doi: 10.1007/pl00007597. [DOI] [PubMed] [Google Scholar]
- 9.Cox G, Crossley J, Xing Z. Macrophage engulfment of apoptotic neutrophils contributes to the resolution of acute pulmonary inflammation in vivo. Am J Respir Cell Mol Biol. 1995;12:232–7. doi: 10.1165/ajrcmb.12.2.7865221. [DOI] [PubMed] [Google Scholar]
- 10.Ravichandran KS. “Recruitment signals” from apoptotic cells: invitation to a quiet meal. Cell. 2003;113:817–20. doi: 10.1016/s0092-8674(03)00471-9. [DOI] [PubMed] [Google Scholar]
- 11.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 235:172–89. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park D, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–4. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 13.Park SY, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15:192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- 14.Friggeri A, et al. Participation of the receptor for advanced glycation end products in efferocytosis. J Immunol. 2011;186:6191–8. doi: 10.4049/jimmunol.1004134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He M, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011;12:358–64. doi: 10.1038/embor.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. 2005;15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86:573–6. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- 18.Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26:381–7. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Muller S, et al. New EMBO members’ review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J. 2001;20:4337–40. doi: 10.1093/emboj/20.16.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin YH, et al. HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J Immunol. 2009;183:6244–50. doi: 10.4049/jimmunol.0900390. [DOI] [PubMed] [Google Scholar]
- 21.Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–7. doi: 10.4049/jimmunol.180.4.2531. [DOI] [PubMed] [Google Scholar]
- 22.Campana L, Bosurgi L, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 for stromal cell-derived factor-1/CXCL12-dependent migration of macrophages and dendritic cells. J Leukoc Biol. 2009;86:609–15. doi: 10.1189/jlb.0908576. [DOI] [PubMed] [Google Scholar]
- 23.Liu G, et al. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol. 2008;181:4240–6. doi: 10.4049/jimmunol.181.6.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H. Pivotal advance: analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin) J Leukoc Biol. 2007;81:49–58. doi: 10.1189/jlb.0306200. [DOI] [PubMed] [Google Scholar]
- 25.Park JS, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 26.Erlandsson Harris H, Andersson U. Mini-review: the nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol. 2004;34:1503–12. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- 27.Banerjee S, Friggeri A, Liu G, Abraham E. The C-terminal acidic tail is responsible for the inhibitory effects of HMGB1 on efferocytosis. J Leukoc Biol. 88:973–9. doi: 10.1189/jlb.0510262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–67. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 29.Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays. 2001;23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 30.Ditsworth D, Zong WX, Thompson CB. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. 2007;282:17845–54. doi: 10.1074/jbc.M701465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffin RJ, et al. Resistance-modifying agents. 5. Synthesis and biological properties of quinazolinone inhibitors of the DNA repair enzyme poly(ADP-ribose) polymerase (PARP) J Med Chem. 1998;41:5247–56. doi: 10.1021/jm980273t. [DOI] [PubMed] [Google Scholar]
- 32.Friggeri A, et al. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol. 299:C1267–76. doi: 10.1152/ajpcell.00152.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 2006;16:189–97. doi: 10.1016/j.tcb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 34.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964–74. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 36.Andersson U, Tracey KJ. HMGB1 Is a Therapeutic Target for Sterile Inflammation and Infection. Annu Rev Immunol. 2010;23:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wisniewski JR, Szewczuk Z, Petry I, Schwanbeck R, Renner U. Constitutive phosphorylation of the acidic tails of the high mobility group 1 proteins by casein kinase II alters their conformation, stability, and DNA binding specificity. J Biol Chem. 1999;274:20116–22. [PubMed] [Google Scholar]
- 38.Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177:7889–97. doi: 10.4049/jimmunol.177.11.7889. [DOI] [PubMed] [Google Scholar]
- 39.Ugrinova I, Mitkova E, Moskalenko C, Pashev I, Pasheva E. DNA bending versus DNA end joining activity of HMGB1 protein is modulated in vitro by acetylation. Biochemistry. 2007;46:2111–7. doi: 10.1021/bi0614479. [DOI] [PubMed] [Google Scholar]
- 40.Ugrinova I, Pasheva EA, Armengaud J, Pashev IG. In vivo acetylation of HMG1 protein enhances its binding affinity to distorted DNA structures. Biochemistry. 2001;40:14655–60. doi: 10.1021/bi0113364. [DOI] [PubMed] [Google Scholar]
- 41.Evankovich J, et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 285:39888–97. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonaldi T, et al. Monocytic cells hyper-acetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–60. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banerjee S, Friggeri A, Liu G, Abraham E. The C-terminal acidic tail is responsible for the inhibitory effects of HMGB1 on efferocytosis. J Leukoc Biol. 2010;88:973–9. doi: 10.1189/jlb.0510262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.