

Abstract
The pathogenesis of sepsis is characterized by overwhelming inflammatory responses that lead to tissue damage and organ failure. Toll-like receptor (TLR) signaling is crucial for induction of hyperinflammatory responses and tissue injury during sepsis. Genipin, an aglycon of geniposide, has antiinflammatory and antimicrobial activities. The purpose of this study was to test the hypothesis that genipin reduces multiple organ dysfunction and mortality during sepsis through inhibition of TLR signaling. Male ICR were subjected to sepsis by cecal ligation and puncture (CLP) or endotoxemia by lipopolysaccharide (LPS). Various doses of genipin (1, 2.5 and 5 mg/kg) or a vehicle were administered intravenously immediately after CLP or intraperitoneally after LPS treatment. In another set of survival tests, mice were treated with 2.5 mg/kg of genipin 0 and 24 h after CLP. Genipin was found to improve survival and to attenuate multiple organ dysfunction. Genipin attenuated production of proinflammatory cytokines and release of high-mobility group box 1 (HMGB1). Genipin prevented TLR2 and TLR4, myeloid differentiation factor 88 and the Toll/interleukin-1 receptor domain-containing adaptor protein, inducing interferon-β overexpression. Phosphorylation of mitogen-activated protein kinases and interferon regulatory factor 3 and translocation of nuclear factor (NF)-κB were prevented by genipin. Moreover, genipin attenuated increases in serum tumor necrosis factor-α and HMGB1 in LPS-induced endotoxemia. Pam3CSK4- and LPS-mediated production of nitrites and proinflammatory cytokines was suppressed by genipin in RAW264.7 cells. Genipin attenuated mortality and organ injuries during sepsis through interference with TLR signaling. Therefore, genipin might be useful as a potential therapeutic agent for treatment of sepsis.
INTRODUCTION
Sepsis, leading to multiple organ failure, remains a leading cause of mortality and morbidity in intensive care units. An uncontrolled hyperinflammatory response and inappropriate cytokine response during early sepsis have been proposed as the cause of multiple organ dysfunction syndrome during sepsis. Control of inflammation during early sepsis may therefore reduce organ injury and prevent death after septic insult.
Complex Toll-like receptor (TLR) signaling and associated downstream regulators play a crucial role in the innate immune system as the first line of defense against pathogens (1). TLR2 and TLR4 have been regarded as the main sensors for recognition of pathogen-associated molecular patterns from gram-positive and gram-negative bacteria, respectively. TLR2 and TLR4 mRNA expression in the lung and liver of septic mice showed a significant increase, and the signal transduction inhibitors of TLRs and downregulation of TLRs demonstrated improved survival in murine models of sepsis (2,3). In addition, monocytic expression of TLR2 and TLR4 in septic patients was also significantly upregulated, compared with expression in healthy individuals (4). Downstream TLR signaling occurs via two major pathways: the myeloid differentiation factor 88 (MyD88)- dependent pathway and the Toll/ interleukin (IL)-1 receptor domain, containing the adaptor protein–inducing interferon (IFN)-β (TRIF) pathway, which finally activates production of proinflammatory mediators (5).
Genipin is an aglycon of geniposide, the major active compound of gardenia fruit, which has long been used in traditional medicine formulations for treatment of inflammation and hepatic disorders (6). Genipin inhibited carrageenan-induced rat paw edema, croton oil–induced ear edema in mice and changes in vascular permeability induced by acetic acid (6,7). In murine macrophage cells, genipin blocked nitric oxide production on stimulation by lipopolysaccharide (LPS)/IFN and inhibited LPS-induced degradation of the inhibitor of nuclear factor (NF)-κB-α (IκB-α) and NF-κB activation. Genipin also reduced the lethality induced by d-galactosamine/LPS-induced fulminant hepatic failure through prevention of oxidative stress, apoptosis and NF-κB nuclear translocation (8).
Therefore, this study was conducted for investigation of the effect of genipin on septic injury and the specific molecular mechanisms of protection, particularly on the TLR signaling pathways.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM), Dulbecco’s phosphate-buffered saline (PBS), penicillin/streptomycin (10,000 U/mL/10,000 μg/mL, respectively) and fetal bovine serum (FBS) were obtained from Gibco BRL, Life Technologies (Grand Island, NY, USA). Pam3CSK4, a TLR2 agonist, was obtained from InvivoGen (San Diego, CA, USA). LPS (Escherichia coli serotype 0127:B8), a TLR4 agonist, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-tetrazolium bromide (MTT) and all the other materials required for culturing cells were purchased from Sigma (St. Louis, MO, USA). All the other chemicals used in this study were of reagent grade.
Animals
Male ICR mice, weighing 27–29 g, were supplied by Orient Bio (Seongnam, Korea). The animals were housed in cages located in temperature-controlled rooms with a 12:12 h light–dark cycle and received water and food ad libitum. All animal procedures were approved by the Sungkyunkwan University Animal Care Committee and were performed in accordance with the guidelines of the National Institutes of Health.
Cecal Ligation and Puncture
Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) according to the method described by Chaudry et al. (9). After anesthetization with ether, a 1-cm ventral midline incision was performed. The cecum was then carefully exposed, ligated just distal to the ileocecal valve to avoid intestinal obstruction and punctured twice using a 20-gauge needle. The punctured cecum was squeezed to expel a small amount of fecal material and returned to the abdominal cavity, and the abdominal incision was closed in two layers. All animals received subcutaneous administration of 1 mL normal saline immediately after the operation (that is, fluid resuscitation). Sham-operated animals were subjected to laparotomy and intestinal manipulation; however, the cecum was neither ligated nor punctured.
Administration of Genipin and Experimental Design
In survival experiments, mice received intravenous administration of genipin (1, 2.5 and 5 mg/kg) or saline (vehicle) via tail vein immediately (0 h) or 0 and 24 h after CLP. Mortality was recorded for up to 10 d after the procedure; survivors were followed for 3 wks to ensure that no late mortalities had occurred. On the basis of a survival test, a dose of 2.5 mg/ kg was chosen for further biochemical studies. The animals were randomly assigned to the following four groups (each group, n = 6–8): (a) vehicle-treated sham (sham), (b) shame treated with 2.5 mg/ kg (genipin), (c) vehicle-treated CLP (CLP), and (d) CLP treated with 2.5 mg/ kg genipin (CLP + genipin). Lung and liver tissue were isolated at 6 and 24 h after CLP and immediately stored at −75°C until assayed. Blood samples were collected from the abdominal inferior vena cava at various different time points after CLP.
Endotoxemia
Endotoxemia was induced by intraperitoneal injection of LPS (10 mg/kg). In survival tests, 1, 2.5 and 5 mg/kg genipin was intraperitoneally administered to mice immediately and 24 after LPS treatment. Mortality was recorded for up to 10 d after the LPS injection. On the basis of a survival test, a dose of 2.5 mg/kg was chosen for further biochemical studies. Blood samples were collected from the abdominal inferior vena cava at 1 and 24 h after LPS treatment.
Assessment of Hepatic, Renal and Heart Damage
Under anesthesia, blood samples were collected at 1, 3, 6, 12, 24 and 48 h after CLP. Serum was separated by centrifugation at 7,700g for 10 min at 4°C. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST), blood urea nitrogen (BUN), creatinine and lactate dehydrogenase (LDH) levels were determined using a Hitachi 7600 automatic analyzer (Hitachi, Tokyo, Japan).
Histological Analysis
Twenty-four hours after CLP, tissue samples of liver and lung were removed for histological analysis. Each sample was fixed by immersion in 10% neutral-buffered formalin. The sample was then embedded in paraffin, sliced into 5-μm sections and stained with hematoxylin and eosin for a blinded histological assessment. For lung injury, vascular congestion, edema and inflammatory cell infiltration were assessed and, for liver injury, hepatocellular necrosis, portal inflammation and inflammatory cell infiltration were evaluated. Each parameter was graded on a scale of 0–3, with 0 meaning “absent,” 1 meaning “mild,” 2 meaning “moderate,” and 3 meaning “severe.” The total injury score was expressed as the sum of the scores for all parameters, with the maximum being 9. Histological changes were evaluated in random, nonconsecutive and 200× histological fields (Olympus BX51/ Olympus DP71, Olympus, Japan).
Cell Culture and Treatment
Mouse macrophage cell line RAW264.7 cells were obtained from American Type of Culture Collection (Manassas, VA, USA). The cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin and maintained at 37°C in a humidified atmosphere with 5% CO2. Experiments were carried out 24 h after the cells were seeded. Cells were plated at a density of 3 × 105 cells/mL in six-well plates with 3 mL culture medium, incubated for 24 h, pretreated with genipin (1, 10, 50 and 100 μmol/L) or a vehicle (dimethyl sulfoxide [DMSO]) for another 2 h and then challenged with Pam3CSK4 or LPS (1 μg/mL). After 24 h incubation, the culture media and cell lysates were harvested for further analysis.
Cell Viability
Cells (1 × 104 cells/well) were seeded in a 96-well plate and incubated at 37°C for 24 h. The cells were treated with various concentrations of genipin or a vehicle (DMSO) and were incubated at 37°C for an additional 24 h. After incubation, 100 μL MTT solution (5 mg/mL in PBS) was added to each well, and the plates was further incubated for 3 h at 37°C. Subsequently, 100 μL DMSO was added to each well to solubilize any deposited formazan. The optical density of each well was measured at 450 nm using a microplate reader (Emax; Molecular Devices, Sunnyvale, CA, USA).
Nitrite Assay
RAW264.7 cells were plated at a density of 2 × 105 cells/well in a 24-well cell culture plate, incubated for 24 h, pre-treated with genipin (1, 10, 50 and 100 μmol/L) or a vehicle for another 2 h and then challenged with Pam3CSK or LPS (1 μg/mL) for an additional 24 h. Equal volumes of culture medium and Griess reagent (1% sulfanilamide in 5% phosphoric acid and 0.1% naphthylethyl-enediamine dihydrochloride in distilled water) were mixed, and the absorbance of the mixture at 540 nm was determined with a microplate reader (Emax).
Cytokine Levels
Concentrations of tumor necrosis factor (TNF)-α, IL-1β and IL-6 were measured using commercially available enzyme-linked immunosorbent assay (ELISA) kits (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s instructions.
High-Mobility Group Box 1 (HMGB1) Preparation
Serum and culture medium were filtered and concentrated through Centricon YM-100 and YM-10 (Millipore, Billerica, MA, USA) with a fixed angle (35°) at 7,500g for 15 min, 4°C. The concentrated samples were then subjected to sodium dodecyl sulfate– polyacrylamide gel electrophoresis (SDS-PAGE).
Total, Cytosolic and Nuclear Protein Extraction
Isolated liver and lung tissues and cells were homogenized in PRO-PREP™ Protein Extraction Solution (iNtRON Biotechnology, Seongnam, Korea) in a microcentrifuge tube. After standing in a cold ice-bath for a period of 30 min for cell lysis, the whole homogenate was centrifuged at 13,000g for 5 min. NE-PER® (Pierce Biotechnology, Rock-ford, IL, USA) was used for extraction of nuclear and cytosolic fractions according to the manufacturer’s instructions. Protein concentrations were determined using the BCA Protein Assay kit (Pierce Biotechnology).
Western Blot Analysis
Protein samples were loaded on 7.5–15% polyacrylamide gels and were then separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes using the Semi-Dry Trans-Blot Cell (Bio-Rad Laboratories, Hercules, CA, USA). After transfer, the membranes were washed with Tris-buffered saline with 0.1% Tween-20 (TBS/T) and blocked for 1 h at room temperature with 5% skim milk powder in TBS/T. Blots were then incubated overnight at 4°C with primary antibodies. After washing three times for 5 min each in TBS/T, the membranes were incubated with appropriate secondary antibodies for 1 h at room temperature, followed by detection using an ECL detection system (iNtRON Biotechnology), according to the manufacturer’s instructions. Intensity of the immunoreactive bands was determined using Total-Lab TL120 software (Nonlinear Dynamics, Newscastle, UK). Primary antibodies against mouse TLR2, TLR4, MyD88, NF-κB/p65 and IκB-α (Santa Cruz Biotechnology, Santa Cruz, CA, USA); TRIF and high-mobility group box (HMGB)-1 (Abcam, Cambridge, MA, USA); p38, extracellular signal–regulated kinase (ERK), c-Jun N-terminal kinase (JNK), IFN regulatory factor (IRF)-3, phospho-p38, phospho-ERK, and phospho-JNK; and phospho-IRF3 (Cell Signaling Technology, Beverly, MA, USA) were used and the signals were standardized to β-actin (Sigma) for whole lysate and lamin B1 (Abcam) for the nuclear fraction.
Statistical Analysis
Survival data were analyzed by the Kaplan-Meier curve and log-rank test. All other data were analyzed by two-way analysis of variance, and the Bonferroni test was used for post hoc comparisons. Differences between the groups were considered statistically significant at P < 0.05. Results are presented as mean ± standard error of the mean (SEM).
RESULTS
Effect of Genipin on CLP-Induced Mortality
In the vehicle-treated CLP group, the survival rate on the first day of observation was 60% and reached a stable 20% survival on the fifth day after CLP. Log-rank analysis of 10-d survival demonstrated that genipin at doses of 2.5 and 5 mg/kg significantly improved survival of septic mice, compared with sham animals (P = 0.0119 and P = 0.0435, respectively; Figure 1A). Repeated administration of 2.5 mg/kg genipin at 24-h intervals resulted in a further increase of the survival rate at 10 d after CLP (Figure 1B).
Figure 1.
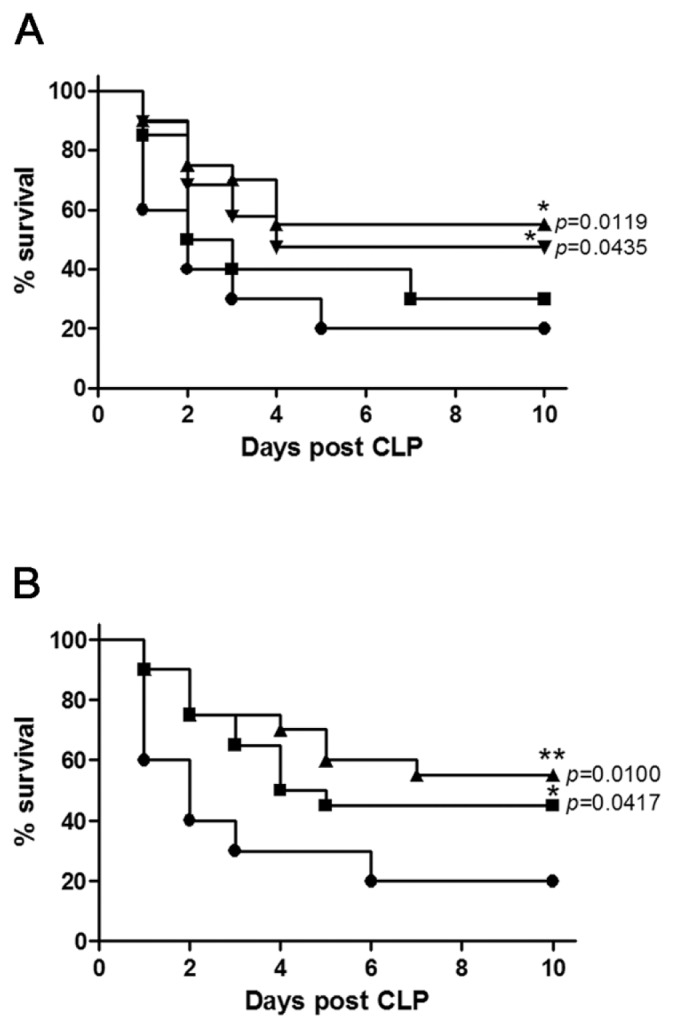
Genipin protects mice against sepsis-induced lethality. (A) Mice were intravenously administered various doses of genipin or vehicle immediately after CLP (n = 20). Significantly different (*P < 0.05) from CLP. (B) Mice were intravenously administered 2.5 mg/kg genipin at 0 h or 0 and 24 h after CLP (n = 20). Significantly different (*P < 0.05, **P < 0.01) from saline. A: ●, CLP; ■, CLP + 1 mg/kg genipin; ▲, CLP + 2.5 mg/kg genipin; ▼, CLP + 5 mg/kg genipin; B: ●, saline; ■, 0 h; ▲, 0 and 24 h.
Effect of Genipin on Organ Injuries Induced by CLP
Serum ALT activity in the sham group remained at 9.8 ± 0.8 to 16.1 ± 1.5 U/L throughout the experiments. Whereas no significant change was observed in ALT activity until 3 h after CLP, serum ALT activity began to increase from 6 h after CLP, peaked at 202.8 ± 23.8 U/L at 24 h after CLP and then declined to 88.3 ± 4.2 U/L at 48 h after CLP. Increases in serum ALT activity that occurred at 12 and 24 h after CLP were attenuated by genipin. Serum AST activity in the sham group remained at the basal level until 48 h after surgery. Serum AST activity began to increase from 3 h after CLP, peaked at 375.6 ± 50.8 U/L at 24 h after CLP and then declined to 196.0 ± 23.3 U/L at 48 h after CLP. Genipin attenuated elevations in serum AST activity that occurred at 12 and 24 h after CLP. The serum BUN level averaged 11.4 ± 0.7 mg/dL in the sham group. The serum BUN level began to increase from 6 h after CLP; this elevation persisted until 12 h (66.9 ± 10.6 mg/dL) and, thereafter, showed a gradual decrease until 48 h after CLP. Elevations in the serum BUN level that occurred at 12 and 24 h after CLP were attenuated by genipin. Serum creatinine level showed elevation from 3 h after CLP, was maximal at 0.43 ± 0.05 mg/dL at 24 h after CLP and, thereafter, decreased to 0.25 ± 0.02 mg/dL at 48 h after CLP. Genipin attenuated increases in the serum creatinine level that occurred at 12 and 24 h after CLP. Serum LDH activity began to increase from 1 h after CLP and further increased until 24 h after CLP (2691.0 ± 482.4 IU/L) and, thereafter, declined to 940.0 ± 201.5 U/L at 48 h after CLP. Genipin attenuated the increases occurring at 12 and 24 h after CLP (Figure 2).
Figure 2.

Effect of genipin on the serum ALT (A), AST (B), BUN (C), creatinine (D), and LDH (E) level. Mice were intravenously administered 2.5 mg/kg genipin immediately after CLP. The results are presented as mean ± SEM (n = 8). Significantly different (*P < 0.05, **P < .01) from sham. Significantly different (+P < 0.05, ++P < 0.01) from CLP. ●, Sham; ■, genipin; ▲, CLP; ▼, CLP + genipin.
Histological Analysis
Histological features revealed normal cell structure in the liver and lung of sham-operated animals. Liver sections isolated 24 h after CLP showed increase in inflammatory cell infiltration, as well as moderate portal inflammation and hepatocellular necrosis. CLP induced marked histopathological changes, including edema, interstitial inflammation and inflammatory cell infiltration in lung tissues obtained from septic animals at 24 h after CLP. These pathological changes in both lung and liver tissues were attenuated by genipin (Figure 3).
Figure 3.

Histological micrographs of liver (A, B, C, D) and lung (F, G, H, I) tissues stained with hematoxylin and eosin (magnification 200×): sham (A, F), genipin (B, G), CLP (C, H), and CLP + genipin (D, I). Mice were intravenously administered 2.5 mg/kg genipin immediately after CLP. Twenty-four hours after CLP, liver and lung tissues were obtained. Semi-quantitative pathology scores in liver and lung (E, K) were determined by a scoring system as described in Materials and Methods. Representative images were chosen from the different experimental groups. Significantly different (**P < 0.01) from sham. Significantly different (+P < 0.05, ++P < 0.01) from CLP.
Effect of Genipin on CLP-Induced Serum Cytokine Levels
In the sham group, the serum level of TNF-α was between 31.3 ± 2.1 and 32.5 ± 2.6 pg/mL at all experimental time points. The serum TNF-α level began to increase from 1 h after CLP, peaked at 373.6 ± 41.7 pg/mL 3 h after CLP and showed a gradual decrease until 12 h after CLP, followed by a slight elevation at 24 h after CLP. Genipin induced marked suppression of increases in the serum TNF-α level that occurred at 3, 6 and 24 h after CLP (Figure 4A). Serum concentrations of IL-1β and IL-6 were very low in the sham group at all time points (IL-1β, 30.2 ± 2.2 to 34.6 ± 0.9 pg/mL; IL-6, 22.0 ± 1.3 to 31.0 ± 2.1 pg/mL). In the CLP group, the serum IL-1β level began to increase from 3 h after CLP, peaked at 136.5 ± 11.4 pg/mL at 6 h after CLP and, thereafter, showed a gradual decline until 24 h after CLP. Genipin attenuated increases that occurred at 6, 12 and 24 h after CLP (Figure 4B). The serum IL-6 level showed a significant increase at 3, 6 and 12 h after CLP, compared with that of the sham group (8.0 ± 1.0, 19.7 ± 2.2 and 22.5 ± 1.9 ng/mL, respectively). Genipin induced marked suppression of these increases (Figure 4C).
Figure 4.

Effect of genipin on the serum TNF-α (A), IL-1β (B), IL-6 (C) and HMGB1 (D) levels. Mice were intravenously administered 2.5 mg/kg genipin immediately after CLP. (A–C) The serum was collected at various time points and assayed for TNF-α, IL-1β and IL-6 levels by use of ELISA kits. (D) Twenty-four hours after CLP, the serum was obtained and assayed for HMGB1 levels by Western blot analysis. The results are presented as mean ± SEM (n = 8). Significantly different (**P < 0.01) from sham. Significantly different (++P < 0.01) from CLP. ●, Sham; ■, genipin; ▲, CLP; ▼, CLP + genipin.
Effect of Genipin on CLP-Induced Serum HMGB1 Release
The serum level of HMGB1 increased approximately 6.6-fold that of the sham group at 24 h after CLP. Genipin attenuated this increase (Figure 4D).
Effect of Genipin on CLP-Induced TLR2 and TLR4, MyD88 and TRIF Protein Expression
In both lung and liver, the levels of TLR2 and TLR4 and MyD88 protein expression showed a significant increase at 6 and 24 h after CLP, compared with levels of the sham group, which was attenuated by genipin. The level of TRIF protein expression showed a marked increase at 6 and 24 h after CLP in liver and 24 h after CLP in lung, respectively. Genipin attenuated increases in the level of TRIF protein expression that occurred at 24 h after CLP in both lung and liver (Figures 5 and 6).
Figure 5.

Effect of genipin on the TLR2 (A, B) and TLR4 (C, D) protein expression levels 6 and 24 h after CLP. Left and right panels show the data from liver and lung tissues, respectively. Mice were intravenously administered 2.5 mg/kg genipin immediately after CLP. The results are presented as mean ± SEM (n = 6). Significantly different (*P < 0.05, **P < 0.01) from sham. Significantly different (+P < 0.05, ++P < 0.01) from CLP. AU, arbitrary units.
 , Sham;
, Sham;
 , genipin; ▤, CLP; ▥, CLP + genipin.
, genipin; ▤, CLP; ▥, CLP + genipin.
Figure 6.

Effect of genipin on the MyD88 (A, B) and TRIF (C, D) protein expression levels 6 and 24 h after CLP. Left and right panels show the data from liver and lung tissues, respectively. Mice were intravenously administered 2.5 mg/kg genipin immediately after CLP. The results are presented as mean ± SEM (n = 6). Significantly different (*P < 0.05, **P < 0.01) from sham. Significantly different (+P < 0.05, ++P < 0.01) from CLP. AU, arbitrary units.
, Sham;
, genipin; ▤, CLP; ▥, CLP + genipin.
Effect of Genipin on CLP-Induced Mitogen-Activated Protein Kinase Activation
In the lung, p38, ERK and JNK were markedly phosphorylated at 6 and 24 h after CLP. Genipin induced markedly suppressed phosphorylation of p38 and ERK. In the liver, phosphorylation of p38, ERK and JNK was significantly elevated at 6 h after CLP, which was prevented by genipin. At 24 h after CLP in the liver, phosphorylation of ERK and JNK was markedly increased. Genipin prevented these phosphorylations (Figure 7).
Figure 7.

Effect of genipin on the phosphorylation of p38 (A, B), ERK (C, D) and JNK (E, F) 6 and 24 h after CLP. Left and right panels show the data from liver and lung tissues, respectively. Mice were intravenously administered 2.5 mg/kg genipin immediately after CLP. The results are presented as mean ± SEM (n = 6). Significantly different (*P < 0.05, **P < 0.01) from sham. Significantly different (+P < 0.05, ++P < 0.01) from CLP.
, Sham;
, genipin; ▤, CLP; ▥, CLP + genipin.
Effect of Genipin on CLP-Induced NF-κB/p65 Nuclear Translocation, IκB-α Expression and IRF3 Phosphorylation
Translocation of NF-κB/p65 to the nucleus in both lung and liver showed a significant increase at 6 and 24 h after CLP (3.9- and 2.4-fold in the lung and 7.0- and 5.4-fold in the liver, compared with the sham group, respectively). These increases were blocked by genipin. The level of cytosolic IκB-α protein expression in both lung and liver showed a marked reduction at 6 and 24 h after CLP (78.1% and 70.1% in the lung and 67.9% and 64.1% in the liver, compared with the sham group, respectively), which was prevented by genipin (Figure 8). Phosphorylation of IRF3 in the liver increased 3.1- and 3.4-fold the levels in the sham group at 6 and 24 h after CLP, respectively. In the lung, phosphorylation of IRF3 showed an increase at 24 h after CLP. Genipin suppressed phosphorylation of IRF3 that occurred at 24 h after CLP in both lung and liver (Figure 9).
Figure 8.

Effect of genipin on the nuclear NF-κB translocation and cytosolic IκB-α protein expression levels 6 and 24 h after CLP. Left and right panels show the data from liver and lung tissues, respectively. Mice were intravenously administered 2.5 mg/kg genipin immediately after CLP. The results are presented as mean ± SEM (n = 6). Significantly different (*P < 0.05, **P < 0.01) from sham. Significantly different (+P < 0.05, ++P < 0.01) from CLP. AU, arbitrary units.
, Sham;
, genipin; ▤, CLP; ▥, CLP + genipin.
Figure 9.
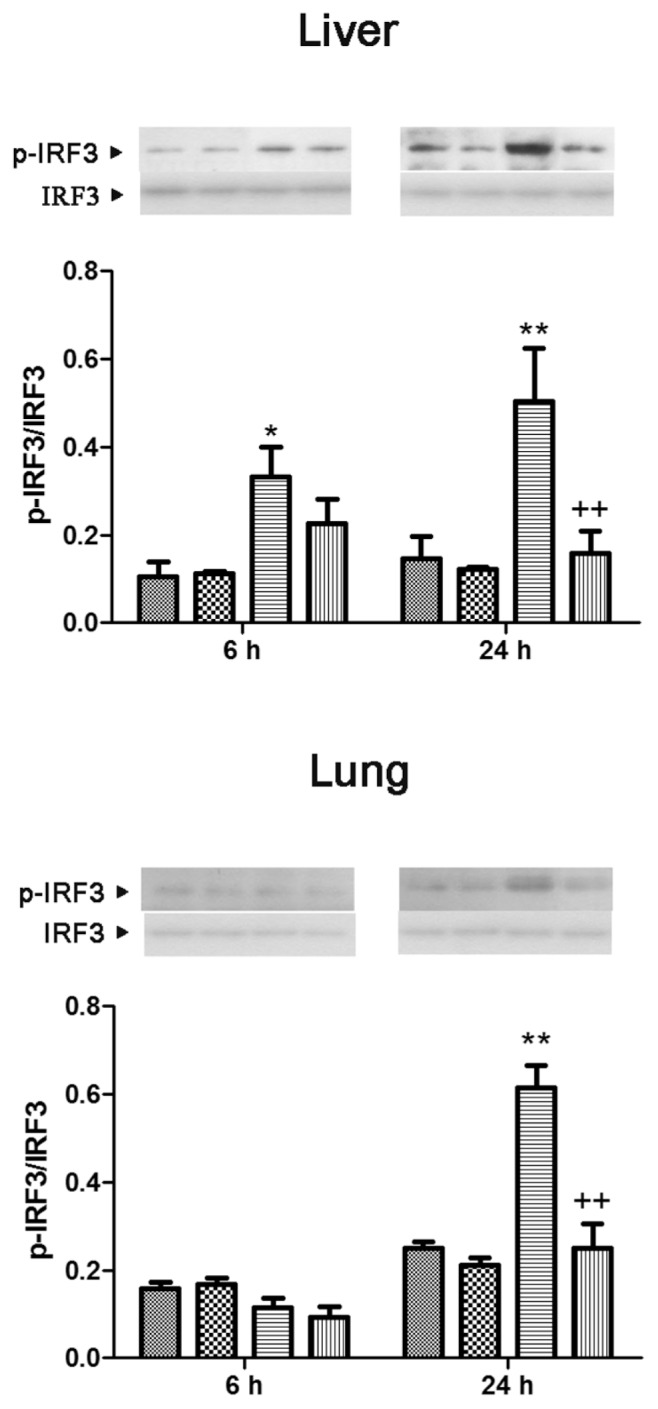
Effect of genipin on the phosphorylation of IRF3 6 and 24 h after CLP. Upper and lower panels show the data from liver and lung tissues, respectively. Mice were intravenously administered 2.5 mg/kg genipin immediately after CLP. The results are presented as mean ± SEM (n = 6). Significantly different (*P < 0.05, **P < 0.01) from sham. Significantly different (++P < 0.01) from CLP.
, Sham;
, genipin; ▤, CLP; ▥, CLP + genipin.
Effect of Genipin on LPS-Induced Mortality
The survival rate on the first day of observation was 60% and reached a stable 20% survival on the sixth day in vehicle-treated LPS-challenged mice. Genipin, 2.5 and 5 mg/kg, significantly improved survival of LPS-treated mice, compared with vehicle-treated animals (P = 0.0067 and P = 0.0390, respectively). Genipin, 1 mg/kg, did not confer the protection against LPS-induced lethality (Figure 10A).
Figure 10.

Effect of genipin on the mortality (A), serum TNF-α (B) and HMGB1 (C) level in LPS-induced endotoxemia. LPS, 10 mg/kg, was intraperitoneally given to mice to induce endotoxemia. Mice were intraperitoneally administered 1, 2.5 and 5 mg/kg genipin immediately after LPS injection in a survival test (n = 20). During the survival test, 2.5 mg/kg genipin was chosen for further biochemical studies. Serum level of TNF-α and HMGB1 was assessed 1 and 24 h after LPS injection, respectively. The results are presented as mean ± SEM (n = 6). Significantly different (**P < 0.01) from control. Significantly different (++P < 0.01) from LPS. A: ●, LPS; ■, LPS + 1 mg/kg genipin; ▲, LPS + 2.5 mg/kg genipin; ▼, LPS + 5 mg/kg genipin.
Effect of Genipin on LPS-Induced Serum TNF-α and HMGB1 Levels
The serum level of TNF-α in the control group was 10.04 ± 2.0 pg/mL. One hour after LPS injection, the serum level of TNF-α markedly increased (66.48 ± 7.1 pg/mL), which was prevented by 2.5 mg/kg genipin (Figure 10B). The serum level of HMGB1 24 h after the LPS challenge increased by approximately 5.7-fold that of the control group. Genipin significantly attenuated this increase (Figure 10C).
Effect of Genipin on Nitrites and Proinflammatory Cytokine Production in Pam3CSK4- or LPS-Treated RAW264.7 Cells
Genipin did not display any cellular toxicity on RAW264.7 cells up to 100 μmol/L (Figure 11A). As shown in Figure 11B, Pam3CSK4 and LPS increased nitrite production by approximately 11.4-and 27.3-fold, respectively, and genipin markedly suppressed nitrite production at the concentration of 50 and 100 μmol/L. After 24 h incubation of cells with Pam3CSK4, the production of TNF-α, IL-1β and IL-6 markedly increased (0.06 ± 0.003 to 24.2 ± 1.7 ng/mL, 124.7 ± 3.1 to 233.9 ± 7.8 pg/mL and 38.1 ± 1.5 to 1,662.3 ± 150.2 pg/mL, respectively). LPS also increased the production of TNF-α, IL-1β and IL-6 (0.05 ± 0.003 to 36.3 ± 1.7 ng/mL, 129.4 ± 3.5 to 374.9 ± 7.4 pg/mL and 37.9 ± 1.5 to 3,644.9 ± 270.6 pg/mL, respectively). Genipin significantly suppressed the production of proinflammatory cytokines in a concentration-dependent manner (Figures 11C–E).
Figure 11.

Effect of genipin on cell viability (A) and production of nitrites (B), TNF-α (C), IL-1β (D), IL-6 (E) and HMGB1 release (F) in Pam3CSK4- and LPS-treated macrophages. RAW264.7 cells were pretreated with various concentrations of genipin (1–100 μmol/L), followed by stimulation with Pam3CSK4 or LPS (1 μg/mL) for 24 h. The results are presented as mean ± SEM of three independent experiments. Significantly different (*P < 0.05, **P < 0.01) from control. Significantly different (+P < 0.05, ++P < 0.01) from Pam3CSK4 or LPS. AU, arbitrary units. B–E:
, Pam3CSK4;
, LPS.
Effect of Genipin on HMGB1 Release in Pam3CSK4- or LPS-Treated RAW264.7 Cells
HMGB1 release into the culture medium increased approximately 2.4- and 4.3-fold by Pam3CSK4 and LPS, respectively. Genipin markedly suppressed HMGB1 release into the culture medium (Figure 11F).
Effect of Genipin on the NF-κB/p65 Nuclear Translocation and Cytosolic IκB-α Expression in Pam3CSK4- or LPS-Treated RAW264.7 Cells
As shown in Figure 12, the nuclear translocation of NF-κB/p65 showed a significant increase after 24-h incubation with Pam3CSK4 and LPS (1.4- and 1.5-fold, respectively), and genipin markedly inhibited these increases. In contrast, the level of cytosolic IκB-α protein expression showed a marked reduction after 24-h incubation with Pam3CSK4 and LPS (81.2% and 79.2%, respectively), which was inhibited by genipin.
Figure 12.

Effect of genipin on the nuclear translocation of NF-κB/p65 and cytosolic IκB-α protein expression in Pam3CSK4-treated (A) and LPS-treated (B) macrophages. RAW264.7 cells were pretreated with 100 μmol/L genipin, followed by stimulation with Pam3CSK4 or LPS (1 μg/mL) for 24 h. The results are presented as mean ± SEM of three independent experiments. Significantly different (**P < 0.01) from control. Significantly different (++P < 0.01) from Pam3CSK4 or LPS. AU, arbitrary units. A:
, Control;
, genipin; ▤, Pam3CSK4; ▥, genipin + Pam3CSK4; B:
, control;
, genipin; ▤, LPS; ▥, genipin + LPS.
DISCUSSION
Despite significant advances in the development of antimicrobial chemotherapy and supportive strategies, mortality from sepsis remains unacceptably high (10). Inflammatory and immune response during the early stage of sepsis involves a vast array of mediators; therefore, control of this complex inflammatory cascade is critical for management of sepsis. Genipin is the main bioactive compound in exhibition of the pharmacological activities of gardenia fruit, which has long been used as an antiinflammatory agent in folk medicine. Genipin not only reveals remarkable antiinflammatory and antimicrobial effects (11), but also possesses the ability for inhibition of lipid peroxidation and production of nitric oxide (12). Here, we demonstrated the protective effect of genipin against septic injury.
In a survival study, we showed that a single dose of genipin administered immediately after CLP induced markedly improved survival. Twice administration of genipin at a 24-h interval further conferred protection against lethal sepsis. Because widening of the therapeutic window is a critical necessity, we tried delayed administration of genipin after onset of sepsis. Unfortunately, genipin only showed a tendency to increase survival when administered 6 or 24 h after CLP (data not shown). In vivo studies with acute endotoxemia demonstrated that genipin improved survival.
A largely unopposed early proinflammatory response is known to occur in severe sepsis and leads to death (13). Indeed, the serum level of proinflammatory cytokines TNF-α, IL-1β and IL-6 peaked at early time points during sepsis. Early proinflammatory cytokines, such as TNF-α and IL-1β, can also activate monocytes/macrophages for release of HMGB1, a late mediator of lethal sepsis (14). Circulating HMGB1 levels are elevated in a delayed fashion in septic mice and in patients with sepsis and are well correlated with the lethal outcome of human sepsis (15). A number of agents that blocked HMGB1 release, resulting in improved survival in experimental sepsis, have been identified (16,17). In our study, genipin induced marked suppression of the systemic release of early proinflammatory cytokines and subsequent HMGB1 accumulation in polymicrobial sepsis.
Excessive production of proinflammatory cytokines causes capillary leakage, tissue injury and lethal organ failure in cases of severe sepsis. Multiple organ dysfunction syndrome is a serious clinical entity and a common cause of death in the critically ill patient. Overall mortality rates range from 30% to 100%, depending on the number of organ systems involved (18). In our study, LDH, a representative marker of tissue breakdown, particularly in the heart, began to increase from 1 h after CLP, indicating that heart muscle is damaged earlier than in other vital organs during sepsis. The serum level of ALT, AST, creatinine and LDH reached their peak at 24 h after onset of sepsis, whereas the maximal level of BUN appeared at 12 h after CLP. Our results indicate the difference in the vulnerability of the vital organs affected by sepsis. Histology analysis in lung and liver supported development of multiple organ dysfunction syndrome during sepsis. Treatment with genipin not only resulted in a decrease of serum markers of organ dysfunction, but also attenuated pathological changes in both lung and liver. These results suggest that genipin is capable of attenuating multiple organ dysfunction during polymicrobial sepsis.
TLR signaling plays an important role in initiation of the inflammatory response in sepsis. Given their large population of resident macrophages and extensive endothelial surface, the lung and liver play a prominent role and, in turn, are affected by the host immune response to infection (19–21). Recent studies have revealed that upregulated expression of TLR2 and TLR4 in multiple organs, including liver and lung, may play a role in high mortality and pathophysiology of sepsis (22,23). TAK-242, a novel small-molecule cyclohexane derivative, which selectively inhibits TLR4 signaling, was reported to inhibit increases in serum cytokine levels and to improve survival in an E. coli–induced sepsis model (24). In our study, TLR2 and TLR4 protein expression were significantly upregulated in both lung and liver at 6 and 24 h after CLP. Of particular interest, TLR2 and TLR4 protein expression showed a more dramatic increase in the lung than in the liver. Genipin attenuated increases in TLR2 and TLR4 protein expression in lung and liver, suggesting that the protective effect of genipin against septic injury might be mediated by suppression of TLR2 and TLR4 overexpression.
TLR2 signaling occurs exclusively through MyD88, whereas TLR4 can activate both MyD88-dependent and MyD88-independent pathways. Weighardt et al. (25) showed that genetic deficiency of MyD88 partially protected mice from the lethal effects of polymicrobial sepsis and that the hyperinflammatory response to sepsis was strongly attenuated in the absence of MyD88. MyD88 is the central adaptor protein for signal transduction of most TLRs and interacts with TLRs, leading to activation of the mitogen-activated protein kinase (MAPK) cascade and NF-κB, causing expression of proinflammatory mediators (26). In the present study, the MyD88 protein expression in both lung and liver showed an increase at 6 and 24 h after CLP. Genipin attenuated the increase in MyD88 protein expression in both lung and liver.
Among MAPKs, activation of p38 and JNK was shown to contribute to sepsis-induced end organ injury (27,28). Removal of ERK by specific inhibitors resulted in reduced activation of NF-κB, suppressed transcription of NF-κB–dependent genes and protection against LPS-induced endotoxemia (29). In our study, p38, ERK and JNK were activated at 6 and 24 h after CLP in the lung. However, in the liver, p38 was activated only at 6 h after CLP, whereas ERK and JNK were activated at both 6 and 24 h after CLP. Maitra et al. (30) reported that p38 in the liver is activated early in sepsis; however, this activation was diminished at 24 h after CLP. Genipin suppressed activation of all three MAPKs in the liver and prevented p38 and EKR activation in the lung. Activation of NF-κB with its translocation to the nucleus is able to induce an increase in TNF-α and IL-6 in septic animals (31). Genipin attenuated the increase in translocation of NF-κB and serum and tissue levels of proinflammatory cytokines. Taken together, our results provide evidence that genipin inhibits overexpression of TLR2 and TLR4; activation of p38, ERK and JNK MAPKs; and translocation of NF-κB. To further clarify the inhibitory effect of genipin on TLR2 and TLR4 signaling, Pam3CSK4 and LPS were exploited to activate TLR2 and TLR4, respectively. These agonists are well known to transmit signals culminating NF-κB activation and the induction of proinflammatory cytokines (32). In our study, genipin markedly suppressed Pam3CSK4-or LPS-induced nuclear translocation of NF-κB/p65 and subsequent production of proinflammatory mediators, nitrites and HMGB1. Moreover, genipin attenuated the increases in serum level of TNF-α and HMGB1 LPS-induced endotoxemia.
TLR4 use a MyD88-independent signaling pathway, using the adapter protein TRIF. TRIF deficiency inhibits production of several cytokines and chemokines and expression of type 1 IFN-regulated genes in macrophages and dendritic cells (33). Microarray analysis of the LPS transcriptome demonstrated that the majority of LPS-inducible genes are regulated in an MyD88-independent fashion, suggesting a crucial role for the TRIF pathway in inflammatory responses (34). Signaling of TRIF to its downstream effector molecules triggers activation of IRF3 and induction of IFN-β. In the present study, increased TRIF protein expression was observed 6 and 24 h after CLP in the liver. However, in the lung, TRIF protein expression showed an increase at 24 h after CLP, showing that TRIF in the lung was upregulated in a delayed manner during sepsis. Activation of IRF3 showed similar patterns with TRIF expression. Of particular interest, genipin markedly suppressed overexpression of TRIF and activation of IRF3 at 24 h after CLP in both the lung and liver, which resulted in decreased expression of IFN-β mRNA (data not shown). Our results suggest that genipin could prevent TRIF/IRF3/IFN-β signaling as well as MyD88-dependent pathways.
CONCLUSION
In conclusion, genipin attenuated mortality and organ injury in sepsis through inhibition of TLR-dependent inflammatory signaling pathways. Thus, we propose that genipin might be useful as a potential therapeutic medication for treating sepsis.
ACKNOWLEDGMENTS
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (2010-0028646).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.van der Poll T, Opal SM. Host-pathogen interactions in sepsis. Lancet Infect Dis. 2008;8:32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- 2.Xie J, Lv R, Yu L, Huang W. Hydroxyethyl starch 130/0.4 inhibits production of plasma proinflammatory cytokines and attenuates nuclear factor-kappaB activation and Toll-like receptors expression in monocytes during sepsis. J Surg Res. 2010;160:133–8. doi: 10.1016/j.jss.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 3.Cinel I, Opal SM. Molecular biology of inflammation and sepsis: a primer. Crit Care Med. 2009;37:291–304. doi: 10.1097/CCM.0b013e31819267fb. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol. 2004;136:312–9. doi: 10.1111/j.1365-2249.2004.02433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Famakin BM, et al. Disruption of downstream MyD88 or TRIF Toll-like receptor signaling does not protect against cerebral ischemia. Brain Res. 2011;1388:148–56. doi: 10.1016/j.brainres.2011.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koo HJ, Lim KH, Jung HJ, Park EH. Antiinflammatory evaluation of gardenia extract, geniposide and genipin. J Ethnopharmacol. 2006;103:496–500. doi: 10.1016/j.jep.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 7.Koo HJ, et al. Antiinflammatory effects of genipin, an active principle of gardenia. Eur J Pharmacol. 2004;495:201–8. doi: 10.1016/j.ejphar.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi S, et al. Genipin prevents fulminant hepatic failure resulting in reduction of lethality through the suppression of TNF-alpha production. Hepatol Res. 2005;33:298–305. doi: 10.1016/j.hepres.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Chaudry IH, Wichterman KA, Baue AE. Effect of sepsis on tissue adenine nucleotide levels. Surgery. 1979;85:205–11. [PubMed] [Google Scholar]
- 10.Matsuda A, et al. Dehydroepiandrosterone modulates Toll-like receptor expression on splenic macrophages of mice after severe polymicrobial sepsis. Shock. 2005;24:364–9. doi: 10.1097/01.shk.0000180624.36811.97. [DOI] [PubMed] [Google Scholar]
- 11.Ishiguro K, Yamaki M, Takagi S. Studies on iridoid-related compounds, II. The structure and antimicrobial activity of aglucones of galio-side and gardenoside. J Nat Prod. 1983;46:532–6. doi: 10.1021/np50028a018. [DOI] [PubMed] [Google Scholar]
- 12.Nam KN, et al. Genipin inhibits the inflammatory response of rat brain microglial cells. Int Immunopharmacol. 2010;10:493–9. doi: 10.1016/j.intimp.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 13.Walley KR, Lukacs NW, Standiford TJ, Strieter RM, Kunkel SL. Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect Immun. 1996;64:4733–8. doi: 10.1128/iai.64.11.4733-4738.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W, Tang Y, Li L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine. 2010;51:119–26. doi: 10.1016/j.cyto.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 16.Ulloa L, et al. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci U S A. 2002;99:12351–6. doi: 10.1073/pnas.192222999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–21. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 18.Wu JY, et al. Therapeutic effects of melatonin on peritonitis-induced septic shock with multiple organ dysfunction syndrome in rats. J Pineal Res. 2008;45:106–16. doi: 10.1111/j.1600-079X.2008.00567.x. [DOI] [PubMed] [Google Scholar]
- 19.Bone RC. The pathogenesis of sepsis. Ann Intern Med. 1991;115:457–69. doi: 10.7326/0003-4819-115-6-457. [DOI] [PubMed] [Google Scholar]
- 20.Matuschak GM, Rinaldo JE. Organ interactions in the adult respiratory distress syndrome during sepsis: role of the liver in host defense. Chest. 1988;94:400–6. doi: 10.1378/chest.94.2.400. [DOI] [PubMed] [Google Scholar]
- 21.Holtzman MJ. Arachidonic acid metabolism: implications of biological chemistry for lung function and disease. Am Rev Respir Dis. 1991;143:188–203. doi: 10.1164/ajrccm/143.1.188. [DOI] [PubMed] [Google Scholar]
- 22.Williams DL, et al. Modulation of tissue Toll-like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality. Crit Care Med. 2003;31:1808–18. doi: 10.1097/01.CCM.0000069343.27691.F3. [DOI] [PubMed] [Google Scholar]
- 23.Zou L, et al. Toll-like receptor 2 plays a critical role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med. 2010;38:1335–42. doi: 10.1097/CCM.0b013e3181d99e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takashima K, et al. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol. 2009;157:1250–62. doi: 10.1111/j.1476-5381.2009.00297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weighardt H, et al. Identification of a TLR4- and TRIF-dependent activation program of dendritic cells. Eur J Immunol. 2004;34:558–64. doi: 10.1002/eji.200324714. [DOI] [PubMed] [Google Scholar]
- 26.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–22. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 27.Yang M, Wu J, Martin CM, Kvietys PR, Rui T. Important role of p38 map kinase/NF-kappaB signaling pathway in the sepsis-induced conversion of cardiac myocytes to a proinflammatory phenotype. Am J Physiol. 2008;294:H994–1001. doi: 10.1152/ajpheart.01044.2007. [DOI] [PubMed] [Google Scholar]
- 28.Chen LC, Gordon RE, Laskin JD, Laskin DL. Role of TLR-4 in liver macrophage and endothelial cell responsiveness during acute endotoxemia. Exp Mol Pathol. 2007;83:311–26. doi: 10.1016/j.yexmp.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang B, et al. Temporal control of NF-kappaB activation by ERK differentially regulates interleukin-1beta-induced gene expression. J Biol Chem. 2004;279:1323–9. doi: 10.1074/jbc.M307521200. [DOI] [PubMed] [Google Scholar]
- 30.Maitra SR, et al. Modulations of signal transduction pathways during sepsis and the effects of insulin and mifepristone. Acad Emerg Med. 2003;10:1–8. doi: 10.1111/j.1553-2712.2003.tb01968.x. [DOI] [PubMed] [Google Scholar]
- 31.Baeuerle PA, Baltimore D. NF-kappaB: Ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 32.Akira S, Kawai T. Toll-like receptors and their crosstalk with other innate receptors in infection and immuniny. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 34.Bjorkbacka H, et al. The induction of macrophage gene expression by LPS predominantly utilizes MyD88-independent signaling cascades. Physiol Genomics. 2004;19:319–30. doi: 10.1152/physiolgenomics.00128.2004. [DOI] [PubMed] [Google Scholar]