

Abstract
The glycopeptide antibiotics are the most important class of drugs used in the treatment of resistant bacterial infections including those caused by methicillin-resistant Staphylococcus aureus (MRSA). After more than 50 years of clinical use, the emergence of glycopeptide resistant Gram-positive pathogens such as vancomycin-resistant enterococci (VRE) and vancomycin-resistant Staphylococcus aureus (VRSA) presents a serious global challenge to public health at a time few new antibiotics are being developed. This has led to renewed interest in the search for additional effective treatments including the development of new derivatives of the glycopeptide antibiotics. General approaches have been explored for modifying glycopeptide antibiotics, typically through the derivatization of the natural products themselves or more recently through chemical total synthesis. In this Perspective, we consider recent efforts to redesign glycopeptide antibiotics for the treatment of resistant microbial infections, including VRE and VRSA, and examine their future potential for providing an even more powerful class of antibiotics that are even less prone to bacterial resistance.
Antibiotic introduction in the early 20th century ushered in a new era in the treatment of microbial infections, providing the medical community with powerful drugs in its battle against disease. In addition to being among the first drugs introduced, antibiotics are also among the most successful, saving countless lives, extending life spans, and permitting previously deadly medical procedures. They represent 5% of the global drug market, and provide $42 billion in annual sales (2009) (1). One would be hard pressed to identify another drug class that enjoys such safe and widespread use, provides so successful health outcomes at such a modest cost, and has such a large economic impact. It is magical to awake from a night’s sleep nearly cured following a day of antibiotic treatment when feeling so miserable the day before. However, and from the very outset, this new revolution came with the concomitant induction of bacterial resistance to these treatments. The first reports of resistance followed only one year after the initial introduction of penicillin. Similarly, the introduction of methicillin in 1959, the next-generation drug for the treatment of penicillin-resistant strains, was followed only two years later by the emergence of methicillin-resistant Staphylococcus aureus (MRSA) (2). This cycle continues to be repeated again and again as new antibiotics are discovered and introduced into widespread use.
The glycopeptide antibiotics have long stood as an exception to this phenomenon. The lack of significant levels of clinical resistance to the two most commonly deployed members, vancomycin and teicoplanin, led to their adoption as drugs of last resort for the treatment of otherwise resistant and deadly infections. Recent years however have finally brought the onset of resistance to these important drugs. Herein, we review the origin of the past success with the glycopeptides antibiotics and the science behind the development of derivatives that address the emerging problem of acquired resistance in pathogenic bacteria, which we believe will provide an even more powerful future class of antibiotics than Nature could devise.
Resistance to Glycopeptide Antibiotics is a Growing Serious Public Health Problem
Vancomycin is the leading member of the class of clinically important glycopeptide antibiotics. Discovered at Eli Lilly, vancomycin was first disclosed in 1956 (3) and introduced into the clinic in 1958, although its structure was not established until nearly 30 years later in 1983 (4). Following the emergence of MRSA, it became the drug of choice to treat resistant bacterial infections and it is also used for the treatment of patients on dialysis, undergoing cancer chemotherapy, or allergic to β-lactam antibiotics (5). Today, more than 60% of the ICU S. aureus infections are MRSA, and its movement from a hospital-acquired to a community-acquired infection has intensified the impact of such resistant bacterial infections (6). The onset of vancomycin resistance was long-delayed in comparison to all other antibiotics. Even after its first three decades of use, there was no notable resistance to vancomycin reported, and some even speculated that the development of resistance might be impossible (7). Vancomycin resistant phenotypes were first reported in enterococci (VRE) in 1987, many years after the introduction of the drug into widespread clinical use, and today >30% of the ICU Enterococcus faecalis infections are VRE. Following the emergence of resistance in enterococci, which is now genetically transferred vertically, concern arose over the emergence of vancomycin-resistant S. aureus (VRSA) as a result of horizontal gene transfer from resistant enterococci. The first cases of fully vancomycin-resistant strains were reported in 2002 (8), and there have been an increasing number of cases of VRSA in the United States confirmed by the CDC (9). A majority of VRSA has been found in patients co-infected with VRE, implicating horizontal gene transfer as the current method for acquiring vancomycin resistance (10). As the prevalence of VRSA increases and as it establishes vertical gene transfer of resistance, new antibiotics with the longevity and dependability of vancomycin will be required to contain their impact. This need is arising at the same time that antibiotic discovery efforts are being discontinued at most major pharmaceutical companies. The reasons for this decline in antibiotic development are largely economic, resulting from a combination of patient short term use, the restricted use of new antibiotics with activity against resistant bacteria, and the increased regulatory criteria for approval.
Molecular Basis of Vancomycin Resistance
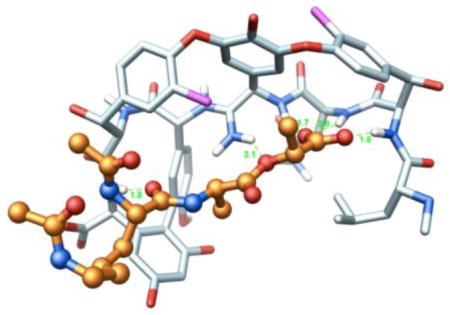
The mechanism of action of glycopeptide antibiotics involves the inhibition of bacterial cell wall synthesis by binding and sequestration of the integral precursor peptidoglycan peptide terminus D-alanine-D-alanine (D-Ala-D-Ala) in the developing cell wall, Figure 1 (11). This precursor is tightly bound by the antibiotic, physically preventing transpeptidation and transglycosylation, arresting cell wall cross-linking and maturation, and leading to cell lysis. In the two most prominent manifestations of resistance (VanA and VanB), this cell wall precursor is remodeled to D-alanine-D-lactate (D-Ala-D-Lac), incorporating an ester linkage in place of the amide of the natural ligand (12). Vancomycin-resistant bacteria sense the antibiotic challenge and subsequently remodel their precursor peptidoglycan terminus from D-Ala-D-Ala to D-Ala-D-Lac. Normal synthesis of lipid intermediate I and II, containing the D-Ala-D-Ala termini, continues but a late stage remodeling to D-Ala-D-Lac ensues to avoid the action of vancomycin. The binding affinity of the antibiotic for this altered ligand is reduced 1000-fold, resulting in a corresponding 1000-fold loss in antimicrobial activity. One key, but subtle insight to emerge from this characterization of vancomycin-resistant bacteria is that the most direct efforts to redesign vancomycin for their treatment should target compounds that not only bind D-Ala-D-Lac, but that also maintain binding to D-Ala-D-Ala.
Figure 1.

Late stage bacterial cell wall synthesis and vancomycin binding to D-Ala-D-Ala.
The mechanism by which the vancomycin challenge is sensed is well understood (13), entailing a two component signaling cascade (VanS/VanR). Initiated by a ligand-induced dimerization of a cell surface His-kinase (VanS), it activates the signaling transcription factor VanR by phosphorylation and dimerization. In turn, the activated VanR translocates to the nucleus where it binds to DNA and induces the expression of three genes encoding three enzymes (VanH, VanA, and VanX) that remodel the peptidoglycan precursor from D-Ala-D-Ala to D-Ala-D-Lac. The species responsible for inducing VanS dimerization and activation has been the subject of intense study. Compelling cases have been made for either direct recognition of the antibiotic itself (14) or the aberrant buildup of a key cell wall precursor(s) (15), and it is conceivable that which of the two inducing VanS ligands used is organism dependent (16).
This sensing mechanism and apparent resistance was most likely co-opted from the vancomycin producing organisms, which use it to avoid the antibiotic effects (17), rather than being orchestrated by sensitive bacteria upon continued treatment. As such, one could make the case that pathogenic bacteria have not yet independently evolved a unique and effective resistance mechanism to glycopeptide challenges.
Semisynthetic Glycopeptides in the Clinic
Because of their structural complexity, nearly all new structures have been derived by semisynthetic modification of the natural products (18). Many of the most significant modifications have introduced hydrophobic moieties into the glycopeptide structure, which often yields increased activity. To date, no semisynthetic compounds have modified the pocket region of the molecules responsible for binding D-Ala-D-Ala in efforts to recover affinity for D-Ala-D-Lac. Instead, the modifications are quite remote from the binding pocket. Strategically placed hydrophobic groups have been shown to increase antibiotic dimerization in solution, and also to confer increased membrane anchoring ability, both of which could conceivably lead to increased binding of the peptidoglycan terminus (19). Additionally, the direct inhibition of transglycosylase enzymes, mediated by a glycopeptide modified carbohydrate, has been proposed as a second mechanism by which the lipophilic glycopeptides with reduced or impaired D-Ala-D-Lac or D-Ala-D-Ala binding properties exhibit antimicrobial effects (20,21).
One such modified glycopeptide is telavancin (22), a clinically approved (2009) semisynthetic derivative of vancomycin used to treat complicated skin infections that are suspected or confirmed to be MRSA. It bears a hydrophobic N-decylamino group to grant increased activity against resistant organisms and a hydrophilic phosphonic acid side chain that provides improved pharmacokinetic properties, Figure 2. Telavancin has been shown to function both through the traditional glycopeptide mechanism of inhibition of cell wall synthesis by binding to D-Ala-D-Ala and also through the disruption of bacterial membrane integrity, a mechanism typically not seen for other glycopeptides (23).
Figure 2.

Structures of oritavancin, telavancin, and teicoplanin.
The inclusion of a modified sugar bearing a chlorobiphenyl group has been explored in a variety of positions, most notably in the glycopeptide derivative oritavancin (24). Oritavancin, the N-(p-chlorobiphenyl)methyl derivative of the naturally occurring glycopeptide chloroeremomycin, possesses the vancomycin core but different sugar residues. It was originally developed by Eli Lilly and it currently is being advanced through phase III clinical trials for possible FDA approval, Figure 2 (25). Studies on the mechanism of action for oritavancin and related compounds have shown that the chlorobiphenyl side chain promotes antibiotic dimerization and membrane anchoring and affords antimicrobial activity against resistant organisms, including VanA and VanB VRE, despite a lack of improved binding for D-Ala-D-Lac in solution.
It is conceivable such semisynthetic changes to vancomycin might avoid bacterial sensing of the antibiotic challenge and this likely accounts for their VanB VRE activity (like teicoplanin) (18) or that some may entail a second mechanism(s) of action. However, it is notable that both of these derivatives also simply increase the antibiotic potency, typically as much as 100-fold. While increasing the bacterial sensitivity to the antibiotics, VanA vancomycin-resistant bacterial strains (MIC = 10 μg/mL) remain 1000-fold less sensitive than susceptible strains (MIC = 0.01 μg/mL), indicating that the intrinsic resistance is still induced, but is overcome by the increased potency of the semisynthetic derivatives.
Fundamental Basis for Redesign of the Vancomycin Binding Pocket
In spite of all the modifications made to vancomycin, analogues bearing alterations to the core structure, including changes to the critical binding pocket region itself, have been essentially unexplored. With the structural complexity of glycopeptides, a modification to the peptide backbone would likely only be accessible through total synthesis. Our efforts on the total synthesis of glycopeptide antibiotics, which first provided the vancomycin aglycon (26,27), and were subsequently extended to provide the teicoplanin (28,29) and ristocetin (30) aglycons and the complestatins (31,32,33), provided us a unique opportunity to explore such deep-seated modifications of the natural products inaccessible by typical semisynthetic efforts.
Vancomycin binds D-Ala-D-Ala through a rich array of hydrophobic van der Waals contacts and a network of five H-bonds between the ligand and the central pocket formed by the glycopeptide backbone (34). Resistance arises when this hydrogen-bonding network is disrupted by a single atom substitution in the ligand (NH → O) that serves to not only remove the central H-bond but also introduces a repulsive lone pair/lone pair interaction between the vancomycin residue 4 carbonyl and D-Ala-D-Lac ester oxygens. In an effort that established the contribution of each of these effects to the net 1000-fold loss in affinity, we prepared a model of the peptidoglycan ligand incorporating a methylene unit, wherein the amide of the natural ligand is now a ketone, and its binding to vancomycin was determined, Figure 3 (35). This examination established that the majority (100-fold) of the 1000-fold reduction in binding was a result of the destabilizing lone pair interaction introduced by the substitution of the ester oxygen. Thus, an initial synthetic redesign of vancomycin could focus principally on removing this destabilizing interaction rather than reintroduction of the lost H-bond and this would be sufficient to compensate for most of the lost binding affinity with D-Ala-D-Lac. An analogue excising the offending vancomycin residue 4 amide carbonyl, and replacing it with a methylene group, would accomplish this goal and was the target of our initial efforts.
Figure 3.

Schematic representation of the interaction of vancomycin with model ligands and measured binding data.
Given the state of the art in organic synthesis, the incorporation of such a deep-seated modification to the vancomycin aglycon to provide [Ψ[CH2NH]Tpg4]vancomycin aglycon could be achieved initially and most directly through total synthesis. Our experience developed in the course of the total syntheses of the naturally occurring glycopeptide antibiotics provided a solid foundation on which to base the efforts. Incorporating a modular strategy based on our synthesis of the vancomycin aglycon, the synthesis of [Ψ[CH2NH]Tpg4]vancomycin aglycon was accomplished providing a modified antibiotic bearing a methylene at residue 4 (36). Consistent with expectations and relative to vancomycin aglycon, this analogue exhibited a 40-fold increase in affinity for D-Ala-D-Lac and a 35-fold reduction in affinity for D-Ala-D-Ala, providing the first modified glycopeptide with dual, balanced binding properties, Figure 4. This methylene derivative of vancomycin aglycon also exhibited activity against vancomycin-resistant bacteria, displaying a potency (MIC = 31 μg/mL, VanA E. faecalis) reflecting these binding characteristics. Thus, the removal of a single atom from the antibiotic countered the bacterial resistance derived from the single atom exchange in the bacterial cell wall peptidoglycan precursor, provided the first rationally redesigned vancomycin prepared to directly address the origin of resistance, and established the foundation for our continued study.
Figure 4.

Key vancomycin residue 4 modifications.
Second-generation Vancomycin Modifications – A Divergent Synthetic Approach
Following the initial success in the redesign of the vancomycin aglycon to achieve this dual binding by the removal of the lone pair repulsion between vancomycin and D-Ala-D-Lac, we sought to access a key series of additional analogues in a search for improved binding affinities and antimicrobial activities. Such a full exploration of the molecular interaction of vancomycin and its two targets would require the synthesis of a number of different analogues bearing modifications to the pocket region. As selective semisynthetic modification of vancomycin at the residue 4 site is not yet possible, we designed a divergent (37) total synthesis that would proceed through a key intermediate that allows a late-stage diversification into a series of analogues.
The strategy replaced the residue 4 amide with the corresponding thioamide, which could then be selectively modified at a late stage in the presence of the multiple amides. After a screen of potential stages at which to incorporate the thioamide, we found the most efficient reaction was that performed on an early stage precursor using Lawesson’s reagent, which proceeded with complete selectively for the sterically more accessible of two possible amides. From this initial thionation stage, a complex total synthesis was elaborated resulting in the preparation of [Ψ[C(=S)NH]Tpg4]vancomycin aglycon, Scheme 1 (38). In addition to early stage residue 4 thioamide introduction, allowing differentiation of one of seven amide bonds central to the vancomycin core structure, the approach relied on two aromatic nucleophilic substitution reactions for formation of the 16-membered diaryl ethers in the CD/DE ring systems, an effective macrolactamization for closure of the 12-membered biaryl AB ring system, and the defined order of CD, AB, and DE ring closures. This order of ring closures follows their ease of thermal atropisomer equilibration, permitting the recycling of any newly generated unnatural atropisomer under progressively milder thermal conditions where the atropoisomer stereochemistry already set is not impacted.
Scheme 1.

Summary of synthetic strategy.
This vancomycin residue 4 thioamide displayed revealing properties in its own right. The single atom substitution of sulfur for oxygen was sufficient to completely disrupt binding to both D-Ala-D-Ala and D-Ala-D-Lac and the compound lacked any antimicrobial activity, Figure 4. Although the weaker H-bonding of a thioamide is likely contributing to this lowered affinity for D-Ala-D-Ala, the magnitude of the loss suggests something more fundamental is responsible. We have suggested that the thiocarbonyl increased bond length and larger van der Waals radii of sulfur are sufficient to sterically displace and completely disrupt the intricate binding of D-Ala-D-Ala. This behavior of the residue 4 thioamide serves as a sharp contrast to the corresponding residue 4 amidine detailed below, illustrating the near perfect isosteric amidine replacement for an amide. In addition, the inactivity of the residue 4 thioamide defines a key intermediate to be targeted with producing non-pathogenic micoorganisms that avoids the otherwise suicidal production of the amidine itself. Thus, the properties of the residue 4 thioamides, which might appear disappointing on the surface, represent an important key observation made in our efforts that will serve to advance the field.
A Redesigned Glycopeptide with Dual Binding and Potent Antimicrobial Activity
With the availability of [Ψ[C(=S)NH]Tpg4]vancomycin aglycon, the stage was set to develop the site-specific single-step introduction of key modifications at a site critical to the interaction between vancomycin and the peptidoglycan terminus. Although the methylene derivative exhibited the desired dual binding properties, we sought to explore analogues that could further enhance D-Ala-D-Lac binding while simultaneously maintaining binding for D-Ala-D-Ala. We turned our focus to [Ψ[C(=NH)NH]Tpg4]vancomycin aglycon, wherein the residue 4 amide of vancomycin is replaced with an amidine. In retrospect and with the results in hand, this choice of residue 4 amidine may appear obvious, but its projected behavior was not straightforward to anticipate. It was not clear whether the residue 4 amidine would further enhance D-Ala-D-Lac binding by not only removing the destabilizing lone pair interaction but by additionally serving as a H-bond donor to the weak acceptor ester oxygen. More significantly and because an amidine would be expected to be protonated, it was not clear whether it would maintain its ability to serve as an effective H-bond acceptor needed to bind D-Ala-D-Ala. Finally and because the altered properties of the residue 4 thioamide were so dramatic, it was not clear whether an amidine could serve as an effective amide replacement at a site so sterically sensitive to modification. After an extensive, albeit initial, exploration of approaches, the residue 4 thioamide was selectively converted to the corresponding amidine in a single step using a unique, previously unexplored AgOAc-promoted reaction (50–85%) that could be conducted on a fully deprotected and fully elaborated vancomycin aglycon, Scheme 2 (39).
Scheme 2.

Single-step synthetic conversion of a residue 4 thioamide to the corresponding amidine and its dual binding behavior toward D-Ala- D-Lac and D-Ala- D-Ala.
This new analogue improved binding to the model D-Ala-D-Lac ligand by nearly 600-fold over that of vancomycin, which is also a more than 10-fold increase over the corresponding methylene derivative, Figure 4. Moreover, its binding affinity for the model D-Ala-D-Ala ligand is only approximately 2-fold less than the vancomycin aglycon itself and 15-fold greater than the methylene derivative, indicating that the amidine functions well as a H-bond acceptor for the amide NH in the model ligand. Importantly, it displays effective, balanced binding affinity for both model ligands at a level that is within 2- to 3-fold that exhibited by vancomycin aglycon for D-Ala-D-Ala. Accurately reflecting these binding properties, it exhibits potent antimicrobial activity (MIC = 0.31 μg/mL, VanA E. faecalis) against VanA VRE, the most stringent of vancomycin-resistant bacteria, being equipotent to the activity that vancomycin and its aglycon display against sensitive bacterial strains. Beautifully, this represents a complementary single atom exchange in the antibiotic (O→NH) to counter a corresponding single atom exchange in the cell wall precursors of resistant bacteria (NH→O). A key additional feature is that the modified antibiotic also maintains its ability to bind the unaltered peptidoglycan D-Ala-D-Ala while also binding the remodeled D-Ala-D-Lac by virtue of its ability to serve as either a H-bond donor or H-bond acceptor.
The clinical impact of such redesigned glycopeptide antibiotics is likely to be important, charting a rational approach forward in the development of antibiotics for the treatment of vancomycin-resistant bacterial infections. Since the single atom exchange described here is a deep-seated change that entails the selective transformation of one of seven amides in the vancomycin core structure, this was accomplished initially by total synthesis. In addition to its use in the exploration of other residue 4 modifications that are under investigation, further improvements in their total synthesis, the development of semisynthetic approaches to selective residue 4 modifications, and the use of engineered or coerced (feeding) microorganisms can be expected to improve access to such modified glycopeptide antibiotics. Since the latter attempts to directly produce the residue 4 amidines will likely simply kill the producing organism, the production of the inactive residue 4 thioamides and their subsequent single-step synthetic conversion to the amidines, as we detail, will likely be the most effective of such approaches.
Combining Peripheral Modifications and Binding Pocket Redesign – a Next Frontier
With the rational synthetic redesign of the glycopeptide antibiotic binding pocket now accessible, an exciting next phase is the combination of these deep-seated structural changes with the modifications to the peripheral regions of the molecules present in the best currently available therapeutic candidates. As discussed, the activity of the latter compounds is not derived from altered peptidoglycan terminus binding although they can be used to sometimes prevent the induction of the D-Ala-D-Lac remodeling (e.g., VanB sensitivity). Rather, they often simply increase antibiotic activity against resistant organisms largely as a result of an overall increase in potency by as much as 100-fold against both resistant and sensitive organisms. It is reasonable to expect that the incorporation of such peripheral modifications into the structure of a vancomycin already modified to achieve dual, effective binding for both D-Ala-D-Ala and D-Ala-D-Lac will increase the resulting compounds’ activity against both sensitive and resistant bacteria to truly remarkable potencies and efficacies. Aside from the intrinsic merits of such candidate structures as new therapeutics, the increased potencies would have an important impact on lowering their production costs and supplies needed for clinical exploration or introduction.
Why Focus on the Glycopeptide Antibiotics?
Have these efforts simply resuscitated an old antibiotic that is in decline or can we expect a more special behavior? Their past history suggests there are several key features of this class of antibiotics, endowing it with a “resistance to resistance”, that make it a uniquely important class that can be even further improved. First, it binds not to a protein or nucleic acid target, but to a near ubiquitous small molecule substrate integral to bacterial cell wall formation that is both fundamental to the bacteria’s survival and incapable of resistance derived from spontaneous genetic mutation. Second, the glycopeptide antibiotics inhibit cell wall synthesis by multiple mechanisms, inhibiting transpeptidase-catalyzed cell wall cross-linking by substrate sequestration and inhibiting transglycosylase-catalyzed lipid intermediate II incorporation by either indirect means (D-Ala-D-Ala binding) or potentially through direct carbohydrate binding to the enzyme. It is statistically difficult to imagine organisms evolving to overcome two or more such mechanisms simultaneously. Additionally, the glycopeptide antibiotics likely occupy multiple cell wall binding sites beyond D-Ala-D-Ala, including D-Ala-Gly and Gly-Gly, which conceivably could serve to further impede cell wall synthesis. Finally, the class benefits from targeting a cell surface (vs intracellular) target avoiding the additional intracellular-mediated common mechanisms of resistance (e.g., bacterial chemical modification and deactivation, efflux, blocked entry) (40). Most significant is the fact that the resistance that has emerged (VanA and VanB) has not been through pathogenic bacteria evolution of a mechanism of resistance, but was likely coopted from the glycopeptide producing non-pathogenic organisms that use it to avoid the antibiotic effects during production. Thus, other than the reduced susceptibility (ca. 10-fold) derived from a minor D-Ala-D-Ser (VanC) VRE variant and that derived from a thickened bacterial cell wall presenting more target sites (VISA, 10-fold) (41–43), pathogenic bacteria themselves have not yet devised an effective resistance strategy to the glycopeptide antibiotics even after more than 50 years of heavy clinical and vetinary use. The development of modified glycopeptides that fundamentally address the intrinsic resistance derived from either inducible or constitutive D-Ala-D-Lac remodeling should not only overcome the emerging problem of acquired resistance in pathogenic bacteria, but will likely provide a more powerful future class of antibiotics than even Nature could devise. Finally, understanding the sources of vancomycin’s lasting utility (the past) also define the characteristics that future new, alternative, or replacement antibiotics should embody (44).
Acknowledgments
We are especially grateful for the financial support of the National Institutes of Health (CA041101 to D.L.B.) and the Skaggs Institute for Chemical Biology. J.G.P. is the recipient of a NIH postdoctoral fellowship (CA144333), A.O. is the recipient of a JSPS fellowship, and R.C.J. is a Skaggs Fellow.
Keywords
- Glycopeptide antibiotics
a large class of natural products that include vancomycin and teicoplanin that has antibiotic activity and is composed of a tricyclic or tetracyclic heptapeptide substituted with carbohydrates
- Resistant bacteria
pathogenic bacteria that are insensitive to treatment with common antibiotics
- VanA and VanB resistant bacteria
resistant bacteria that are capable of remodeling their peptidoglycan precursor peptide terminus from D-Ala-D-Ala to D-Ala-D-Lac to avoid a glycopeptide antibiotic challenge. VanA denotes resistance to all naturally occurring glycopeptide antibiotics, whereas VanB denotes a strain still sensitive to teicoplanin, but not vancomycin
References
- 1.Hamad B. The antibiotic market. Nat Rev Drug Discovery. 2010;9:675–676. doi: 10.1038/nrd3267. [DOI] [PubMed] [Google Scholar]
- 2.Parker MT, Jevons MP. A survey of methicillin resistance in Staphylococcus aureus. Postgrad Med J. 1964;40:170–178. doi: 10.1136/pgmj.40.suppl.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCormick MH, Stark WM, Pittenger GE, Pittenger RC, McGuire JM. Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot Annu. 1955–1956:606–611. [PubMed] [Google Scholar]
- 4.Harris CM, Kopecka H, Harris TM. Vancomycin: structure and transformation to CDP- 1. J Am Chem Soc. 1983;105:6915–6922. [Google Scholar]
- 5.Nagarajan R, editor. Glycopeptide Antibiotics. Marcel Dekker Inc; New York: 1994. [Google Scholar]
- 6.Walsh CT, Fischbach MA. New ways to squash superbugs. Sci Amer. 2009;301:44–51. doi: 10.1038/scientificamerican0709-44. [DOI] [PubMed] [Google Scholar]
- 7.Cooper GL, Given DB. Vancomycin, A Comprehensive Review of 30 Years of Clinical Experience. Park Row Publications; Indianapolis, IN: 1986. The development of vancomycin; pp. 1–5. [Google Scholar]
- 8.Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.CDC. Staphylococcus aureus (VRSA) May, 2010. Reminds clinical laboratories and healthcare infection preventionists of their role in the search and containment of vancomycin-resistant. [Google Scholar]
- 10.Zhu W, Murray PR, Huskins WC. Dissemination of an Enterococcus Inc18-like VanA plasmid associated with vancomycin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2010;54:4214–4320. doi: 10.1128/AAC.00185-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perkins HR. Vancomycin and related antibiotics. Pharmacol Ther. 1982;16:181–197. doi: 10.1016/0163-7258(82)90053-5. [DOI] [PubMed] [Google Scholar]
- 12.Bugg TDH, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. Molecular basis of vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry. 1991;30:10408–10415. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- 13.Healy VL, Lessard IAD, Roper DI, Knox JR, Walsh CT. Vancomycin resistance in enterococci: reprogramming of the D-Ala-D-Ala ligases in bacterial peptidoglycan biosynthesis. Chem Biol. 2000;7:R109–R119. doi: 10.1016/s1074-5521(00)00116-2. [DOI] [PubMed] [Google Scholar]
- 14.Koteva K, Hong H-J, Wang XD, Nazi I, Hughes D, Naldrett MJ, Buttner MJ, Wright GD. A vancomycin photoprobe identifies the histidine kinase VanSsc as a vancomycin receptor. Nat Chem Biol. 2010;6:327–329. doi: 10.1038/nchembio.350. [DOI] [PubMed] [Google Scholar]
- 15.Hong HJ, Hutchings MI, Buttner MJ. Vancomycin resistance VanS/VanR two-component systems. Adv Exp Med Biol. 2008;631:200–213. doi: 10.1007/978-0-387-78885-2_14. [DOI] [PubMed] [Google Scholar]
- 16.Arthur M, Quintiliani R., Jr Regulation of VanA- and VanB-type glycopeptide resistance in enterococci. Antimicrob Agents Chemother. 2001;45:375–381. doi: 10.1128/AAC.45.2.375-381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marshall CG, Lessard IA, Park I, Wright GD. Glycopeptide antibiotic resistance genes in glycopeptide-producing organisms. Antimicrob Agents Chemother. 1998;42:2215–2220. doi: 10.1128/aac.42.9.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahne D, Leimkuhler C, Lu W, Walsh CT. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 19.Allen NE, Hobbs JN, Jr, Nicas TI. Inhibition of peptidoglycan biosynthesis in vancomycin-susceptible and -resistant bacteria by a semisynthetic glycopeptide antibiotic. Antimicrob Agents Chemother. 1996;40:2356–2362. doi: 10.1128/aac.40.10.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge M, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding D-Ala-D-Ala. Science. 1999;284:507–511. doi: 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- 21.Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Vancomycin analogues active against VanA-resistant strains inhibit bacterial transglycosylase without binding substrate. Proc Natl Acad Sci USA. 2003;100:5658–5663. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corey GR, Stryjewski ME, Weyenberg W, Yasothan U, Kirkpatrick P. Telavancin. Nat Rev Drug Discovery. 2009;8:929–930. doi: 10.1038/nrd3051. [DOI] [PubMed] [Google Scholar]
- 23.Higgins DL, Chang R, Debabov DV, Leung J, Wu T, Krause KM, Sandvik E, Hubbard JM, Kaniga K, Schmidt DE, Jr, Gao Q, Cass RT, Karr DE, Benton BM, Humphrey PP. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2005;49:1127–1134. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicas TI, Mullen DL, Flokowitsch JE, Preston DA, Snyder NJ, Zweifel MJ, Wilkie SC, Rodriguez MJ, Thompson RC, Cooper RD. Semisynthetic glycopeptide antibiotics derived from LY264826 active against vancomycin-resistant enterococci. Antimicrob Agents Chemother. 1996;40:2194–2199. doi: 10.1128/aac.40.9.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen NE, Nicas TI. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol Rev. 2003;26:511–532. doi: 10.1111/j.1574-6976.2003.tb00628.x. [DOI] [PubMed] [Google Scholar]
- 26.Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL. Diastereoselective total synthesis of the vancomycin aglycon with ordered atropisomer equilibrations. J Am Chem Soc. 1999;121:3226–3227. [Google Scholar]
- 27.Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O. Total synthesis of the vancomycin aglycon. J Am Chem Soc. 1999;121:10004–10011. [Google Scholar]
- 28.Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng J-H, Mori Y, Rogel O, Castle SL, McAtee JJ. Total synthesis of the teicoplanin aglycon. J Am Chem Soc. 2000;122:7416–7417. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 29.Boger DL, Kim SH, Mori Y, Weng J-H, Rogel O, Castle SL, McAtee JJ. First and second generation total synthesis of the teicoplanin aglycon. J Am Chem Soc. 2001;123:1862–1871. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 30.Crowley BM, Mori Y, McComas CC, Tang D, Boger DL. Total synthesis of the ristocetin aglycon. J Am Chem Soc. 2004;126:4310–4317. doi: 10.1021/ja039795a. [DOI] [PubMed] [Google Scholar]
- 31.Garfunkle J, Kimball FS, Trzupek JD, Takazawa S, Shimamura H, Tomishima M, Boger DL. Total synthesis of chloropeptin II (complestatin) and chloropeptin I. J Am Chem Soc. 2009;131:16036–16038. doi: 10.1021/ja907193b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimamura H, Breazzano SP, Garfunkle J, Kimball FS, Trzupek JD, Boger DL. Total synthesis of complestatin: development of a Pd(0)-mediated indole annulation for macrocyclization. J Am Chem Soc. 2010;132:7776–7783. doi: 10.1021/ja102304p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breazzano SP, Boger DL. Synthesis and stereochemical determination of complestatin A and B (neuroprotectin A and B) J Am Chem Soc. 2011;133:18495–18502. doi: 10.1021/ja208570q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams DH, Bardsley B. The vancomycin group of antibiotics and the fight against resistant bacteria. Angew Chem Int Ed. 1999;38:1172–1193. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 35.McComas CC, Crowley BM, Boger DL. Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-bond and repulsive lone pair contributions. J Am Chem Soc. 2003;125:9314–9315. doi: 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- 36.Crowley BM, Boger DL. Total synthesis and evaluation of [Ψ[CH2NH]Tpg4]vancomycin aglycon: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J Am Chem Soc. 2006;128:2885–2892. doi: 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boger DL, Brotherton CE. Total synthesis of azafluoranthene alkaloids: rufescine and imelutine. J Org Chem. 1984;49:4050–4055. [Google Scholar]
- 38.Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL. Total synthesis of [Ψ[C(=S)NH]Tpg4]vancomycin aglycon, [Ψ[C(=NH)NH]Tpg4]vancomycin aglycon, and related key compounds: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J Am Chem Soc. 2012;134:1284–1297. doi: 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie J, Pierce JG, James RC, Okano A, Boger DL. A redesigned vancomycin engineered for dual D-Ala-D-Ala and D-Ala-D-Ala binding exhibits potent antimicrobial activity against vancomycin-resistant bacteria. J Am Chem Soc. 2011;133:13946–13949. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright GD. Molecular mechanisms of antibiotic resistance. Chem Commun. 2011;47:4055–4061. doi: 10.1039/c0cc05111j. [DOI] [PubMed] [Google Scholar]
- 41.Walsh TR, Howe RA. The prevalence and mechanisms of vancomycin resistance in Staphylococcus aureus. Ann Rev Microbiol. 2002;56:657–675. doi: 10.1146/annurev.micro.56.012302.160806. [DOI] [PubMed] [Google Scholar]
- 42.Courvalin P. Vancomycin resistance in Gram-positive cocci. Clin Infect Dis. 2006;42:S25–S34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 43.Hegstad K, Mikalsen T, Coque TM, Werner G, Sundsfjord A. Mobile genetic elements and their contribution to the emergence of antimicrobial resistant Enterococcus faecalis and Enterococcus faecium. Clin Microbil Infect. 2010;16:541–554. doi: 10.1111/j.1469-0691.2010.03226.x. [DOI] [PubMed] [Google Scholar]
- 44.Walker S, Chen L, Helm J, Hu Y, Rew Y, Shin D, Boger DL. Chemistry and biology of ramoplanin: a lipoglycodepsipeptide with potent antibiotic activity. Chem Rev. 2005;105:449–476. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]