

Abstract
Transforming growth factor-beta1 (TGF-β1) and vascular endothelial growth factor (VEGF), both angiogenesis inducers, have opposing effects on vascular endothelial cells. TGF-β1 induces apoptosis; VEGF induces survival. We have previously shown that TGF-β1 induces endothelial cell expression of VEGF, which mediates TGF-β1 induction of apoptosis through activation of p38 mitogen-activated protein kinase (MAPK). Because VEGF activates p38MAPK but protects the cells from apoptosis, this finding suggested that TGF-β1 converts p38MAPK signaling from prosurvival to proapoptotic. Four isoforms of p38MAPK - α, β, γ and δ – have been identified. Therefore, we hypothesized that different p38MAPK isoforms control endothelial cell apoptosis or survival, and that TGF-β1 directs VEGF activation of p38MAPK from a pro-survival to a proapoptotic isoform. Here we report that cultured endothelial cells express p38 α, β and γ. VEGF activates p38 β, whereas TGF-β1 activates p38 α. TGF-β1 treatment rapidly induces p38 α activation and apoptosis. Subsequently, p38 α activation is downregulated, p38 β is activated, and the surviving cells become refractory to TGF-β1 induction of apoptosis and proliferate. Gene silencing of p38 α blocks TGF-β1 induction of apoptosis, whereas downregulation of p38 β or p38 γ expression results in massive apoptosis. Thus, in endothelial cells p38 α mediates apoptotic signaling, whereas p38 β and p38 γ transduce survival signaling. TGF-β1 activation of p38 α is mediated by VEGF, which in the absence of TGF-β1 activates p38 β. Therefore, these results show that TGF-β1 induces endothelial cell apoptosis by shifting VEGF signaling from the prosurvival p38 β to the proapoptotic p38 α.
Keywords: TGF-β1, VEGF, p38MAPK isoforms, endothelial cells, apoptosis
INTRODUCTION
Angiogenesis, the formation of capillaries from preexisting blood vessel, is mediated by a variety of cytokines and growth factors with paracrine or autocrine modes of action. Vascular endothelial growth factor (VEGF) and transforming growth factor-beta 1 (TGF-β1), potent angiogenesis inducers, act on vascular endothelial cells through different mechanisms [1–3].
VEGF (VEGF A), the prototype member of a family of four growth factors (VEGF A - D), upregulates endothelial cell proliferation and migration, and protects endothelial cells from apoptosis [1]. VEGF exerts its activity through two tyrosine kinase receptors, VEGFR-1 (flt-1) and VEGFR-2 (flk-1). VEGFR-2 has been implicated in endothelial cell proliferation and survival, and VEGFR-1 in chemotaxis and vascular permeability [4, 1]. Protein kinase B (PKB, or Akt) and mitogen-activated protein kinases (MAPK) are components of the signaling mechanism activated by VEGFR-2 [5].
TGF-β1, the prototype member of a superfamily of five multifunctional growth factors, is a potent proliferation inhibitor for most cell types, and an important regulator of tissue morphogenesis [3, 6]. TGF-β1 induces vessel formation in vitro and in vivo [7–11]; however, it inhibits endothelial cell proliferation and migration [12], and downregulates VEGFR-2 expression [13, 14]. Notably, TGF-β1 induces endothelial cell apoptosis [15, 12] by inhibiting expression of the anti-apoptotic protein Bcl-2 [16] and activating p38 MAPK (p38MAPK) [17].
Thus, although both VEGF and TGF-β1 induce angiogenesis, they have opposing effects on endothelial cells. Remarkably, TGF-β1 is a potent inducer of apoptosis, whereas VEGF protects the cells from apoptosis. It has therefore been proposed that TGF-β1 induces angiogenesis in vivo through an indirect mechanism, by recruiting inflammatory cells that in turn secrete VEGF and/or other angiogenesis inducers. However, TGF-β1 induces endothelial cell expression of VEGF in vitro and in vivo [18–20] and, surprisingly, VEGF is required for induction of endothelial cell apoptosis through VEGFR-2 activation of p38MAPK [19, 20].
p38MAPK, a class of MAPK activated by environmental stresses, growth factors and cytokines, controls cell functions including proliferation and apoptosis, differentiation and senescence [21, 22]. Four isoforms of p38MAPK - α, β, γ and δ - have been identified in mammalian cells. Despite >60% sequence homology and >90% identity within their kinase domains, the p38MAPK isoforms show notable differences in tissue expression, upstream activators and downstream effectors. p38α and p38β are ubiquitous; p38γ and p38δ are tissue-specific [22]. In addition, the p38 isoforms have been described in different cell compartments, where they can have opposing effects on the same substrate [21]. The specific function(s) of the individual isoforms in physiology and pathology are largely unknown. The genetic deficiency of p38α in mice results in embryonic lethality, with aberrant placental development and abnormal angiogenesis in the yolk sac and embryo [23]. In contrast, disruption of the other isoforms generates no apparent phenotype [21]. Numerous studies have implicated p38MAPK in induction of endothelial cell apoptosis by a variety of agents [24–27]. However, the p38 isoforms involved have not been characterized.
Here we report that in vascular endothelial cells p38α mediates proapoptotic signaling from inducers of apoptosis such as TGF-β1, whereas p38β relays survival signaling from prosurvival factors including VEGF, and that TGF-β1 induces endothelial cell apoptosis by converting VEGF signaling from the pro-survival p38β to the pro-apoptotic p38α.
MATERIALS AND METHODS
Materials
Human purified or recombinant TGF-β1, recombinant human VEGF, and antibodies to p38α, p38γ and p38δ were purchased from R&D Systems (Minneapolis, MN); antibodies to human cleaved caspase 3 and cleaved PARP from Cell Signaling Technologies (Beverly, MA); mouse and rabbit non-immune IgG from Sigma-Aldrich (St. Louis, MO), antibodies to p38, α-tubulin and cyclin E from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Human recombinant fibroblast growth factor-2 (FGF-2) was purchased from Gibco BRL (Rockville, MD); antibody to active p38 from Promega Corp. (Madison, WI), monoclonal antibody to p38β from Zymed Laboratories (San Francisco, CA); recombinant p38α from R&D Systems, p38 β Biovision, (Mountain View, CA), and p38γ from Abnova, (Walnut, CA).
Cells and Media
Bovine capillary endothelial cells (BCE) were as described [28], and used at passages 6–15. Human umbilical vein endothelial (HUVE; Clonetics, San Diego, CA) cells were grown in medium (EBM2; Clonetics) containing 2% fetal calf serum (FCS) and the growth supplements provided by the company, and were used at passages 3–5. BCE or HUVE cells were starved overnight in their medium supplemented with 0.5% DCS or FCS, respectively, after which either TGF-β1 (1 ng/ml) or VEGF (30 ng/ml) or FGF-2 (10 ng/ml) was added and incubation was continued for the indicated time. The time of addition of TGF-β1 was considered as time 0.
Western blotting was performed as described [19].
Bromodeoxyuridine (BrdU) uptake was performed as described [19].
Immunoprecipitation
Cells were lysed in 10 mM Tris-HCl pH 7.4 containing 150 mM NaCl, 1% Triton-X-100, 1 mM Pefabloc (Roche), 1 mM leupeptin, 1 mM Na3VO4, and 2 mM CaCl2. (plus 100 μM peroxyvanadate for phosphorylation studies ). One hundred micrograms of cell extract protein was pre-cleared at 4°C for 30 min with 0.5 μg of non-immune IgG coupled to 10 μl of protein A+G agarose beads (Santa Cruz Biotechnology). Pre-cleared extracts were centrifuged for 60 s at 300 x g, and the supernatant was immunoprecipitated overnight at 4°C with 10 μl of protein A+G agarose beads and 0.75 μg of antibody per 100 μg of protein. After washing three times with lysis buffer the beads were boiled in reducing Laemmli buffer for 5 min and loaded onto SDS/polyacrylamide gels.
siRNA transfection
Subconfluent HUVE cells were transiently transfected as described [19] and used for the experiments 48 h after transfection.
Reverse transcription-polymerase chain reaction (RT-PCR)
The following primers were synthesized by IDT DNA technologies (Coralville, IA) based on the published sequences: GAPDH (forward: 5′-CCC ACT CTT CCA CCT TCG-3′; reverse: 5′-TCC TTG GAG GCC ATG TAG GCC AT-3′); p38α (forward: 5′-GCA GGG ACC TTC TCA TAG AT-3′; reverse: 5′-GAG GGA TAG CCT CAG ACC-3′); p38β (forward: 5′-CTG CAA GGA AAG GCC CTC-3′; reverse: 5′-CAG GCA ATG CCT CAC TGC-3′); p38γ (forward: 5′-GAT TAC TGG GAA GAT CCT G-3′; reverse: 5′-CGT CAC AGA GCC GTC TCC-3′); p38δ (forward: 5′-GAC ACT CTT CAA GGG CAA G-3′; reverse: 5′-GCC ATC AAT CAC TGC AGC-3′). Complementary DNA (cDNA) was synthesized from 1 μg of total RNA using SuperScript II RT (Invitrogen, Carlsbad, CA) and oligo-dT 3′ primer. Two μl of cDNA was amplified by PCR as described.
Proximity ligation assay (PLA) [29]
HUVE cells grown on gelatin-coated glass coverslips were treated with TGF-β1 (1 ng/ml) or control medium for 6 h or 72 h and fixed. The cells were then incubated with isoform-specific antibodies to p38α or β together with antibody to phospho-p38 overnight at 4° C with gentle agitation. PLUS and MINUS secondary PLA probes against rabbit and mouse IgG (Olink Bioscience) were added, and the cells were incubated at 37° C for 1 h with gentle agitation, followed by incubation with ligation mix for 30 min at 37° C. Amplification mix was then applied for 100 min at 37° C. The coverslips were mounted on microscope slides with Doulink Mounting Medium with Dapi, and the cells photographed under a fluorescence microscope.
In vitro angiogenesis assay in 3D collagen gel [30]
Collagen type I (Becton Dickinson, Bedford, MA), was placed into 24-well tissue culture plates (40 μl/well) and allowed to gel for 30 min at 37° C. HUVE cells (2.5 × 104 cells/well) were seeded into the wells and incubated at 37° C for 1 h in growth medium. Subsequently, the medium was removed and 40 μl/well of collagen were added. After incubation at 37° C for 30 min, 100 μl of medium supplemented with 0.5% FCS and 1 ng/ml of TGF-β1 was added, and the plates were incubated at 37° C. TGF-β1 (1 ng/ml) was added every other day. After 5–7 days, the culture medium was removed; the cultures were washed with PBS, stained with toluidine blue, and photographed under an inverted microscope. The number of tube-like structures forming anastomoses was counted as described [20].
Statistical Analysis
Student’s t-tests on the equality of means were performed using Stata 8.
RESULTS
We have previously shown that TGF-β1 induces endothelial cell apoptosis in vitro and in vivo through VEGF/VEGFR-2 activation of p38MAPK [20, 19]. In the absence of TGF-β1, VEGF activates p38MAPK but protects endothelial cells from apoptosis [19]. Therefore, our finding raised the question: How does TGF-β1 convert VEGF signaling from anti- to pro-apoptotic? Because p38MAPK exists in four isoforms with different biological functions, we hypothesized that TGF-β1 shifts VEGF activation of p38MAPK from one isoform to another.
To investigate this hypothesis we characterized endothelial cell expression of the p38 isoforms, and the effect of TGF-β1 and VEGF on their activation. By RT-PCR endothelial cells expressed p38α, β and γ mRNAs, whereas p38δ mRNA was barely detectable (Fig. 1A). Western blotting with p38MAPK isoform-specific antibodies showed expression of the corresponding proteins (Fig. 1B and C).
Fig. 1. Endothelial cell expression of p38 isoforms.
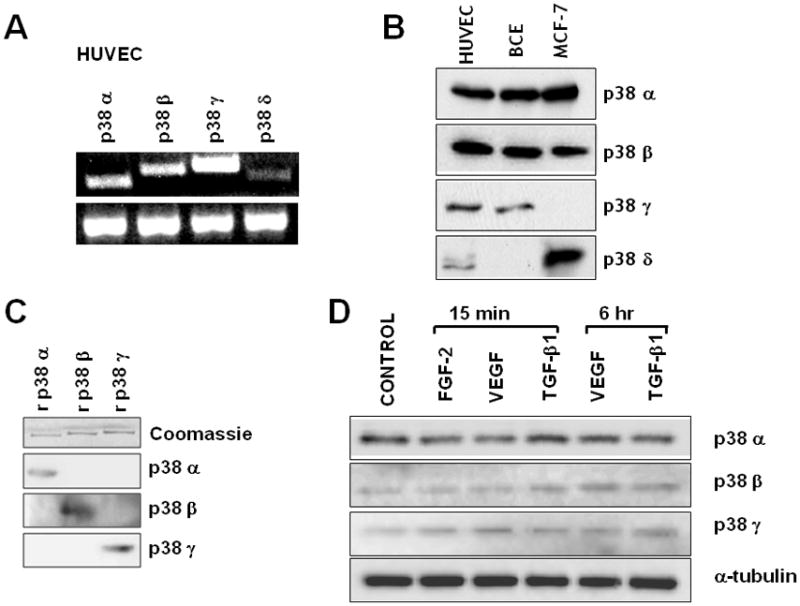
A. RT-PCR analysis of HUVE cell expression of p38MAPK isoforms. G3PDH is shown as a loading control in the lower panel. B. Western blotting analysis of HUVE and BCE cell extracts expression of p38MAPK isoforms. Human MCF-7 mammary carcinoma cells are shown as a control for p38δ. The blot was probed sequentially with the different antibodies. C. Recombinant (r) purified p38α, p38β and p38γ analyzed by Western blotting with antibodies to the three isoforms. Coomassie blue staining of the SDS/polyacrylamide gel is shown as a loading control. D. Western blotting analysis of p38 isoform expression in HUVE cells incubated with either FGF-2 (10 ng/ml) or VEGF (30 ng/ml) or TGF-β1 (1 ng/ml) for the indicated times. Cells receiving no addition (CONTROL) were used as control. The blot was probed sequentially with antibodies to p38α, β and γ, and to α-tubulin as a loading control. These experiments were repeated twice with comparable results.
Neither VEGF nor TGF-β1 treatment of endothelial cells significantly altered p38α, β or γ expression (Fig. 1D), suggesting that the activity of these kinases is regulated at post-transcriptional level. Isoform-specific antibodies to phosphorylated p38 do not exist because the isoforms’ phosphorylation sequences are identical. Therefore, we immunoprecipitated extracts of TGF-β1- or VEGF-treated cells with antibodies to the individual p38 isoforms, and analyzed the immunoprecipitates by Western blotting with an antibody that recognizes all phosphorylated isoforms. The results (Fig. 2A) showed that untreated endothelial cells had no active p38α but showed comparable levels of active p38β and p38γ, Treatment with TGF-β1 selectively activated p38α. Conversely, VEGF selectively activated p38β. FGF-2, which protects endothelial cells from apoptosis, strongly upregulated p38β activation; in contrast, UVB irradiation, which induces apoptosis, activated p38α.
Figure 2. Control of p38 isoform activation in endothelial cells.
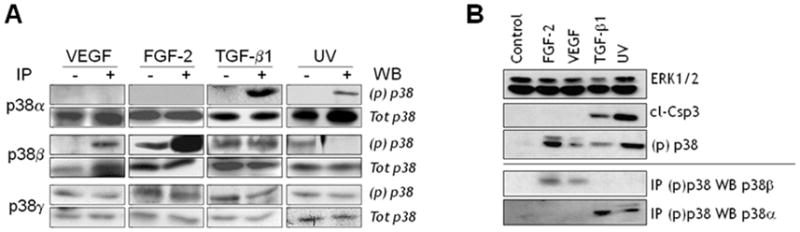
A. Endothelial cells were incubated with either FGF-2 (10 ng/ml) or VEGF (30 ng/ml) or TGF-β 1 (1 ng/ml) for 6 h, or irradiated with UVB (20 mJ/cm2). Cell extracts were immunoprecipitated with antibodies to p38α, β or γ, and analyzed by Western blotting with the corresponding antibodies and with antibody to phospho-p38 [(p)p38]. B. Endothelial cells were treated with the indicated reagents as described above. Cell extracts were immunoprecipitated with antibody to phospho-p38 and analyzed by Western blotting with antibodies to p38α or p38β (lower panel). Extracts were also analyzed by Western blotting for cleaved caspase-3 (cl-Csp3), an apoptosis marker, and p38 activation [(p)p38] (upper panel). ERK2: loading control.
Although the anti-p38α and p38β antibodies did not show cross-reactivity by Western blotting (Fig. 1C), they might cross-react by immunoprecipitation. Therefore, we performed reverse experiments in which cell extracts were immunoprecipitated with antibody to phospho-p38 and the immunoprecipitates analyzed by Western blotting with antibodies to p38α or p38β. The results (Fig. 2, B) showed that treatment with FGF-2 or VEGF selectively activated p38β, whereas TGF-β1 or UVB irradiation activated p38α. Western blotting analysis of caspase 3 activation, a marker of apoptosis, showed that TGF-β1 and UVB induced cell death, whereas FGF-2 and VEGF had no such effect (Fig. 2B). Therefore, these findings suggested that in endothelial cells p38α mediates apoptotic signaling, whereas p38β relays prosurvival signaling. Our previous studies [19, 20] have shown that in endothelial cells TGF-β1 activation of p38MAPK is abolished by downregulation of VEGFR-2, demonstrating that endothelial cell VEGF mediates p38MAPK activation by TGF-β1. In addition, TGF-β1 does not induce apoptosis in endothelial cells that do not express VEGF in response to TGF-β1 [19]. Because in the absence of TGF-β1 VEGF activates p38β, our present results suggested that TGF-β1 induces endothelial cell apoptosis by shifting VEGF activation of p38MAPK from the β to the α isoform.
The apoptotic effect of TGF-β1 is rapid (3 h – 12 h) and followed by refractoriness of the surviving cells to TGF-β1 induction of apoptosis up to 96 h [20]. We found that TGF-β1 treatment of endothelial cells for 72–96 h resulted in increased BrdU uptake and cyclin E expression between 24 h and 96 h (Figs. 3 and 4), coincident with the formation of cord-like structures (Fig. 3) [20, 7]. A low level of apoptosis (caspase-3 activation) was also detected between 72 h and 96 h of TGF-β1 treatment, concurrent with cell proliferation (Figs. 3 and 4). The level of BrdU uptake induced by TGF-β1 was comparable to that induced by 10% FCS, and occurred in cells grown in starvation medium (0.5% serum) for 72 h – 96 h, a condition that induces endothelial cell apoptosis [31]. Thus, TGF-β1 has a dual effect on endothelial cells: rapid and transient induction of apoptosis, followed by sustained proliferation of the surviving cells.
Figure 3. TGF-β1 induction of endothelial cell proliferation and capillary morphogenesis.
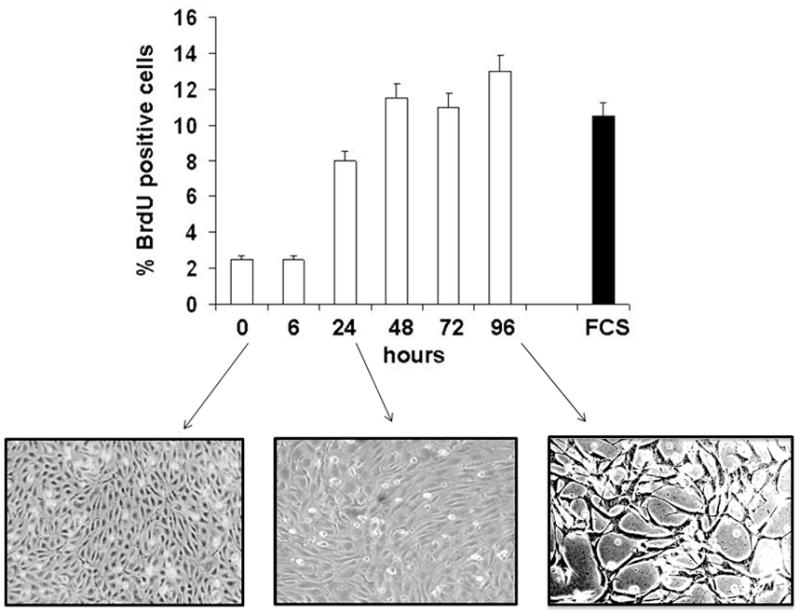
BrdU uptake by BCE cells treated with TGF-β1 (1 ng/ml) for the indicated times. Cells incubated with 10% FCS are shown as positive control. The cells were photographed at the indicated times to show morphological changes. Magnification: 100 X.
Figure 4. TGF-β 1 induction of endothelial cell apoptosis and proliferation correlates with activation of distinct p38 isoforms.
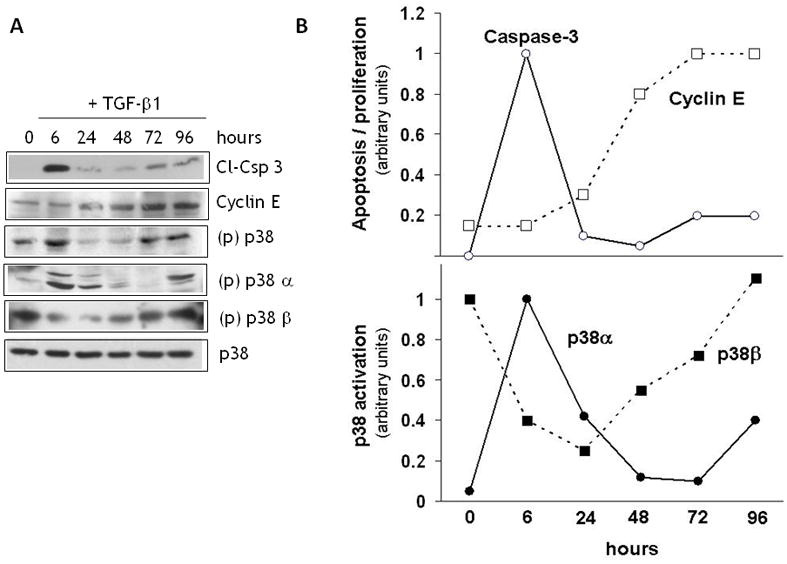
A. Western blotting analysis of caspase 3 cleavage, cyclin E expression, and activation of total p38 (all isoforms), p38α and p38β in endothelial cells treated with TGF-β1 (1 ng/ml) for the indicated times. Total p38 [(p)p38] activation was analyzed by Western blotting with antibody to phospho-p38. Activation of p38α and p38β [(p)p38α and (p)p38] was characterized by immunoprecipitation with antibody to phospho-p38 followed by Western blotting with antibodies to p38α or p38β. Total p38MAPK (p38) is shown as a loading control. Representative blots from triplicate experiments are shown. B. Densitometric analysis of the Western blot bands shown in panel A.
Based on these findings, we hypothesized that the cell proliferation and refractoriness to apoptosis that follow TGF-β1-induced apoptosis are mediated by a p38MAPK isoform(s) other than the one that mediates apoptosis. Therefore, we analyzed p38MAPK isoform activation in endothelial cells treated with TGF-β1 for different times. Consistent with our previous findings [20, 19], Western blotting analysis with an antibody that recognizes all phosphorylated p38 isoforms showed that TGF-β1 induced p38MAPK activation between 3 h and 9 h of treatment (Fig. 4A), concomitantly with the onset of apoptosis, and subsequently between 72 h and 96 h, a time when addition of TGF-β1 fails to induce apoptosis [20] and cell proliferation occurs (Figs. 3 and 4). This observation suggested that the early induction of p38MAPK activation (3–6 h) involves the α isoform, which is associated with apoptosis, and the late activation (72–96 h) entails the β isoform, which mediates survival signaling. Therefore, we analyzed the cell extracts by immunoprecipitation with antibody to phospho-p38MAPK followed by Western blotting with p38α or p38β antibodies. The results (Fig. 4A and B) showed that p38α activation occurred between the initial 6–24 h of treatment, coincident with apoptosis, while p38β activation decreased. Subsequently, the level of active p38α decreased, and p38β activation increased between 48 h and 96 h, concurrently with cell proliferation (Fig. 3 and 4) and refractoriness to TGF-β1 induction of apoptosis [20]. Activation of p38α - to a level lower than at 6 h - also occurred at 72–96 h, coincident with the low level of caspase-3 activation observed at this time of incubation.
To confirm these findings we used the in situ proximity ligation assay [29] with antibodies to p38α or p38β together with phospho-p38 antibodies, which allowed the detection of phosphorylated p38α and p38β in individual cells. The results showed that cells treated with TGF-β1 for 6 h stained positively for p38α but not for p38β. Conversely, TGF-β1 treatment for 72 h resulted in activation of p38β but not of p38α (Fig. 5). Thus, altogether these results indicated that p38α mediates TGF-β1 induction of apoptosis, whereas p38β is associated with TGF-β1-induced cell proliferation and refractoriness to apoptosis.
Figure 5. Proximity ligation assay (PLA) of p38α and p38β activation by TGF-β1.
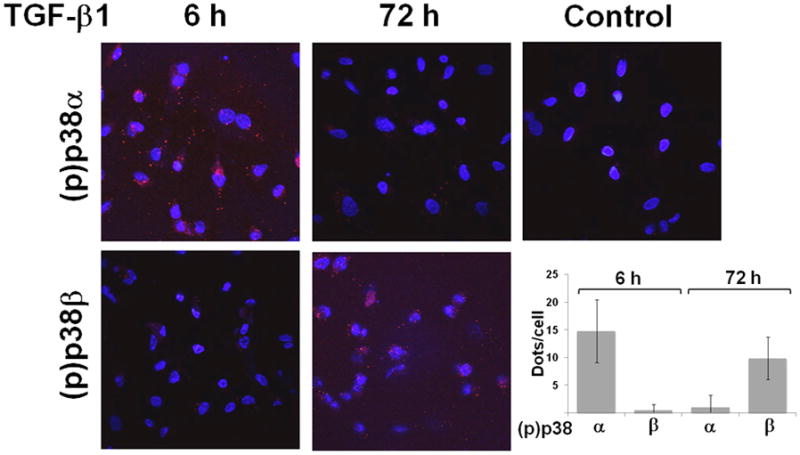
HUVE cells treated with TGF-β1 (1 ng/ml) for the indicated times, or with control medium for 72 h. Phosphorylation of the p38 isoforms is revealed by red dots). The cells were counterstained with DAPI to evidence the nuclei. Magnification: 600 X. The graph shows m±s.e. of red dots/cell counted in 20–30 cells from three independent experiments.
We therefore used siRNAs to selectively downregulate the p38α, β and γ isoforms, and characterized their effect on apoptosis. Transient transfection with siRNA to one p38 isoform downregulated the corresponding isoform without affecting expression of the other isoforms (Fig. 6A), showing that the inhibitory action of the siRNA did not reflect nonspecific effects of the siRNAs or the transfection conditions. Transient transfection with p38α siRNA or a mixture of siRNAs to all p38 isoforms abolished TGF-β1 induction of apoptosis. Conversely, transfection with p38β or γ siRNA induced apoptosis both in untreated and TGF-β1-treated cells (Fig. 6B). Thus, in endothelial cells p38α transduces apoptotic signaling by apoptosis inducers such as TGF-β1, and p38β transduces survival signaling by pro-survival factors such as FGF-2 or VEGF. p38γ also mediates survival signaling but its activation is not controlled by VEGF or TGF-β1.
Figure 6. Control of endothelial cell apoptosis and survival by p38 α, β and γ.
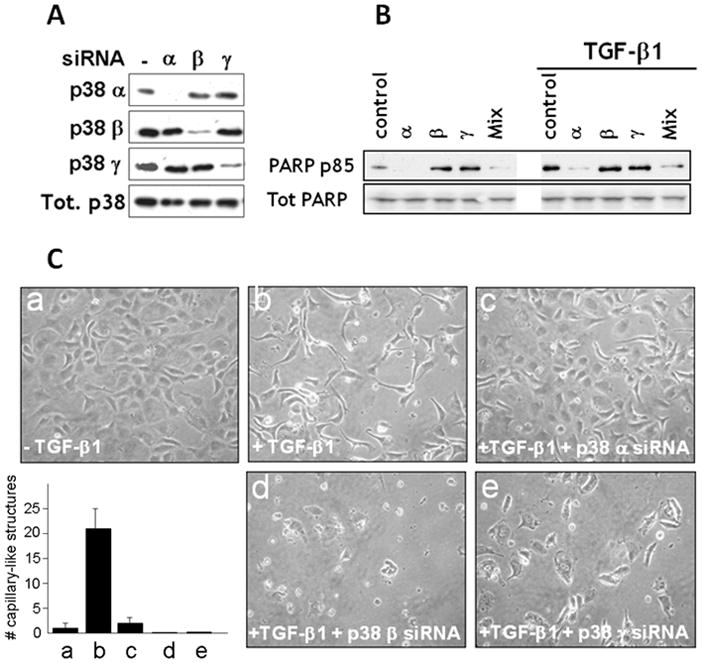
A. Western blotting analysis of p38 isoform expression in HUVE cells transfected with siRNAs to either p38α or β or γ. A blot with an antibody to all p38 isoforms (Tot. p38) is shown as a loading control. B. HUVE cells transfected with siRNAs to either p38α or β or γ, or with a mixture thereof (Mix), or mock transfected (control) and incubated with TGF-β 1 (1 ng/ml) or control medium for 6 h were analyzed by Western blotting with antibody to cleaved PARP (PARP p85) or to total PARP as a loading control. C. Confluent HUVE cells grown in gelatin-coated dishes and either non transfected and untreated (a) or transfected with transfection reagent alone (b) or siRNAs to either p38α (c) or β (d) or γ (e). The cells were photographed after 96 h incubation in the absence (a) or presence (b-d) of TGF-β 1 (1 ng/ml). Bar: 100 μm. Histogram. The results were quantitated as described [20]. Mean ± SE of triplicate samples are shown. *: p < 0.05 (sample vs. a). These experiments were repeated three times with comparable results.
The morphological analysis of the siRNA-transfected cells showed effects consistent with the biochemical characterization of apoptosis (Fig. 6C). As described [7, 20], confluent endothelial cells treated with TGF-β1 for 72 – 96 h formed structures that resemble precapillary cords (Fig. 6C, panels a and b). However, TGF-β1-treated cells transfected with p38α siRNA retained a confluent monolayer with no apparent morphological changes (Fig. 6C, panel c). In contrast, transfection with p38β or γ siRNAs caused massive cell death resulting in sparse, disrupted cord-like structures (Fig. 6C, panels d and e). Notably, downregulation of these p38 isoforms blocked the proliferative effect of TGF-β1 that follows the transient induction of apoptosis (Figs. 3 and 4), and induced massive apoptosis at a time of TGF-β1 treatment (72 – 96 h) when endothelial cells are refractory to TGF-β1 induction of apoptosis [20].
To investigate the relative contribution of p38α and p38β to blood vessel formation, we used an in vitro angiogenesis assay (Fig. 7). For this purpose HUVE cells were either mock transfected or transiently transfected with siRNAs to the α, β or γ isoforms of p38, and grown in 3D collagen gels in the presence of 1 ng/ml of TGF-β1. Within 5 – 7 days mock transfected cells formed a network of cord-like structures as described [30]. In contrast, with cells transfected with the siRNAs to the p38MAPK isoforms this effect was dramatically blocked. Cells transfected with p38α siRNA showed very rudimentary cord-like structures, larger than those obtained with mock transfected cells and consisting of flattened cells that in several areas maintained a confluent monolayer. Conversely, cells transfected with siRNAs to p38β or γ showed sparse, disrupted capillary-like structures that failed to form a network (Fig. 7). Numerous areas of the collagen gels only contained isolated clusters of globular cells.
Figure 7. In vitro angiogenesis assay in 3D collagen gel.
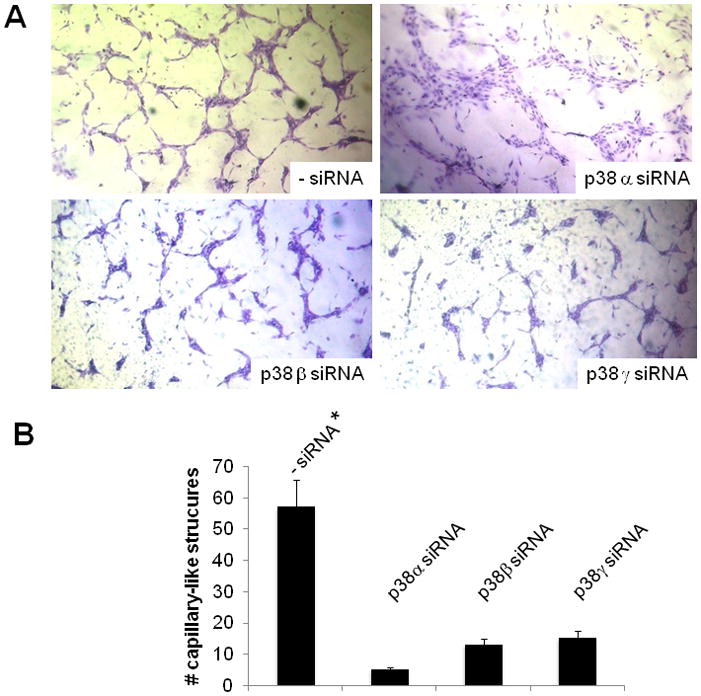
A. HUVE cells transfected with the indicated siRNAs, or mock transfected (- siRNA), and grown in 3 D collagen gels for 5 days in the presence of TGF-β1 (1 ng/ml). Magnification: 100 X. B. Results of the experiments shown in panel A quantitated as described under Materials and Methods. Mean ± SE of triplicate samples are shown. *: p < 0.05 (sample vs. a). These experiments were repeated three times with comparable results.
Thus, the p38MAPK isoforms appear to have opposing roles in TGF-β1 induction of in vitro angiogenesis, which mirror their effects on endothelial cell apoptosis and survival. Because p38α activation by TGF-β1 is mediated by VEGF [20, 19], which in the absence of TGF-β1 activates p38β, our results show that TGF-β1 converts VEGF signaling from activation of the prosurvival p38β to the proapoptotic p38α.
DISCUSSION
We have previously shown that TGF-β1 induces endothelial cell apoptosis through VEGF/VEGFR-2-mediated activation of p38MAPK [19]. Because in the absence of TGF-β1 VEGF activates p38MAPK but protects endothelial cells from apoptosis, our finding suggested that TGF-β1 modifies VEGF signaling from anti- to pro-apoptotic. The data reported in this paper show that TGF-β1 induces endothelial cell apoptosis by shifting VEGF activation of p38MAPK from the β to the α isoform. In endothelial cells the β isoform of p38MAPK mediates prosurvival signaling induced by growth factors such as VEGF and FGF-2, which protect endothelial cells from apoptosis. Conversely, p38α mediates apoptotic signaling from inducers of endothelial cell apoptosis such as TGF-β1 and UVB. p38γ also mediates endothelial cell survival, but is not controlled by VEGF or TGF-β1.
Numerous studies implicate p38MAPK in apoptosis. In endothelial cells p38MAPK activation mediates apoptosis induction by a variety of agents [24–26, 32, 27, 33, 34], and VEGF blocks endothelial cell apoptosis by decreasing p38MAPK phosphorylation [35, 27]. However, the p38MAPK isoforms involved in endothelial cell apoptosis have not been characterized. In cell types other than endothelial cells, p38MAPK activation in response to a variety of stimuli induces either proliferation or growth arrest/apoptosis, depending on the cell type [21, 36–38]. The specific p38 isoforms involved in these processes remain largely unidentified. In human fibroblasts activation of p38γ, but not of other p38 isoforms, is required for induction of G2 arrest by gamma irradiation [39], suggesting that the individual p38 isoforms can have different effects on cell fate. Both p38α and β have been implicated in the pathogenesis of myocardial hypertrophy, where they may play different roles; p38β being more potent in inducing hypertrophy, and p38α in other functions such as apoptosis [40]. Similarly, p38α but not p38β is required for ischemic preconditioning of the myocardium [41]. Our study is the first to identify opposing roles for the α and β isoforms of p38MAPK in the control of endothelial cell survival and apoptosis.
Our data also show several features of the mechanism by which TGF-β1 controls endothelial cell death and proliferation. Under most culture conditions, TGF-β1 is a potent inhibitor of endothelial cell proliferation and migration, and an inducer of apoptosis [42, 43]. However, TGF-β1 also stimulates endothelial cell proliferation in vitro and in vivo [43, 10, 44]. We found that the apoptotic effect of TGF-β1 on endothelial cells is rapid and transient, and is followed by a long period during which the surviving cells are refractory to apoptosis induction by TGF-β1 [20] and proliferate. These opposing effects of TGF-β1 are mediated by the selective activation of two different p38 isoforms. The rapid induction of apoptosis results from downregulation of p38β and upregulation of p38α activation. Subsequently, downregulation of p38α and parallel, sustained increase in p38β activation provide survival signaling. Based on our data we cannot conclude whether TGF-β1 induction of endothelial cell proliferation is also mediated by p38β, because siRNA-mediated downregulation of p38 β expression results in rapid and massive apoptosis. Our previous findings that TGF-β1 induces prolonged upregulation of endothelial cell VEGF and FGF-2 expression and activates ERK1/2 by a VEGF/VEGFR-2-dependent mechanism [19] suggest that the proliferative effect of TGF-β1 could be mediated by ERK1/2, while the sustained activation of p38β provides endothelial cells with survival signaling. Thus, TGF-β1 controls both endothelial cell apoptosis and survival through the selective activation of the α and β isoforms of p38MAPK, respectively.
The effect of TGF-β1 on endothelial cells is mediated by two type-I receptors (TGF-βRI): ALK (Activin receptor-Like Kinase)-5, the predominant type I receptor that mediates TGF-β1 responses, and ALK-1, whose expression is restricted to endothelial cells. ALK-5 and ALK-1 mediate TGF-β signaling through distinct Smad proteins: Smad2/Smad3 and Smad1/Smad5, respectively [11]. Although TGF-β1 primarily activates Smad2 and -3, in primary endothelial cells it also activates Smad1 and -5 through a heterodimeric receptor complex consisting of ALK5 and ALK1 [45]. The ALK5 and ALK1 pathways mediate opposing effects; ALK5 inhibits, while ALK1 stimulates cell migration and proliferation [46]. In addition ALK5, but not ALK1, mediates TGF-β1 induction of endothelial cell apoptosis [47]. However, ALK-1/Smad1 signaling mediates TGF-β1 induction of VEGF expression in endothelial cells [18], which is required for TGF-β1 induction of apoptosis [19]. Based on the data presented we cannot conclude whether ALK1 and ALK5 activate different p38 isoforms. It is tempting to speculate that signaling by both receptors is required for TGF-β1 induction of apoptosis, and that downregulation of ALK5 is responsible for the ensuing cell proliferation (Fig. 4) [20]. The relative contribution of TGFβRs to activation of the different isoforms of p38MAPK and control of endothelial cell apoptosis warrants further investigation.
Our conclusions are partly based on results of siRNA-mediated gene silencing experiments. It has been reported that siRNAs can inhibit angiogenesis by a nonspecific effect mediated by toll-like receptor 3 (TLR3), a double-stranded RNA receptor that generates apoptotic signaling in endothelial cells [48, 49]. This effect requires siRNAs over 21 nt in length, and has been obtained with non-chemically modified molecules. We used 19-bp siRNAs with dinucleotide 3′ DNA overhangs, a chemical modification to prevent nonspecific effects. Indeed, in our studies we did not observe nonspecific apoptotic effects or p38MAPK activation induced by our siRNAs. Our VEGFR-1 and VEGFR-2 siRNAs did not induce apoptosis [19, 20], nor did our siRNAs to p38α or a mixture of siRNAs to p38α, β and γ (Fig. 6B). Similarly, our siRNAs to VEGFR-1 and VEGFR-1, or control siRNA to lamin A did not induce p38MAPK activation [19]. Therefore, our siRNAs did not have nonspecific effects on p38MAPK activation or endothelial cell apoptosis mediated by TLR3.
Our results are in contrast with previous data showing that inhibition of p38MAPK blocks endothelial cell apoptosis [19, 35, 27]. Because p38β and γ mediate survival signaling, their inhibition should result in apoptosis. This discrepancy is explained by the fact that all currently available inhibitors of p38MAPK inhibit both p38α and p38β, and therefore abolish both pro- and anti-apoptotic signaling. In addition, the apoptotic effect of p38α activation appears to be prevalent. Indeed, siRNA-mediated downregulation of all the p38 isoforms expressed by endothelial cells blocks TGF-β1 induction of apoptosis as effectively as downregulation of p38α alone. In contrast, selective inhibition of p38β or p38γ expression results in apoptosis (Fig. 6B and C).
Based on our data we cannot conclude whether the proliferative effect of TGF-β1 on endothelial cells is mediated by VEGF or other endothelial cell-derived growth factors. TGF-β1-induced upregulation of VEGF lasts at least 24 h [19], a timing coincident with the onset of cell proliferation. Although TGF-β1 downregulates endothelial cell expression of VEGFR-2, this effect does not exceed 40–50% [13, 14]. In addition, TGF-β1 upregulates endothelial cell expression of FGF-2 [19], which can also provide a mitogenic stimulus. Therefore, both VEGF and FGF-2 could mediate TGF-β1 induction of endothelial cell proliferation through sustained activation of the ERK1/2 or other signaling pathways.
The coordinated mechanism of control of endothelial cell apoptosis, survival and proliferation by TGF-β1 – VEGF activation of p38α and β has profound effects on in vitro angiogenesis. Downregulation of p38α-mediated apoptosis blocks the formation of capillary-like structures in 3D collagen gel. Similarly, induction of excess apoptosis by downregulation of p38β – mediated survival signaling blocks endothelial cell sprouting and organization into a capillary-like network. These findings are consistent with our previous observation that inhibition of endothelial cell apoptosis blocks angiogenesis in vitro and in vivo [20].
We have previously shown that VEGF mediates TGF-β1 induction of endothelial cell apoptosis through activation of p38MAPK [19]. This conclusion was based on our findings that inhibition of VEGF or downregulation of VEGFR-2 blocks TGF-β1 activation of p38MAPK, and inhibition of p38MAPK expression abrogates TGF-β1 induction of apoptosis [19]. Our present data show that VEGF activates the prosurvival p38β, whereas TGF-β1 activates the proapoptotic p38α. Because TGF-β1 activates p38α through VEGF, which in the absence of TGF-β1 activates p38β, our findings show that TGF-β1 shifts VEGF signaling from p38β to p38α.
Our results therefore generate the following model for the control of endothelial cell apoptosis and survival by TGF-β1 and VEGF (Fig. 8). In the absence of TGF-β1, VEGF binding to VEGFR-2 induces endothelial cell activation of p38β and cell survival (left panel). TGF-β1 induces endothelial cell expression of VEGF, and switches VEGF/VEGFR-2-activated signaling to the proapoptotic p38α. This effect involves crosstalk between TGF-β1 and VEGF signaling pathways that can occur at multiple levels upstream of p38MAPK. VEGF/VEGFR-2-mediated activation of p38α is necessary for TGF-β1 induction of endothelial cell apoptosis; however, parallel TGF-β1-initiated signaling acting in concert with p38α may also be required. Subsequently (right panel), p38α activation is downregulated and p38β activation upregulated. This effect results in abrogation of apoptosis and increased prosurvival signaling.
Figure 8. Control of endothelial cell apoptosis and survival by TGF-β 1 - VEGF interaction.
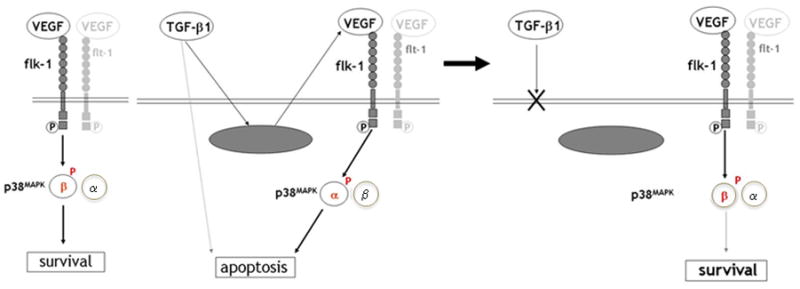
In the absence of TGF-β 1 VEGF induces activation of p38 β through VEGFR-2 and promotes endothelial cell survival (left panel). TGF- β1 induces VEGF expression. Crosstalk between VEGF- and TGF-β 1-specific signaling pathways converts VEGF/VEGFR-2 activation of p38 β into activation of the pro-apoptotic p38 α. Activation of p38 α is required for TGF- β1 induction of apoptosis; however, parallel TGF-β 1 signaling may also be necessary (dotted line). After the initial induction of apoptosis endothelial cells become refractory to the apoptotic effect of TGF-β 1, and VEGF/VEGFR-2 signaling reverts to activation of the prosurvival p38 β (right panel).
The signaling mechanism(s) that control the selective activation of the α or β isoform of p38 are largely unknown, as is the crosstalk between TGF-β1 and VEGF signaling that mediates the shift of VEGF activation of p38 from the β to the α isoform. Understanding these mechanisms can shed light on the complex control of vascular endothelial cell survival and death during angiogenesis. Importantly, our findings provide indications for the development of novel pharmacological approaches to inhibit tumor angiogenesis. We found that, whereas the effect of TGF-β1 on apoptosis is transient (Fig. 4) and followed by refractoriness of the cells to TGF-β1 induction of apoptosis [20], inhibition of p38β and/or p38γ signaling results in sustained induction of apoptosis by TGF-β1 (Fig. 6). Presently, all synthetic inhibitors of p38 block both the α and the β isoforms. Novel inhibitors that selectively inhibit p38β activity (or activation) could block the prosurvival effect of TGF-β1 (and VEGF) on endothelial cells which follows apoptosis induction. These inhibitors could thus transform the transient apoptotic effect of TGF-β1 into a sustained, massive effect. Mice genetically deficient in p38β show no major phenotype if unchallenged [50], indicating that selective inhibition of p38β would represent a feasible and safe approach to anti-tumor angiogenesis therapy.
Acknowledgments
Financial support: NIH grants R01 HL070203, R01 HL070203-03S1, 5R01 CA136715, R21 RAG033735A and funds from Department of Cardiothoracic Surgery, NYU School of Medicine and Dr. Leo A. Shifrin and Roslyn Myers Breast Cancer Discovery Fund [P.M.], R21 AG028785 [G.P.], RC1 HL10035 [G.F]
Work supported by NIH grants R01 HL070203, R01 HL070203-03S1, 5R01 CA136715, R21 RAG033735A, funds from Department of Cardiothoracic Surgery, NYU School of Medicine and Dr. Leo A. Shifrin and Roslyn Myers Breast Cancer Discovery Fund [P.M.], R21 AG028785 [G.P.], and RC1 HL10035 [G.F].
Footnotes
Conflicts of interest: none
References
- 1.Ferrara N. Vascular endothelial growth factor. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:789–91. doi: 10.1161/ATVBAHA.108.179663. [DOI] [PubMed] [Google Scholar]
- 2.Tian M, Neil JR, Schiemann WP. Transforming growth factor-beta and the hallmarks of cancer. Cellular signalling. 2011;23:951–62. doi: 10.1016/j.cellsig.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bierie B, Moses HL. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine & growth factor reviews. 2010;21:49–59. doi: 10.1016/j.cytogfr.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 5.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. The Biochemical journal. 2011;437:169–83. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 6.Santibanez JF, Quintanilla M, Bernabeu C. TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clinical science. 2011;121:233–51. doi: 10.1042/CS20110086. [DOI] [PubMed] [Google Scholar]
- 7.Choi ME, Ballermann BJ. Inhibition of capillary morphogenesis and associated apoptosis by dominant negative mutant transforming growth factor-beta receptors. J Biol Chem. 1995;270:21144–50. doi: 10.1074/jbc.270.36.21144. [DOI] [PubMed] [Google Scholar]
- 8.Madri JA, Pratt BM, Tucker AM. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J Cell Biol. 1988;106:1375–84. doi: 10.1083/jcb.106.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83:4167–71. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang EY, Moses HL. Transforming growth factor beta 1-induced changes in cell migration, proliferation, and angiogenesis in the chicken chorioallantoic membrane. J Cell Biol. 1990;111:731–41. doi: 10.1083/jcb.111.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlova VV, Liu Z, Goumans MJ, Ten Dijke P. Controlling angiogenesis by two unique TGF-beta type I receptor signaling pathways. Histology and histopathology. 2011;26:1219–30. doi: 10.14670/HH-26.1219. [DOI] [PubMed] [Google Scholar]
- 12.Pollman MJ, Naumovski L, Gibbons GH. Vascular cell apoptosis: cell type-specific modulation by transforming growth factor-beta1 in endothelial cells versus smooth muscle cells. Circulation. 1999;99:2019–26. doi: 10.1161/01.cir.99.15.2019. [DOI] [PubMed] [Google Scholar]
- 13.Minami T, Rosenberg RD, Aird WC. Transforming growth factor-beta 1-mediated inhibition of the flk-1/KDR gene is mediated by a 5′-untranslated region palindromic GATA site. J Biol Chem. 2001;276:5395–402. doi: 10.1074/jbc.M008798200. [DOI] [PubMed] [Google Scholar]
- 14.Mandriota SJ, Menoud PA, Pepper MS. Transforming growth factor beta 1 down-regulates vascular endothelial growth factor receptor 2/flk-1 expression in vascular endothelial cells. J Biol Chem. 1996;271:11500–5. doi: 10.1074/jbc.271.19.11500. [DOI] [PubMed] [Google Scholar]
- 15.Pollman MJ, Naumovski L, Gibbons GH. Endothelial cell apoptosis in capillary network remodeling. J Cell Physiol. 1999;178:359–70. doi: 10.1002/(SICI)1097-4652(199903)178:3<359::AID-JCP10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 16.Tsukada T, Eguchi K, Migita K, Kawabe Y, Kawakami A, Matsuoka N, et al. Transforming growth factor beta 1 induces apoptotic cell death in cultured human umbilical vein endothelial cells with down-regulated expression of bcl-2. Biochem Biophys Res Commun. 1995;210:1076–82. doi: 10.1006/bbrc.1995.1766. [DOI] [PubMed] [Google Scholar]
- 17.Hyman KM, Seghezzi G, Pintucci G, Stellari G, Kim JH, Grossi EA, et al. Transforming growth factor-beta1 induces apoptosis in vascular endothelial cells by activation of mitogen-activated protein kinase. Surgery. 2002;132:173–79. doi: 10.1067/msy.2002.125304. [DOI] [PubMed] [Google Scholar]
- 18.Bostrom K, Zebboudj AF, Yao Y, Lin TS, Torres A. Matrix GLA protein stimulates VEGF expression through increased transforming growth factor-beta1 activity in endothelial cells. J Biol Chem. 2004;279:52904–13. doi: 10.1074/jbc.M406868200. [DOI] [PubMed] [Google Scholar]
- 19.Ferrari G, Pintucci G, Seghezzi G, Hyman K, Galloway AC, Mignatti P. VEGF, a prosurvival factor, acts in concert with TGF-beta1 to induce endothelial cell apoptosis. Proc Natl Acad Sci U S A. 2006;103:17260–5. doi: 10.1073/pnas.0605556103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J Cell Physiol. 2009;219:449–58. doi: 10.1002/jcp.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007;1773:1358–75. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. The Biochemical journal. 2010;429:403–17. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 23.Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, et al. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6:109–16. [PubMed] [Google Scholar]
- 24.Ohta T, Eguchi R, Suzuki A, Miyakaze S, Ayuzawa R, Kaji K. Hypoxia-induced apoptosis and tube breakdown are regulated by p38 MAPK but not by caspase cascade in an in vitro capillary model composed of human endothelial cells. J Cell Physiol. 2007;211:673–81. doi: 10.1002/jcp.20975. [DOI] [PubMed] [Google Scholar]
- 25.Chai W, Liu Z. p38 mitogen-activated protein kinase mediates palmitate-induced apoptosis but not inhibitor of nuclear factor-kappaB degradation in human coronary artery endothelial cells. Endocrinology. 2007;148:1622–8. doi: 10.1210/en.2006-1068. [DOI] [PubMed] [Google Scholar]
- 26.Kumar P, Miller AI, Polverini PJ. p38 MAPK mediates gamma-irradiation-induced endothelial cell apoptosis, and vascular endothelial growth factor protects endothelial cells through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J Biol Chem. 2004;279:43352–60. doi: 10.1074/jbc.M405777200. [DOI] [PubMed] [Google Scholar]
- 27.Gratton JP, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC. Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. J Biol Chem. 2001;276:30359–65. doi: 10.1074/jbc.M009698200. [DOI] [PubMed] [Google Scholar]
- 28.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141:1659–73. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 30.Koh W, Stratman AN, Sacharidou A, Davis GE. In vitro three dimensional collagen matrix models of endothelial lumen formation during vasculogenesis and angiogenesis. Methods Enzymol. 2008;443:83–101. doi: 10.1016/S0076-6879(08)02005-3. [DOI] [PubMed] [Google Scholar]
- 31.Gerber H-P, Dixit V, Ferrara N. Vascular Endothelial Growth Factor Induces Expression of the Antiapoptotic Proteins Bcl-2 and A1 in Vascular Endothelial Cells. J Biol Chem. 1998;273:13313–16. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 32.Yue TL, Ni J, Romanic AM, Gu JL, Keller P, Wang C, et al. TL1, a novel tumor necrosis factor-like cytokine, induces apoptosis in endothelial cells. Involvement of activation of stress protein kinases (stress-activated protein kinase and p38 mitogen-activated protein kinase) and caspase-3-like protease. J Biol Chem. 1999;274:1479–86. doi: 10.1074/jbc.274.3.1479. [DOI] [PubMed] [Google Scholar]
- 33.Xu ZR, Hu L, Cheng LF, Qian Y, Yang YM. Dihydrotestosterone protects human vascular endothelial cells from H(2)O(2)-induced apoptosis through inhibition of caspase-3, caspase-9 and p38 MAPK. European journal of pharmacology. 2010;643:254–9. doi: 10.1016/j.ejphar.2010.06.039. [DOI] [PubMed] [Google Scholar]
- 34.Jiang H, Liang C, Liu X, Jiang Q, He Z, Wu J, et al. Palmitic acid promotes endothelial progenitor cells apoptosis via p38 and JNK mitogen-activated protein kinase pathways. Atherosclerosis. 2010;210:71–7. doi: 10.1016/j.atherosclerosis.2009.10.032. [DOI] [PubMed] [Google Scholar]
- 35.Yilmaz A, Kliche S, Mayr-Beyrle U, Fellbrich G, Waltenberger J. p38 MAPK inhibition is critically involved in VEGFR-2-mediated endothelial cell survival. Biochem Biophys Res Commun. 2003;306:730–6. doi: 10.1016/s0006-291x(03)01064-7. [DOI] [PubMed] [Google Scholar]
- 36.Ambrosino C, Nebreda AR. Cell cycle regulation by p38 MAP kinases. Biol Cell. 2001;93:47–51. doi: 10.1016/s0248-4900(01)01124-8. [DOI] [PubMed] [Google Scholar]
- 37.Delston RB, Matatall KA, Sun Y, Onken MD, Harbour JW. p38 phosphorylates Rb on Ser567 by a novel, cell cycle-independent mechanism that triggers Rb-Hdm2 interaction and apoptosis. Oncogene. 2011;30:588–99. doi: 10.1038/onc.2010.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smeeton J, Zhang X, Bulus N, Mernaugh G, Lange A, Karner CM, et al. Integrin-linked kinase regulates p38 MAPK-dependent cell cycle arrest in ureteric bud development. Development. 2010;137:3233–43. doi: 10.1242/dev.052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Mcgowan CH, Zhao M, He L, Downey JS, Fearns C, et al. Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol Cell Biol. 2000;20:4543–52. doi: 10.1128/mcb.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.New L, Han J. The p38 MAP kinase pathway and its biological function. Trends Cardiovasc Med. 1998;8:220–8. doi: 10.1016/s1050-1738(98)00012-7. [DOI] [PubMed] [Google Scholar]
- 41.Sicard P, Clark JE, Jacquet S, Mohammadi S, Arthur JS, O’keefe SJ, et al. The activation of p38 alpha, and not p38 beta, mitogen-activated protein kinase is required for ischemic preconditioning. Journal of molecular and cellular cardiology. 2010;48:1324–8. doi: 10.1016/j.yjmcc.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawdey M, Podor TJ, Loskutoff DJ. Regulation of type 1 plasminogen activator inhibitor gene expression in cultured bovine aortic endothelial cells. Induction by transforming growth factor-beta, lipopolysaccharide, and tumor necrosis factor-alpha. J Biol Chem. 1989;264:10396–401. [PubMed] [Google Scholar]
- 43.Madri JA, Bell L, Merwin JR. Modulation of vascular cell behavior by transforming growth factors beta. Mol Reprod Dev. 1992;32:121–6. doi: 10.1002/mrd.1080320207. [DOI] [PubMed] [Google Scholar]
- 44.Iruela-Arispe ML, Sage EH. Endothelial cells exhibiting angiogenesis in vitro proliferate in response to TGF-beta 1. J Cell Biochem. 1993;52:414–30. doi: 10.1002/jcb.240520406. [DOI] [PubMed] [Google Scholar]
- 45.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, Ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo J. 2002;21:1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–28. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 47.Ota T, Fujii M, Sugizaki T, Ishii M, Miyazawa K, Aburatani H, et al. Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-beta in human umbilical vein endothelial cells. J Cell Physiol. 2002;193:299–318. doi: 10.1002/jcp.10170. [DOI] [PubMed] [Google Scholar]
- 48.Berge M, Bonnin P, Sulpice E, Vilar J, Allanic D, Silvestre JS, et al. Small interfering RNAs induce target-independent inhibition of tumor growth and vasculature remodeling in a mouse model of hepatocellular carcinoma. The American journal of pathology. 2010;177:3192–201. doi: 10.2353/ajpath.2010.100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–7. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beardmore VA, Hinton HJ, Eftychi C, Apostolaki M, Armaka M, Darragh J, et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454–64. doi: 10.1128/MCB.25.23.10454-10464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]