

Abstract
Structure–activity relationship studies on 4-(4-(4-chlorophenyl)-1,4-diazepan-1-yl)-1-(4-fluorophenyl) butan-1-one (SYA 013), a homopiperazine analog of haloperidol has resulted in an understanding of the effect of structural modifications on binding affinity at dopamine and serotonin receptor subtypes. Further exploration, using bioisosteric replacement strategies has led to the identification of several new agents including compounds 7, 8, 11 and 12 which satisfy the initial criteria for further exploration as new antipsychotic agents. In addition, compound 18, a D3 selective tropanol, has been identified as having the potential for further optimization into a useful drug which may combat neuropsychiatric diseases.
Keywords: Antipsychotics, Haloperidol analogs, Structure–activity relationship studies, Atypical antipsychotics, Homopiperazine, Benzothiazole
1. Introduction
There is considerable interest in the development of new antipsychotic drugs partially because of the side effects associated with both typical and atypical antipsychotic drugs currently on the market.1 Atypical antipsychotics were developed to replace typical antipsychotics partially because of the debilitating extrapyramidal side effects. One of the problems in the development of new drugs is the fact that new structural classes of drugs often lead to new types of off-target interactions or side effects when the general population is exposed to the drug. Thus, we hypothesized that there may be advantages in using known drug scaffolds as leads in drug design while attempts are made to eliminate off-target interactions. In this regard, we have focused on the typical antipsychotic drug, haloperidol, taking advantage of the fact that haloperidol binds to several receptors of interest in antipsychotic drug development (D2Ki = 0.89 nM, 5HT1A = 3600 nM; 5HT2A = 120 nM; 5HT2C = 4700 nM) and thus modifications can be made either to enhance or diminish binding at given receptors.2–6 In pursuit of this strategy, we have identified several agents2–6 including a homopiperazine analog of haloperidol, SYA 013 (2),7 with a pharmacological profile similar to those of current atypical antipsychotics but potentially without the EPS associated with haloperidol or the weight gain associated with several atypical antipsychotics.8–10
SYA 013 or 4-(4-(4-chlorophenyl)-1,4-diazepan-1-yl)-1-(4-fluorophenyl)butan-1-one is a haloperidol analog with the piperidinol moiety replaced with a homopiperazine. It binds to the DAD2 receptor (Ki = 43.3 nM), DAD3 (Ki = 158.8 nM), DAD4 (Ki = 6.6 nM), 5HT1A (Ki = 117.4 nM) and 5HT2A (Ki = 23.3 nM) receptor. On the other hand, it has only moderate to weak binding at the 5HT2C receptor (Ki = 1425 nM) and Histamine H1 receptor (Ki = 189 nM). This combination of binding affinities inspired in vivo studies that identified SYA 013 as having atypical antipsychotic properties without the associated catalepsy even at five times the ED50 dose.7 In addition, the low binding affinity to 5HT2C and H1 receptors compared with clozapine for example, predicts that SYA 013 may not induce weight gain. The current study probes the SYA 013 scaffold using bioisosteric replacements to design, synthesize, and conduct structure–activity relationships that may serve as a foundation for new lead compounds active at CNS receptors associated with neuropsychiatric drug development. In particular, it is desirable to obtain compounds that meet the following criteria7: compounds should bind with moderate affinity at the D2 receptor (30 nM < Ki < 150 nM), as avid affinity to this receptor may lead to induction of acute EPS; and a weak or no binding affinity to 5HT2C and histamine H1 receptors, as both receptors are reported to be associated with weight gain and subsequently type II diabetes, often observed with some of the atypical antipsychotics.
2. Chemistry
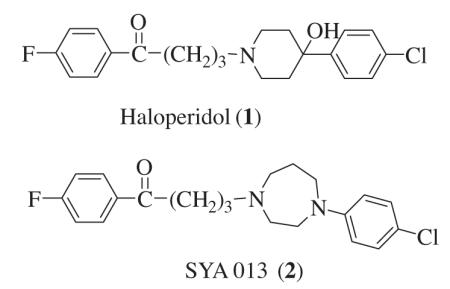
Compounds2, 3, 4, 5, 6 (Chart 1), analogs of haloperidol 1, were reported in previous papers from this lab.6,7 including the synthetic details as well as important intermediates. Alkylating agent 1-(4-chlorobutyl)-4-fluorobenzene 21 was prepared by reductive deoxygenation of 4-chloro-1-(4-fluorophenyl)butan-1-one 20 using Clemmensen’s reaction (Scheme 1). Refluxing a mixture of 20 and amalgamated zinc in aqueous acidic toluene for 5 h, afforded 21 after distillation.
Chart 1.

Structures of compounds 2–6.
Scheme 1.
Reagents and conditions: (a) Zn, HgCl2, Conc HCl, toluene, reflux, 5 h.
Alkylating agents 26 and 27 were prepared from 4-fluorophenol 22 and 4-fluorobenzenethiol 23 (Scheme 2). By refluxing the mixture of 4-fluorophenol 22, 3-chloropropanol, K2CO3, and KI in iPrOH, 3-(4-fluorophenoxy)propan-1-ol 24, was obtained. Methanesulfonation was carried out at room temperature with Et3N as a base afforded 3-(4-fluorophenoxy)propyl methanesulfonate 26. The same procedure was used to prepare 3-((4-fluorophenyl)thio)propyl methanesulfonate 27.
Scheme 2.

Reagents and conditions: (a) KI, K2 CO3, iPrOH, 3-chloropropanol, 80 °C,12 h; (b) MsCl, Et3N, CH2Cl2, 5 °C–rt.
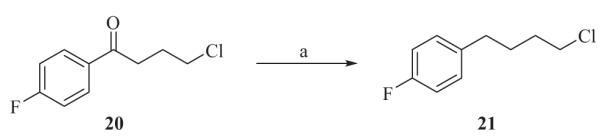
Benzothiazole alkylating agents 32, 33 and 34 were prepared from 2-aminothiophenol with chloroacyl chlorides in toluene at room temperature, as described in Scheme 3.
Scheme 3.
Reagents and conditions: toluene, rt, 16 h.
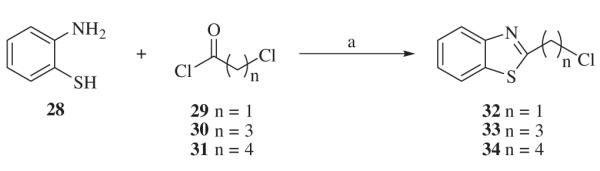
Alkylating agents 1-(benzo[d]thiazol-2-yl)-4-iodobutan-1-one 38 and 1-(benzo[d]thiazol-2-yl)-5-iodopentan-1-one 39 were synthesized from benzothiazole 35, as described in Scheme 4. Deprotonation of benzothiazole 35 with BuLi at 78 −°C followed by treatment with lactone 36 or 37, resulted in the corresponding alcohols. Iodides 38 and 39 were obtained by conversion of the corresponding alcohols into iodides with I2, imidazole, and Ph3P in CH2Cl2.
Scheme 4.
Reagents and conditions: (a) BuLi, –78 °C, THF; (b) I2, imidazole, Ph3P, CH2Cl2,0 °C–rt.
Alkylating agent 42 (Scheme 5) was prepared from 4-chloro-1-(4-fluorophenyl)butan-1-one 20 in a three step conversion. 4-Chloro-1-(4-fluorophenyl)-2-methylenebutan-1-one 40 was obtained by the treatment of 20 with Eschenmoser’s salt in acetic anhydride at 105 °C for 18 h. The yield was 20–26%. Nazarov related cyclization11 was carried out in concentrated H2SO4 at 60 °C to yield indanone 41 in quantitative yield. Protection of the ketone with ethylene glycol afforded compound 42.
Scheme 5.
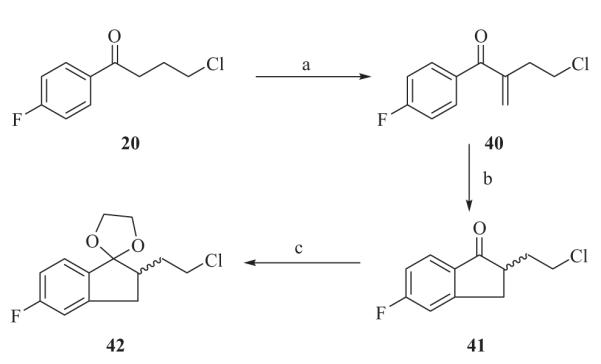
Reagents and conditions: (a) hexamethylenetetramine, Ac2O, 105 °C, 18 h; (b) H2SO4, 60 °C; (c) ethylene glycol, TsOH, toluene, reflux.
1-(4-Chlorophenyl)-1, 4-diazepane 43 was synthesized7 according to our previous published method involving the coupling of homopiperazine with 4-iodochlorobenzene in the presence of CuI. 3-(4-Chlorophenyl)-8-azabicyclo[3.2.1]octan-3-ol 44 was prepared as previously published by us.6
By coupling the alkylating agents 21, 26, 27, 32, 33, 34, 38, 39 and 42 with 1-(4-chlorophenyl)-1,4-diazepane 43, homopiperazine derived analogs 9–17 (Chart 2) were synthesized as shown in Scheme 6. By coupling the alkylating agents 21, 26, 34, and 39 with 3-(4-chlorophenyl)-8-azabicyclo[3.2.1]octan-3-ol 44, compounds 7, 8, 18 and 19 were obtained (Chart 3) as shown in Scheme 7.
Chart 2.

Structures of homopiperazine analogs, 9–17.
Scheme 6.

Reagents and conditions: Method A: (1) KI, K2CO3, DME, reflux, 16 h; (2) TsOH, MeOH, rt. Method B: KI, K2CO3, DME, reflux, 16 h. Method C: K2CO3, DME, reflux, 16 h.
Chart 3.

Structures of tropanol analogs 7, 8, 18 and 19.
Scheme 7.

Reagents and conditions: Method B: KI, K2CO3, DME, reflux, 16 h. Method C: K2CO3, DME, reflux, 16 h.
3. Results and discussion
Based on the binding profile of SYA 013 (Table 1), it is clear that replacement of the piperidinol in haloperidol with the homopiperazine moiety has a significant effect on binding to several receptors of interest. Replacement of the seven-membered homopiperazine ring with the six-membered piperazine ring (3) or its bridged analog, (1R,5S)-3,6-diazabicyclo[3.1.1]heptane, to form compounds 3 and 4, respectively, did not result in improved binding at any of the receptors of interest. However, binding to D2-like receptors was maintained. Bridging the homopiperazine ring with an ethylene group to form compound 5 again failed to improve binding when compared to lead compound 2. We have previously shown that the tropane analog of haloperidol (6) resulted in very potent binding at dopamine receptors.6 Indeed, compound 6 was found to be more potent than haloperidol at the D2 receptor and displayed higher potency than SYA 013 at the 5HT1A (Ki = 27.7 nM) and 5HT2A (Ki = 30.9 nM) receptors as well. Not surprisingly then, it also produced severe catalepsy in animal models. This observation also suggested that binding to 5HT1A or 5HT2A may not necessarily prevent catalepsy in animal models. Based on this profile, we opined that moderating the D2 binding affinity of 6 might lead to reduction of the catalepsy. Thus, compounds 7 and 8 were synthesized to explore the hypothesis that D2 binding can be moderated using butyrophenone bioisosteres identified in our laboratory.12 Both compounds indeed have diminished D2 binding (Ki = 22.0 and 33 nM, respectively), similar to that of SYA 013 as desired. However, the binding affinity at the 5HT1A and 5HT2A receptors significantly reduced as well.
Table 1.
Binding affinity constants of compounds at selected DA, 5-HT and H-1 receptors
| Compd # | >Binding data; Ki ± SEM (nM) |
||||||
|---|---|---|---|---|---|---|---|
| 5-HT1A | 5-HT2A | 5-HT2C | D2 | D3 | D4 | H1 | |
| Cloza | 140 | 8.9 | 17.0 | 130 | 240 | 54 | 1.8 |
| Hala | 3600 | 120 | 4700 | 0.89 | 2.5 | 3.3 | 440 |
| 2a | 117.4 ± 32.6 | 23.6 ± 2.7 | 1425 ± 207 | 43.3 ± 13.3 | 158.8 ± 35.1 | 6.6 ± 0.6 | 188.6 ± 16.0 |
| 3a | 90.9 ± 21.0 | 109.6 ± 16.0 | 3552± 943 | 253.5 ± 38.9 | ND | 17.5 ± 2.0 | 157.6 ±36.0 |
| 4a | ND | ND | ND | 170 | 220 | 520 | ND |
| 5a | 2332 ± 470 | 194.8 ± 53.0 | 3513 ± 912 | 178.4 ± 29.2 | 548.1± 246.0 | 41.8 ± 9.0 | 1014 ± 206 |
| 6a | 27.7 ± 8.0 | 30.9 ± 6.0 | 872.1 ± 178.0 | 1.6 ± 0.1 | 5.1 ± 3.0 | 5.3 ± 1.0 | 8780 ± 1625 |
| 7 | 123.0 ± 12.9 | 236.0 ± 21.6 | MP | 22.0 ± 3.1 | 4.1± 0.6 | 49.0 ± 9.1 | 4,149 ± 924 |
| 8 | 102.0 ± 11.8 | 317.0 ± 29.2 | 2434 ± 583 | 33.0 ± 4.6 | 9.7 ± 1.1 | 9.4 ± 1.5 | 1719 ± 368 |
| 9 | 415.0 ± 48.1 | 411.5 ± 52.5 | 3775 ± 704 | 1021 ± 142 | 590.0 ± 54.3 | 37.0 ± 4.3 | ND |
| 10 | 168 ± 19 | 163.0 ± 20.0 | 2529 ± 223 | 751.1 ± 92.3 | 1,020 ± 25 | 32.1 ± 2.5 | 800.2 ± 69.9 |
| 11 | 73.0 ± 14.7 | 566.6 ± 91.9 | >10,000 | 172.0 ± 28.1 | 208.8 ± 43.4 | 110.0 ± 12.6 | 509.0 ± 71.1 |
| 12 | 91.0 ± 10.5 | 510.3 ±76.9 | 4097 ± 754 | 139.0 ± 25.3 | 3043 ± 176 | 123.0 ± 19.9 | 1247 ± 204 |
| 13 | 263.0 ± 36.5 | 159.0 ± 11.0 | 1420 ± 295 | 258.7 ± 48.4 | 152.0 ± 21.0 | 28.6 ± 3.4 | 61.0 ± 8.5 |
| 14 | 216.0 ± 30.0 | 696.0 ± 51.3 | ND | MP | 863.0 ± 132.9 | 431.0 ± 40.7 | 665.0 ± 47.0 |
| 15 | MP | MP | MP | MP | MP | MP | MP |
| 16 | 495.0 ± 91.0 | 1003 ± 96 | 5920±769 | 1983 ± 163 | 1051 ± 174 | 1215 ± 67 | 129.0 ± 12.0 |
| 17 | MP | 1038b | MP | MP | 777.0 ± 125.7 | 126.0 ± 20.4 | ND |
| 18 | MP | MP | MP | 180.0 ± 29.5 | 16.0 ± 2.6 | 508.0 ± 1.2 | ND |
| 19 | 274.0 ± 52.7 | >10,000 | 4623b | 44.0 ± 3.1 | 13.0 ± 1.8 | 432.0 ± 91.1 | 187.0 ± 34.5 |
Abbreviations: MP, Missed primary assay threshold of 50% inhibition; ND, Not determined; Cloz, Clozapine; Hal, Haloperidol.
Results were previously reported in Ref. 7.
Standard Errors are within 20% of the mean.
Based on the above observations, it was of interest to explore the butyrophenone moiety in SYA 013 for enhancement in binding affinity at the desired receptors. A published report has suggested that a restricted butyrophenone moiety such as in the indenone analog 9 might increase potency at the 5HT2A receptor.13 Synthesis of compound 9 and its evaluation showed a moderate decrease in 5HT1A binding but over 20-fold decrease in 5HT2A receptor binding (Ki = 477.5 nM) compared to SYA 013 suggesting that the aryl cycloalkylamine moiety also plays a significant role in binding to the 5HT receptors. In addition, there were similar decreases in binding at the D2-like receptors. Replacement of the butyrophenone carbonyl group with a sulfur atom (10,D2Ki = 751.1 nM) or with an oxygen atom (11, D2Ki = 172.0 nM) or a methylene group (12, D2Ki = 139.0 nM) have each resulted in lower binding at the D2 receptor. Despite the reduction in binding affinity, compounds 11 and 12 have similar binding affinity to that of clozapine (D2Ki = 130 nM). While compound 10 binds with a lower affinity than SYA 013 at the 5HT1A, both 11 and 12 bind with higher affinity at the 5HT1A receptor. All three compounds have only weak to moderate binding at the 5HT2A receptor. In a previous evaluation, we discovered that the benzothiazole moiety (unpublished observation) could substitute for the 4-fluorophenyl moiety in butyrophenones. Thus, we synthesized compounds 13–15 to explore the effect of the benzothiazole ring on the homopiperazine analog receptor binding. Compound 13 turned out to have a lower binding affinity at all the receptors of interest. Similarly, reducing the chain length between the benzothiazole and the homopiperazine by one, or three methylene groups (compounds 14 and 15, respectively), led to further reductions in binding at all the receptors evaluated. In fact, compound 15 has no binding affinity at any of the receptors of interest. Insertion of a carbonyl group between the benzothiazole ring and the first methylene group in compound 13 to form 16 and in compound 14 to form 17 also did not result in an increase in binding at any of the receptors of interest. A cursory look at the contribution of each pharmacophoric group suggested that the tropanol moiety contributes significantly to binding at the dopamine receptors. So we hypothesized that replacement of the homopiperazine ring in 16 with the tropanol moiety to form compound 18 should improve binding at the dopamine receptors. Evaluation revealed that compound 18 binds to the D2 receptor with an affinity of 180 nM, 16 nM at the D3 and 508 nM at the D4 receptor. Among the compounds synthesized and evaluated, compound 18 with 180/ 16 or 11.3 for D2/D3 receptor selectivity ratio and 508/16 or 31.8 for D4/D3 receptor selectivity ratio is the only compound with selectivity towards the D3 receptor. It is important to note that there is preclinical evidence that selective antagonism at DAD3 receptors reduces the reinforcing efficacy of drugs of abuse, reverses cognitive deficits and shows efficacy in animal models of schizophrenia and Parkinson’s disease and thus, selective DAD3 receptor antagonists may hold promise in the treatment of several neuropsychiatric diseases.14–16 Excision of the carbonyl from compound 18 to form compound 19 produced an increase in binding at the D2 receptor (D2Ki = 44 nM) and no significant changes in binding at the D3 or D4 receptors. There was also diminished binding at the serotonin receptors of interest for both compounds 18 and 19. Thus, it would seem that the benzothiazole-linked aryl tropanols may constitute a new scaffold for the development of selective D3 receptor agents.
Finally, all compounds were evaluated at the histamine H-1 receptors in order to explore their potential to induce weight gain.10 Apart from compounds 13, 16 and 19, (Table 1) all compounds bind with lower affinity than SYA 013 and hence can be considered less likely to induce treatment-emergent weight gain.
In conclusion, the SAR studies identified four of the homopiperazine and tropanol analogs, compounds 7, 8, 11 and 12, satisfy our stated initial criteria for binding to D2 and 5HT1A receptors and compare favorably with clozapine binding at these receptors. In addition, the four compounds have very low binding affinity at5HT2C and histamine H-1 receptors indicating a low propensity for inducing weight gain. Compounds 18 and 19 show selectivity for the D3 receptor and thus may constitute a new scaffold for further exploration.
4. Experimental
4.1. General
Melting points were determined on a Gallenkamp (UK) apparatus and are uncorrected. NMR spectra were obtained on a Varian 300 MHz Mercury Spectrometer. Elemental analyses were carried out by Atlantic Microlab, Inc., Norcross, GA, and are within 0.4% of theory unless otherwise noted. Flash chromatography was performed with Davisil grade 634 silica gel. N,N-dimethylformamide was distilled from CaSO4 and stored over 4 Å molecular sieves. 4-Chloro-4′-fluorobutyrophenone was obtained from Sigma–Aldrich, but was purified by distillation under reduced pressure to a colorless liquid prior to use. Other starting materials were used without further purification.
4.2. Synthesis
4.2.1. Synthesis of methanesulfonates 26 and 27
A mixture of 4-fluorophenol (1.12 g, 10 mmol), 3-chloropropanol (1.4 g, 15 mmol), KI (50 mg), K2 CO3 (2.76 g, 20 mmol) in iPrOH was refluxed under N2 for 12 h. The resulting mixture was diluted with EtOAc (200 mL), and washed with water (50 mL) then brine (50 mL). The organic layer was dried with Na2SO4, filtered and the filtrate was concentrated in vacuo to give 3-(4-fluorophenoxy)propan-1-ol 24 as the product. The product was used for the next step without further purification. 1H NMR (CDCl3): δ 6.96 (2H, t, J = 8.4 Hz), 6.84 (2H, dd, J = 4.5, 9.0 Hz), 4.09 (2H, t, J = 6.0 Hz), 3.85 (2H, m), 2.03 (2H, m).
To a solution of 3-(4-fluorophenoxy)propan-1-ol 24 (1.3 g, 7.6 mmol) and Et3N (3 mL) in CH2Cl2 (10 mL) was added at room temperature MsCl (0.8 mL, 10.3 mmol). The mixture was stirred at room temperature for 12 h followed by direct purification through column chromatography on silica gel to provide 3-(4-fluorophenoxy)propyl methanesulfonate 26, with a yield of 95%. 1H NMR (CDCl3): δ 6.97 (2H, t, J = 8.1 Hz), 6.83 (2H, dd, J = 4.5, 9.0 Hz), 4.44 (2H, t, J = 6.0 Hz), 4.05 (2H, t, J = 6.0 Hz), 2.21 (2H, m).
4.2.2. 3-(4-Fluorophenylthio)propan-1-ol (25)
Yield 72%. 1H NMR (CDCl3): δ 7.35 (2H, dd, J = 5.4, 8.4 Hz), 6.99 (2H, t, J = 8.4 Hz), 3.76 (2H, t, J = 6.0 Hz), 2.98 (2H, t, J = 7.2 Hz), 1.85 (2H, m).
4.2.3. 3-((4-Fluorophenyl)thio)propyl methanesulfonate (27)
Yield 94%. 1H NMR (CDCl3): δ 7.37 (2H, dd, J = 9.0, 4.8 Hz), 7.00 (2H, t, J = 9.0 Hz), 4.30 (2H, t, J = 8.0 Hz), 3.00 (3H, s), 2.78 (2H, t, J = 7.2 Hz), 2.02 (2H, m).
4.3. General procedure for the synthesis of 32, 33 and 34
4.3.1. 2-(4-Chlorobutyl)benzo[d]thiazole (34)
A solution of 2-aminothiophenol 28 (5 g, 39.93 mmol) in toluene (50 ml) and 5-chloropentanoyl chloride (18) (6.81 g, 44 mmol) was added dropwise with cotinuous stirring over a 15 min period resulting in the formation of an off-white precipitate. The mixture was allowed to continue stirring at rt overnight and then partitioned between H2O (100 mL) and EtOAc (200 mL) and the organic layer separated. The organic layer was washed with a saturated solution of NaCl (100 mL), H2O (100 mL) dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography (Combiflash) using EtOAc/Hexanes (1:9) to produce 2-(4-chlorobutyl)benzo[d]thiazole 34 as an oily liquid (5 g, 55.5%). 1H NMR (CDCl3): δ 7.95 (1H, d, J = 6.2 Hz), 7.8 (1H, d, J = 8.7 Hz), 7.46–7.40 (1H, m), 7.35–7.30 (1H, m), 3.52 (2H, t, J = 6.6 Hz), 3.10 (2H, t, J = 7.8 Hz), 1.95–1.77 (2H, m), 1.62–1.54 (2H, m).
4.3.2. 2-(3-Chloropropyl)benzo[d]thiazole, 33
Using 4-chlorobutanoyl chloride, compound 33 was obtained in 72% yield. 1H NMR (CD3OD): δ 8.14 (1H, d, J = 4.1 Hz), 8.02 (1H, d, J = 4.1 Hz), 7.72–7.59 (2H, m), 3.64–3.57 (2H, m), 3.38–3.28 (2H, m), 1.95–1.86 (2H, m).
4.3.3. 2-(Chloromethyl)benzo[d]thiazole (32)
Using 2-chloroacetyl chloride, compound 32 was obtained in 76% yield. 1H NMR (CCCl3): δ 7.96 (1H, d, J = 7.5 Hz), 7.70 (1H, dd, J = 7.8, 09 Hz), 7.48–7.43 (1H, m), 7.39–7.33 (1H, m), 3.32 (2H, t, J = 6.0 Hz).
4.4. General procedure for the synthesis of 38 and 39
4.4.1. 1-(Benzo[d]thiazol-2-yl)-5-iodopentan-1-one, 39
To a stirred solution of benzothiazole 35 (10 g, 74.0 mmol) in dry THF (37 mL) was added drop wise n-BuLi (37 mL 1 M solution in THF) at −78 °C under N2. A clear orange solution was obtained. To this resulting solution was added a solution of lactone 37 (8.14 g, 94.7 mmol) in dry THF (37 mL) at −78 °C and the mixture continuously stirred at −78 °C for 1 h. After removal of the cold bath, the reaction mixture was continuously stirred for 30 min and then quenched with large excess of (0.1 M) HCl (300 mL). The aqueous mixture was extracted with ethyl acetate (3 × 150 mL). The combined organic extract was washed with H2O(2 × 100 mL), saturated NaCl, and dried over Na2SO4. The solution was concentrated in vacuo and the crude product was dissolved in EtOAc (50 mL), and hexane (200 mL) was added. An orange precipitate resulted, was collected, washed with 10% EtOAc in Hexane (200 mL), and dried in vacuo to give the pure product 1-(benzo[d]thiazol-2-yl)-5-hydroxypentan-1-one (6.5 g). The product was used directly without further purification in subsequent reactions. 1H NMR (CDCl3): δ 8.20–8.16 (1H, dd, J = 1.8, 6.9 Hz), 7.99–7.96 (1H, dd, J = 1.5, 7.2 Hz), 7.62–7.52 (2H, m), 3.74–3.72 (2H, t, J = 6.3 Hz), 3.51–3.30 (2H, t, J = 7.2 Hz), 2.00–1.88 (2H, m), 1.76–1.67 (2H, m).
To a solution of triphenylphosphine (TPP) (3.12 g, 11.9 mmol) and imidazole (810 mg) in dichloromethane (30 mL) was added iodine (3.02 g, 11.9 mmol) at 0–5 °C. The reaction mixture was stirred at 0–5 °C for 30 min. A solution of 1-(benzo[d]thiazol-2-yl)-5-hydroxypentan-1-one (2.0 g, 8.5 mmol) in dichloromethane (15 mL) was added drop wise in 5 min. The reaction mixture was stirred at 0–5 °C for another 30 min, and then the ice bath was removed and continuously stirred at room temperature for 12 h. When TLC showed that the reaction was completed, the reaction mixture was treated with water (100 mL). The two layers were separated, and the aqueous layer was extracted with dichloromethane (2 × 50 mL). The combined organic extracts were washed with water (2 × 100 mL), 10% sodium thiosulfate (50 mL), water (100 mL) and saturated NaCl aqueous solution (75 mL), then dried over Na2SO4 and concentrated in vacuo. The crude product was further purified by flash chromatography (Combiflash) using EtOAc/Hexane (1:9) to obtain the pure product 1-(benzo[d]thiazol-2-yl)-5-iodopentan-1-one (39) 1.8 g, in a yield of 61%. 1H NMR (CDCl3): δ 8.20–8.17 (1H, m), 8.00–7.97 (1H, m), 7.61–7.51 (2H, m), 3.34–3.29 (2H, t, J = 6.6 Hz), 3.27–3.23 (2H, t, J = 6.9 Hz), 1.98–1.92 (4H, m).
4.4.2. 1-(Benzo[d]thiazol-2-yl)-4-iodobutan-1-one, 38
Using benzothiazole 35 and lactone 36, compound 38 was obtained (as described for compound 39 above) in 37% yield. 1H NMR (CDCl3): δ 8.21–8.18 (1H, m), 8.00–7.97 (1H, m), 7.62–7.52 (2H, m), 3.47–3.42 (2H, t, J = 6.9 Hz), 3.37–3.32 (2H, t, J = 6.6 Hz), 2.39–2.30 (2H, q, J = 6.9 Hz).
4.4.3. Synthesis of 2′-(2-chloroethyl)-5′-fluoro-2′,3′-dihydrospiro[[1,3]dioxolane-2,1′-indene], 42
A mixture of 20 (10 g, 50 mmol), hexamethylenetetraamine (10.5 g, 75 mmol) in Ac2O (25 mL) was refluxed under N2 for 16 h and allowed to cool to room temperature. The mixture was then diluted with CHCl3 (500 mL), washed with 10% HCl solution (2 × 300 mL), H2O (300 mL) and saturated NaHCO3 (300 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo followed by column chromatography on silica gel affording the pure product as 4-chloro-1-(4-fluorophenyl)-2-methylenebutan-1-one 40, (2.8 g, 26.4% yield). 1H NMR (CDCl3): δ 7.82 (2H, dd, J = 5.7, 9.0 Hz), 7.13 (2H, t, J = 9.0 Hz), 5.99 (1H, s), 5.73 (1H, s), 3.72 (2H, t, J = 6.6 Hz), 2.94 (2H, t, J = 6.0 Hz).
Compound 40 (1.2 g, 5.64 mmol) was dissolved in concentrated H2SO4 (4 mL) and heated at 60 °C for 1 h. After cooling to room temperature, the mixture was diluted with EtOAc (200 mL) and washed with saturated NaHCO3 (2 × 200 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo followed by column chromatography on silica gel affording 2-(2-chloroethyl)-5-fluoro-2,3-dihydro-1H-inden-1-one 41. 1H NMR (CDCl3): δ 7.76 (1H, dd, J = 5.4, 8.4 Hz), 7.08 (2H, m), 3.83 (1H, m), 3.76 (1H, m), 3.42 (1H, dd, J = 7.8, 17.1 Hz), 2.93 (1H, m), 2.83 (1H, dd, J = 4.2, 17.1 Hz), 2.44 (1H, m), 1.91 (1H, m).
A solution of compound 41 (5 g, 23.5 mmol), ethylene glycol (5 mL), and TsOH (100 mg) in toluene (50 mL) was refluxed under N2 for 48 h. Water was removed by azeotropic distillation and the reaction was monitored by 1H NMR. The reaction was quenched by addition of Et3N (1 mL), diluted with EtOAc (250 mL), washed with saturated NaHCO3 (25 mL) followed by water (25 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo to obtain a mixture of 2-(2-chloro-ethyl)-5-fluoro-inden-1-one 41 and its ethylene acetal 42 in a ratio of 1:4. 2-(2-chloroethyl)-5-fluoro-indan-1-one 41 was removed by reducing it to its 2-(2-chloroethyl)-5-fluoro-indan-1-ol with NaBH4 in MeOH followed by column chromatography on silica gel affording 2-(2-chloro-ethyl)-5-fluoro-indan-1-one ethylene acetal 42 (4.5 g, 75% yield). 1H NMR (CDCl3): δ 7.26 (1H, m), 6.92 (2H, m), 4.26 (1H, m), 4.12 (3H, m), 3.71 (1H, m), 3.60 (1H, m), 3.07 (1H, m), 2.72 (1H, m), 2.64 (1H, m), 2.16 (1H, m), 1.94 (1H, m), 1.94 (1H, m).
4.5. Synthesis of compound 9
4.5.1. Method A: 2-(2-(4-(4-chlorophenyl)-1,4-diazepan-1-yl)ethyl)-5-fluoro-2,3-dihydro-1H-inden-1-one, 9
A mixture of 42 (1.2 g, 4.67 mmol), 43 (1.3 g, 5.6 mmol), KI (100 mg, 0.5 mmol), and K2CO3 (1.2 g, 8.75 mmol) in DME (10 mL) was heated to reflux under N2 for 16 h. The mixture was directly purified through column chromatography on silica gel affording 2-{2-[4-(4-chlorophenyl)-[1,4]diazepan-1-yl]-ethyl}-5-fluoro-indan-1-one ethylene acetal. This product was dissolved in wet MeOH, and TsOH added with stirring at rt. After stirring at rt for 12 h, the solution was diluted with EtOAc (450 mL), followed by washing with saturated NaHCO3 (40 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo to dry and followed by column chromatography on silica gel affording 2-(2-(4-(4-chlorophenyl)-1,4-diazepan-1-yl)ethyl)-5-fluoro-2,3-dihydro-1H-inden-1-one. The product was converted to its hydrochloride salt, and recrystallized from MeOH–Et2O affording the HCl salt of 9, in a yield of 25%, mp 176–177 °C. 1H NMR (DMSO-d6): δ 11.16 (1H, br s), 7.71 (1H, dd, J = 5.4, 8.7 Hz), 7.43 (1H, d, J = 8.7 Hz), 7.27 (1H, t, J = 8.7 Hz), 7.19 (2H, d, J = 8.7 Hz), 6.75 (2H, d, J = 8.7 Hz), 3.77 (2H, m), 3.47 (2H, m), 3.34 (3H, m), 3.12 (4H, m), 2.83 (2H, m), 2.37 (1H, m), 2.23 (1H, m), 2.11 (1H, m), 1.86 (1H, m). Calcd for C22H26Cl3FN2O:C 57.47, H 5.70 N 6.09; Found: C 57.94, H 6.04, N 6.02.
4.6. Synthesis of compounds 7, 8, 10–15, 19: General procedure: Method B
4.6.1. 3-(4-Chlorophenyl)-8-(3-(4-fluorophenoxy)propyl)-8-azabicyclo[3.2.1]octan-3-ol (7)
A mixture of 3-(4-fluorophenoxy)propylmethanesulfonate 26 (0.73 g, 3.1 mmol), 3-(4-chlorophenyl)-8-azabicyclo[3.2.1]octan-3-ol 44 (1.0 g, 2.56 mmol), KI (0.2 g), and K2CO3 (1.10 g, 7.69 mmol) in DME (25 ml) was heated to reflux under N2 for 16 h. The mixture was diluted with EtOAc (450 mL), followed by washing with saturated NaHCO3 (100 mL). The organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated in vacuo to dry followed by column chromatography on silica gel affording 3-(4-chloro-phenyl)-8-(3-(4-fluorophenoxy)propyl)-8-azabicyclo[3.2.1]octan-3-ol 7 in an off white semi-solid form. The product was converted to the HCl salt, followed by recrystallization in EtOAc–Et2O to give its HCl salt (722 mg), in a yield of 72.3%, mp 218.4–219.2 °C. 1H NMR (CD3OD): δ 7.40 (2H, m), 7.15 (2H, m), 7.88 (2H, m), 6.77 (2H, m), 3.89 (2H, t, J = 6.3 Hz), 3.21 (3H, m), 2.53 (2H, m), 2.20 (4H, m), 1.88 (4H, m), 1.69 (2H, m). Calcd for C22H26Cl2FNO2:C 61.98, H 6.15, N 3.26; Found: C 61.48, H 6.13, N 3.23.
4.6.2. 3-(4-Chlorophenyl)-8-(4-(4-fluorophenyl)butyl)-8-azabicyclo[3.2.1]octan-3-ol HCl, 8
The product of alkyl chloride 21 and amine 44 was converted into its HCl salt and recrystallized from EtOAc to give the HCl salt of 8 in a yield of 79.5%, mp 223.2–224.9 °C. 1H NMR (CDCl3): δ 7.56 (2H, m), 7.35 (2H, m), 7.23 (2H, m), 7.00 (2H, m), 4.11 (2H, m), 3.32 (2H, m), 3.07 (2H, t, J = 16.5 Hz), 2.67 (4H, m), 2.54 (2H, m), 2.20 (4H, m), 1.79 (4H, m). Calcd for C23H28Cl2FNO:C 65.09, H 6.65, N 3.30; Found: C 64.95, H 6.66, N 3.31.
4.6.3. 1-(4-Chlorophenyl)-4-(3-((4-fluorophenyl)thio)propyl)-1,4-diazepane, 10
The product of the reaction of methanesulfonate 27 and amine 43 was converted to the HCl salt and recrystallized from MeOH–Et2O. Yield 69%, mp 172–173 °C. 1H NMR (DMSO-d6): δ 11.2 (1H, s), 7.63 (1H, s), 7.42 (2H, dd, J = 5.4, 9.0 Hz), 7.17 (4H, m), 6.75 (2H, d, J = 9.0 Hz), 3.75 (2H, m), 3.41 (4H, m), 3.16 (2H, m), 3.08 (2H, m), 2.99 (2H, t, J = 7.2 Hz), 2.38 (2H, m), 2.10 (2H, m), 1.98 (2H, m). Calcd for C20H26Cl3FN2S:C53.16, H 5.80, N 6.20; Found: C 53.39, H 5.98, N 6.22.
4.6.4. 1-(4-Chlorophenyl)-4-(3-(4-fluorophenoxy)propyl)-1,4-di azepane, 11
The product of the methanesulfonate 26 and amine 43 was converted to the HCl salt after crystallization from MeOH–Et2O, in a yield of 53.9%, mp 207–208 °C. 1H NMR (CDCl3): δ 7.23 (4H, m), 6.98 (4H, m), 4.89 (2H, s), 4.432 (2H, s), 3.66 (2H, m), 3.30 (3H, m), 3.19 (2H, t, J = 6.6 Hz), 2.710(2H, t, J = 7.8 Hz), 1.79 (4H, m). Calcd for C20H26Cl3FN2O ·0.25H2O:C 54.56, H 6.07, N 6.36; Found: C 54.58, H 5.902, N 6.00.
4.6.5. 1-(4-Chlorophenyl)-4-(4-(4-fluorophenyl)butyl)-1,4-diaze pane, 12
The product of alkyl chloride 21 and amine 43 was converted to the HCl salt after crystallization from MeOH–Et2O, in a yield of 83.2%, mp 206–207 °C. 1H NMR (CDCl3): δ 7.21 (4H, m), 7.07 (2H, m), 6.75 (2H, m), 4.23 (1H, m), 3.60 (6H, m), 3.00 (6H, m), 2.60 (2H, m), 2.31 (1H, m), 1.95 (2H, m), 1.65 (2H, m). Calcd for C21H27Cl2FN2 ·0.75H2O: C 61.39, H 6.99, N 6.82; Found: C 61.22, H 6.89, N 6.80.
4.6.6. 2-(4-(4-(4-Chlorophenyl)-1,4-diazepan-1-yl)butyl) benzo [d]thiazole, 13
The product of benzothiazole 34 and amine 43 was converted into the tosylate salt, and re-crystallized from MeOH–Et2O, in a yield of 15%, mp 146–147 °C. 1H NMR (DMSO-d6): δ 9.29 (1H, br s), 8.24 (2H, m), 7.64 (2H, m), 7.45 (2H, d, J = 8.1 Hz), 7.21 (2H, d, J = 9.0 Hz), 7.08 (2H, d, J = 8.1 Hz), 6.76 (2H, d, J = 9.0 Hz), 3.77 (1H, m), 3.61 (2H, m), 3.40 (8H, m), 3.23 (3H, m), 2.26 (3H, s), 2.15 (2H, m), 2.07 (2H, m). Calcd for C29H34ClN3O3S2:C 60.87, H 5.99, N 7.34; Found: C 60.82, H 5.78, N 7.35.
4.6.7. 2-(3-(4-(4-Chlorophenyl)-1,4-diazepan-1-yl)propyl)benzo [d]thiazole, 14
The product of benzothiazole 33 and amine 43 was converted to the hydrochloride salt after crystallization from MeOH–Et2O. Yield 48%, mp 97–100 °C. 1H NMR (CD3OD): δ 8.16 (1H, d, J = 4.1 Hz), 8.02 (1H, d, J = 4.1 Hz), 7.72–7.59 (2H, m), 7.21 (2H, d, J = 9.0 Hz), 6.98 (2H, d, J = 4.5 Hz), 3.92 (3H, m), 3.60 (3H, d, J = 5.7 Hz), 2.51–3.41 (7H, m), 2.52–2.34 (5H, m), 1.28–1.25 (1H, m). Calcd for C21H27Cl4N3S·1.5H2O:C 48.29, H 5.21, N 8.04; Found: C 48.26, H 5.64, N 8.09.
4.6.8. 2-((4-(4-Chlorophenyl)-1,4-diazepan-1-yl)methyl)benzo [d]thiazole, 15
The product of the benzothiazole 32 and amine 43 was converted to the HCl salt after recrystallization from MeOH–Et2Oin a yield of 53%, 197–199 °C. 1H NMR (CDCl3): δ 8.08 (2H, t, J = 7.8 Hz), 7.62–7.49 (2H, m), 7.29 (2H, d, J = 4.5 Hz), 7.05 (2H, d, J = 5.1 Hz), 5.02 (2H, s), 4.99 (4H, s), 3.98–3.63 (7H, m). Calcd for C19H21Cl2N3S:C 57.87, H 5.37, N 10.66; Found: C 57.69, H 5.26, N 10.99.
4.6.9. 8-(4-(Benzo[d]thiazol-2-yl)butyl)-3-(4-chlorophenyl)-8-azabicyclo[3.2.1]octan-3-ol, 19
The product of the benzothiazole 34 and amine 44 was converted to the HCl salt after recrystallization from MeOH–Et2Oin a yield of 37%, mp 198–200 °C. 1H NMR (DMSO-d6): δ 10.92 (1H, br s), 8.04 (1H, d, J = 8.1 Hz), 7.92 (1H, d, J = 8.1 Hz), 7.77 (2H, d, J = 8.4 Hz), 7.47 (1H, m), 7.40 (1H, m), 7.34 (2H, d, J = 8.4 Hz), 3.98 (2H, m), 3.15 (2H, m), 3.00 (2H, m), 2.63 (2H, m), 2.46 (4H, m), 2.08 (2H, m), 1.93 (6H, m). Calcd for C24H28Cl2N2OS·0.4H2O:C 61.24, H 6.00, N 5.95; Found: C 61.29, H 6.05, N 5.85.
4.7. Synthesis of compounds 16–18: Method C General procedure
4.7.1. 1-(Benzo[d]thiazol-2-yl)-5-(3-(4-chlorophenyl)-3-hydroxy-8-azabicyclo[3.2.1]octan-8-yl)pentan-1-one, 18
A mixture of 1-(benzo[d]thiazol-2-yl)-5-iodopentan-1-one 39 (0.635 g, 2.5 mmol), 3-(4-chlorophenyl)-8-azabicyclo[3.2.1]octan-3-ol 44 (680 mg, 2.85 mmol), K2CO3 (700 mg, 5.07 mmol) in DME (10 mL) was heated to reflux under N2 for 16 h. The mixture was diluted with EtOAc (400 mL) and washed with brine (50 mL). The organic layer was dried over Na2SO4 and filtered. The filtrate was concentrated in vacuo to dry followed by column chromatography on silica gel affording 1-benzothiazol-2-yl-5-(4-phenyl-piperidin-1-yl)-pentan-1-one. The product was converted into the HCl salt followed by crystallization from MeOH–Et2O (520 mg) in a yield of 43%, mp 202–204 °C. 1H NMR (DMSO-d6): δ 10.46 (1H, br s), 8.24 (2H, m), 7.73 (2H, d, J = 8.7 Hz), 7.64 (2H, m), 7.36 (2H, d, J = 9.0 Hz), 5.47 (1H, s), 4.30 (2H, m), 3.35 (4H, m), 3.14 (2H, m), 3.00 (2H, m), 2.57 (2H, m), 2.09 (2H, m), 1.88 (4H, m), 1.75 (2H, m). Calcd for C25H28Cl2N2O2S·0.8H2O: C 59.36, H 5.58, N 5.54; Found: C 59.46, H 5.82, N 5.57.
4.7.2. 1-(Benzo[d]thiazol-2-yl)-5-(4-(4-chlorophenyl)-1,4-diazepan-1-yl)pentan-1-one, 16
Using 1-(benzo[d]thiazol-2-yl)-5-iodopentan-1-one 39 and amine 43, the free base of compound 16 was obtained and converted to HCl salt followed by crystallization from MeOH–Et2O in a yield of 32%. 1H NMR (CDCl3) δ 8.18–8.15 (2H, d, J = 8.7 Hz), 7.97–7.94 (2H, d, J = 8.7 Hz), 7.59–7.50 (2H, m), 7.13–7.09 (2H, d, J = 9.3 Hz), 6.57–6.54 (2H, d, J = 9 Hz), 3.49–3.39 (4H, m), 3.28 (2H, t, J = 7.2 Hz), 2.74–2.71 (2H, m), 2.59–2.5 (4H, m), 1.96–1.8 (4H, m), 1.64–1.56 (2H, m).
The free base was converted to the HCl salt, mp 242 °C. 1H NMR (CD3OD): δ 8.14–8.05 (2H, m), 7.61–7.51 (2H, m), 7.16 (2H, br s), 6.79 (2H, br s), 3.85–3.45 (13H, m), 2.30 (2H, m), 1.90 (3H, m). Calcd for C23H28Cl3N3OS .3H2O:C 49.78, H 5.09, N 7.57; Found:C 49.68, H 5.28, N 7.43.
4.7.3. 1-(Benzo[d]thiazol-2-yl)-4-(4-(4-chlorophenyl)-1,4-diazepan-1-yl)butan-1-one, 17
The product of 1-(benzo[d]thiazol-2-yl)-4-iodobutan-1-one 38 and amine 43 was converted to the hydrochloride salt followed by crystallization from MeOH–Et2O. Yield 22%, mp 194–195 °C. 1H NMR (DMSO-d6): δ 10.83 (1H, br s), 8.23 (2H, m), 7.64 (2H, m), 7.19 (2H, d, J = 9.0 Hz), 6.76 (2H, d, J = 9.0 Hz), 3.78 (2H, m), 3.50 (2H, m), 3.40 (4H, m), 3.16 (4H, m), 2.35(1H, m), 2.11 (3H, m). Calcd for C22H26Cl3N3OS: C 54.27, H 5.38, N 8.63; Found:C 54.71, H 5.33, N 8.64.
4.8. Receptor binding studies
Binding affinities reported in Table 1 were conducted by the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP). Details of the methods and radioligands used for the binding assays were previously reported.17
Acknowledgments
We gratefully acknowledge the financial support of the National Institute of General Medical Studies (NIGMS) for MBRS Grant No. 1SC1GM088451-01, NIMH Psychoactive Drug Screening Program, and a Title III Grant to Florida A&M University. This work was supported in part by the Pharmaceutical Research Center NIH/NCRR 1 C06-RR12512-01 grant. The authors acknowledge Mrs. Barbara Bricker for editorial assistance.
References and notes
- 1.Baldessarini RJ, Tarazi FI. In: In Goodman s The Pharmacological Basis of Therapeutics. 11th ed Brunton L, Lazo J, Parker K, editors. McGraw-Hill; New York: 2006. pp. 461–500. [Google Scholar]
- 2.Sikazwe DMN, Nkansah NT, Altundas R, Zhu XY, Roth BL, Ablordeppey SY. Bioorg. Med. Chem. 2009;17:1716. doi: 10.1016/j.bmc.2008.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ablordeppey SY, Lyles-Eggleston M, Bricker B, Zhang W, Zhu XY, Goodman C, Roth BL. Bioorg. Med. Chem. Lett. 2006;16:3219. doi: 10.1016/j.bmcl.2006.03.057. [DOI] [PubMed] [Google Scholar]
- 4.Sikazwe DMN, Li S, Mardenborough L, Cody V, Roth BL, Ablordeppey SY. Bioorg. Med. Chem. Lett. 2004;14:5739. doi: 10.1016/j.bmcl.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 5.Lyles-Eggleston M, Altundas R, Xia J, Sikazwe DMN, Fan P, Yang Q, Li S, Zhang W, Zhu X, Schmidt AW, Vanase-Frawley M, Shrihkande M, Villalobos A, Borne RF, Ablordeppey SY. J. Med. Chem. 2004;47:497. doi: 10.1021/jm0301033. [DOI] [PubMed] [Google Scholar]
- 6.Sikazwe DMN, Lyles-Eggleston MD, Li S, Ablordeppey SY. Bioorg. Med. Chem. Lett. 2003;13:3779. doi: 10.1016/j.bmcl.2003.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Ablordeppey SY, Altundas R, Bricker B, Zhu XY, Kumar E. V. Suresh, Jackson T, Khan A, Roth BL. Bioorg. Med. Chem. 2008;16:7291. doi: 10.1016/j.bmc.2008.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reynolds GP, Hill MJ, Kirk SL. J. Psychopharmacol. 2006;20:15. doi: 10.1177/1359786806066040. [DOI] [PubMed] [Google Scholar]
- 9.Reynolds GP, Zhang ZJ, Zhang XB. Lancet. 2002;359:2086. doi: 10.1016/S0140-6736(02)08913-4. [DOI] [PubMed] [Google Scholar]
- 10 (a).Kim SF, Huang AS, Snowman AM, Teuscher C, Snyder SH. PNAS. 2007;104:3456. doi: 10.1073/pnas.0611417104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, et al. Neuropsychopharmacology. 2003;28:519. doi: 10.1038/sj.npp.1300027. [DOI] [PubMed] [Google Scholar]
- 11.Vaidya T, Eisenberg R, Frontier AJ. ChemCatChem. 2011;3:1531. [Google Scholar]
- 12.Peprah K, Zhu XY, Eyunni SVK, Setola V, Roth BL, Ablordeppey SY. Bioorg. Med. Chem. 2012;20:1291. doi: 10.1016/j.bmc.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raviña E, Casariego I, Masaguer CF, Fontenla JA, Montenegro GY, Rivas ME, Loza MI, Enguix MJ, Villazon M, Cadavid MI, Demontis GC. J. Med. Chem. 2000;43:4678. doi: 10.1021/jm0009890. [DOI] [PubMed] [Google Scholar]
- 14.Micheli F, Heidbreder C. Recent Pat CNS Drug Discov. 2006;1:271. doi: 10.2174/157488906778773634. [DOI] [PubMed] [Google Scholar]
- 15.Heidbreder CA, Newman AH. Ann. N Y Acad. Sci. 2010;1187:4. doi: 10.1111/j.1749-6632.2009.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Micheli F. ChemMedChem. 2011;6:1152. doi: 10.1002/cmdc.201000538. [DOI] [PubMed] [Google Scholar]
- 17.Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, Mailman R. Neuropsychopharmacology. 2003;28:1400. doi: 10.1038/sj.npp.1300203. [DOI] [PubMed] [Google Scholar]