

Abstract
Using a combination of whole-genome resequencing and high-density genotyping arrays, genome-wide haplotypes were reconstructed for two of the most important bulls in the history of the dairy cattle industry, Pawnee Farm Arlinda Chief (“Chief”) and his son Walkway Chief Mark (“Mark”), each accounting for ∼7% of all current genomes. We aligned 20.5 Gbp (∼7.3× coverage) and 37.9 Gbp (∼13.5× coverage) of the Chief and Mark genomic sequences, respectively. More than 1.3 million high-quality SNPs were detected in Chief and Mark sequences. The genome-wide haplotypes inherited by Mark from Chief were reconstructed using ∼1 million informative SNPs. Comparison of a set of 15,826 SNPs that overlapped in the sequence-based and BovineSNP50 SNPs showed the accuracy of the sequence-based haplotype reconstruction to be as high as 97%. By using the BovineSNP50 genotypes, the frequencies of Chief alleles on his two haplotypes then were determined in 1,149 of his descendants, and the distribution was compared with the frequencies that would be expected assuming no selection. We identified 49 chromosomal segments in which Chief alleles showed strong evidence of selection. Candidate polymorphisms for traits that have been under selection in the dairy cattle population then were identified by referencing Chief’s DNA sequence within these selected chromosome blocks. Eleven candidate genes were identified with functions related to milk-production, fertility, and disease-resistance traits. These data demonstrate that haplotype reconstruction of an ancestral proband by whole-genome resequencing in combination with high-density SNP genotyping of descendants can be used for rapid, genome-wide identification of the ancestor’s alleles that have been subjected to artificial selection.
Keywords: next generation sequencing, single nucleotide polymorphism, quantitative trait locus
Identification of the molecular genetic basis of complex traits is a recalcitrant problem in modern genetics. In outbred populations and livestock species subjected to selection for economically important quantitative trait loci (QTL), only a handful of causative mutations have been identified (reviewed in ref. 1). For most mapped QTL, genomic regions containing the QTL are quite large, making the identification of candidate genes a matter of guesswork (2). The low resolution of QTL maps is generally a consequence of QTL with small effects and inadequate sample size for fine mapping. Clearly, new strategies are needed to understand how quantitative traits are controlled by genomic features.
Dairy cattle are ideal to test new strategies for finding mutations responsible for variation in complex traits, because the population has been subjected to more than 50 y of intense selection for milk-production traits (3), dense SNP arrays are commercially available (4), and recently the cattle whole-genome sequence assembly was completed (5). Selection is practiced primarily in sires through artificial insemination and progeny testing (6), with the best bulls in the population having thousands of daughters that are used for estimating breeding values for quantitative traits. For example, using quantitative animal-breeding methods, the amount of milk produced by Holstein cows increased from 5,000 pounds per lactation in the 1940s to 19,000 pounds per lactation in 2005 (http://aipl.arsusda.gov/publish/presentations/ADSA05/ADSA05_culling.ppt). Thus, retrospective semen collections such as the US Dairy Bull DNA Repository (7) are invaluable for gaining a molecular understanding of the genomic changes that occur as a result of artificial selection.
Population genetic theory predicts that when alleles are under natural or artificial selection, their frequencies will change over time (8). Presently, even with low-cost high-throughput DNA sequencing, it is not feasible to track the parental origin of alleles on a comprehensive, whole-genome scale over multiple generations; thus our ability to understand how selection affects genome architecture and the underlying mutations that are responsible for QTL effects is limited. A major limitation in studying the effects of selection on the genome is that haplotype structure for most organisms is incomplete or unknown. Until recently, genome-wide reconstruction of haplotypes was limited by the number of available polymorphisms (9, 10). Now, with millions of SNPs available, it is possible to identify haplotypes and linkage disequilibrium at high resolution (11). However, in any given individual, these blocks do not represent a complete catalog of polymorphisms in a diploid genome, because many SNPs and other DNA polymorphisms go undetected by common genotyping procedures. Only whole-genome sequencing has the power to detect the majority of DNA polymorphisms in an individual; these polymorphisms then can be used for the reconstruction of haplotypes. The complete reconstruction of individual haplotypes is important for the identification of disease-causing mutations (12). Furthermore, the tracking of identical-by-descent (IBD) haplotypes in the population will be crucial for dissecting complex traits in human and animal populations (13, 14).
In the present study, we used a DNA resequencing-based strategy for the phasing of alleles in first-order relatives that, when used in combination with high-density SNP genotyping arrays and phenotypic data in descendants, revealed changes in haplotype frequencies over multiple generations. By then imputing the sequence in these regions, using the full-genome sequence of the bulls Pawnee Farm Arlinda Chief (“Chief”) and Walkway Chief Mark (“Mark”), we provide evidence that the method can be used to identify potential causative mutations that underlie QTL regions.
Results
Sequencing and Assembly.
The genomes of Walkway Chief Mark (“Mark”) and Pawnee Farm Arlinda Chief (“Chief”) were sequenced to ∼13.5× and ∼7.3× coverage, respectively. In total, 89.7 million reads comprising 31.8 Gbp were uniquely aligned to the reference genome sequence using Newbler (15), and 27.2 million additional reads (11.3 Gbp) were aligned by BLAT (16). Of these reads, 40.3 Gbp were mapped to individual Btau4.0 chromosomes, and 2.8 Gbp were mapped to unassigned contigs (Table 1). On average, 88.7% and 83.2% of bovine chromosome sequences were covered by the reads originating from Mark and Chief, respectively (SI Appendix, Fig. S1). We excluded 2.7 million Mark reads and 2.0 million Chief reads (∼4.3% of all of the reads mapped to chromosomes) as duplicates. All remaining mapped reads were recalled by the PyroBayes software (17) and used to build a reference genome-based alignment of the combined Mark and Chief genomes (SI Appendix, Table S1). A total of 2,212,379,271 bp, 86.9% of the length of the reference chromosome sequences, was covered ≥1× with the combined Chief and Mark reference-based assembly. All unmapped reads were assembled de novo and remapped against the cattle genome (SI Appendix, Table S2).
Table 1.
Sequencing and mapping statistics
| Statistic | Walkway Chief Mark (Mark) | Pawnee Farm Arlinda Chief (Chief) |
| High-quality reads generated (million) | 106.4 | 59.8 |
| DNA sequenced (Gbp) | 37.9 | 20.5 |
| Average read length (bp) | 356 | 342 |
| Average sequence per plate (Mbp) | 443 | 417 |
| Expected genome coverage* | 13.5x | 7.3x |
| Mapped to chromosomes | ||
| Reads (million) | 71.4 | 37.7 |
| Base pairs (Gbp) | 26.6 | 13.7 |
*Based on 2.8 Gbp, the estimated size of the cattle genome (5).
Identification of SNPs.
A total of 12,434,860 raw autosomal SNPs were detected in the Mark and Chief genomes (SI Appendix, Table S3). Of these, 1,851,126 SNPs were homozygous in Mark and Chief reads but had an allele different from the reference genome allele; these SNPs were termed “homozygous” SNPs. The second group of 10,583,734 autosomal SNPs had two alleles detected in Mark, Chief, or both bulls’ reads; these SNPs were termed “heterozygous” SNPs. After filtering the homozygous and heterozygous sets for quality, there were 1,207,103 high-quality homozygous SNPs (SI Appendix, Table S3). Heterozygous SNPs were filtered additionally for <3× coverage per allele in Mark reads and <2× coverage per allele in Chief reads, resulting in a set of 1,311,454 autosomal heterozygous Mark SNPs and 818,065 autosomal heterozygous Chief SNPs (SI Appendix, Fig. S2 and Table S3).
We then compared the set of 2,058,654 unique SNPs from the cattle dbSNP (build 130) with our SNP sets. The homozygous SNPs had 304,120 (25.2%) positions overlapping with the cattle dbSNP, whereas the set of heterozygous SNPs had 219,071 (15.1%) positions in common with cattle dbSNP. All SNPs were submitted to dbSNP. The set of heterozygous and homozygous SNPs then were located in GLEAN gene predictions, repeats, and segmental duplications. In general, the distribution of heterozygous and homozygous SNPs was similar in all the sets, demonstrating that the Mark and Chief SNPs were not biased to genes, repeats, or segmental duplications (SI Appendix, Fig. S3).
Two final filtering steps were applied before using the SNPs for the reconstruction of haplotypes (SI Appendix). We found that the SNPs removed were overrepresented in duplicated genome regions (43.5% and 34.7% overlap in Chief and Mark, respectively) (18). In the final filtered set there were 1,243,113 heterozygous SNPs in Mark and 757,266 heterozygous SNPs in Chief. Of these, 1,356,094 SNPs were unique and passed the filtering criteria in at least one bull (SI Appendix, Table S3).
Comparison of DNA Sequence to High-Density Genotyping.
Mark and Chief DNA samples were genotyped for 54,001 SNPs using the Illumina SNP50 platform. We obtained 52,138 successful genotype calls for Mark and 52,155 genotype calls for Chief. In addition, 92 progeny-tested sons of Mark were genotyped, and the data were used to reconstruct Mark haplotypes (SI Appendix). Using the segregation of alleles in Mark offspring, we estimated the SNP genotyping error in Mark to be ∼0.3%. Some of the errors were corrected using segregation data, resulting in 15,826 heterozygous SNP50 positions in Mark and 15,448 in Chief. Among the heterozygous and homozygous SNPs detected by DNA sequencing, there were 8,009 and 5,359 shared heterozygous SNP50 SNP positions in Mark and Chief, respectively. When the coverage criteria used for heterozygous SNPs (≥6× total coverage in Mark and ≥4× total coverage in Chief) were applied to the sequence-based SNPs that were homozygous in one of the sires and heterozygous in the other, we found 96.7% and 84.4% overall concordance between Mark and Chief SNPs detected by SNP50 and DNA-based sequencing, respectively.
Reconstruction of Mark and Chief Haplotypes.
The shared chromosome segments inherited by Mark from Chief then were reconstructed using SNPs identified by genome sequencing. This reconstruction was accomplished using 972,479 mapped, high-quality autosomal SNP loci that were informative for segregation from Chief (i.e., all SNPs that were homozygous in one bull but heterozygous in the other). From the difference, the maternal genome of Mark and the nontransmitted chromosome segments of Chief were imputed (SI Appendix, Table S3). The mean spacing between the adjacent SNPs in the haplotypes was ∼2.7 kbp (median, 0.5 kbp). The phasing of alleles in Mark’s genome was confirmed directly by segregation analysis in 92 of his sons using 15,826 heterozygous SNP50 SNPs (SI Appendix, Table S4 and Figs. S4 and S8). Concordance of the in silico-phased alleles imputed from DNA sequencing and the set of overlapping SNP50 alleles was up to 97.0% (SI Appendix, Table S5). The false-negative rate (homozygous in sequence, heterozygous in genotyping) of sequencing-based SNPs compared with SNP50 SNPs was estimated at 15.2% for Chief and at 1.1% for Mark. The false-positive rate (heterozygous in sequence, homozygous in genotyping) was 0.6% for Chief and 1.0% for Mark SNPs.
Signatures of Selection.
We then determined whether Chief’s haplotypes had changed in frequency after more than 40 y of selection in the Australian Holstein-Friesian population. By using an extension of the ChromoPhase algorithm, Chief’s haplotypes were tracked in 1,183 SNP50-genotyped Australian Holstein bulls that were up to seven generations removed from Chief (19). Chief’s haplotype blocks were tracked over his entire genome and in his descendants (SI Appendix, Fig. S5). The average percentage of Chief’s alleles found in his descendants across seven generations ranged from 10 to 30%, resulting in an average tracked haplotype block size of 12.08 Mbp (range, 8.31 Mbp on BTA5 to 19.82 Mbp on BTA9 (SI Appendix, Table S6). It was not possible to determine the chromosomal origin for stretches of the genome where Chief was homozygous. However, such segments still could be tracked. The relatively small fraction of the genome that could not be tracked over multiple generations likely was the result of genotyping and mapping errors that led to incomplete phasing.
The distribution of Chief’s SNP50 alleles in his descendants was analyzed in greater detail to determine if they deviated from expected frequencies. We used gene dropping of Chief’s SNP50 alleles through the pedigree to obtain the distribution of frequencies that would be expected under the null hypothesis of no selection (random drift only). We identified 49 intervals on 16 chromosomes where the frequency of Chief’s haplotypes lay outside the genome-wide 99th percentile of the expected distribution under random drift alone (Fig. 1). It is possible that the selection signals observed are not associated only with Chief’s haplotypes but are an artifact caused by matching alleles that are at high frequency in the general population. This artifact would result in allele frequencies in Chief’s descendants that are similar to the allele frequencies in nondescendants (SI Appendix, Fig. S6). To investigate this possibility, we compared allele frequencies of SNPs at the putative selection sites in descendants and nondescendants of Chief. The proportion of alleles identical to Chief’s among his descendants is shown in Fig. 1. A linear model then was used to determine whether the frequency of tracked alleles in Chief’s descendants was significantly different from the frequency in nondescendants (SI Appendix, Fig. S7). A total of 1,105 SNPs exceeded the significance threshold (P < 0.001) and were distributed over 32 regions, 12 of which overlapped with 11 of the previously identified 49 intervals. The expected number of SNPs exceeding this significance threshold by chance is 35 (35,036 tests performed), thus supporting the hypothesis that the great majority of the alleles within these Chief chromosomal intervals has been subjected to recent selection.
Fig. 1.
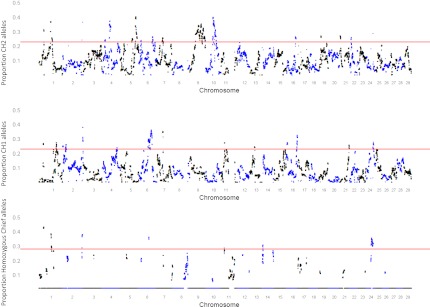
Proportion of Chief alleles originating from his alternative haplotype CH1, CH2, or homozygous in Chief’s genome that are present in the current Australian Holstein population. Red line indicates the threshold at which the fraction of Chief SNPs in the population could be explained by a random distribution of Chief alleles. Single nucleotide polymorphisms on individual chromosomes are demarcated by alternating black and blue colors.
Identification of Candidate Genes Under Selection.
As a proof of principle that our method could be used to detect candidate DNA polymorphisms for traits affected by recent selective sweeps, we examined the gene content of the 49 Chief chromosomal intervals that exhibited deviation from the expected frequency of Chief alleles in the current population of Australian Holsteins and then searched for tracked SNPs that would qualify as causative for a selected trait(s). Forty-two (85.7%) of these intervals overlap with known milk-production QTL reported in CattleQTLdb (20). Of 49 regions, 11 intervals had Chief alleles that differed significantly in their frequency among Chief descendants compared with nondescendants. These regions contained 3,504 SNPs from known RefSeq genes; 82 of these SNPS are exonic, and 29 are nonsynonymous substitutions. Three high-probability QTL candidate genes were found within these intervals: SCARB2, CXCL10, and PLG. Within the remaining 38 intervals with a high proportion of Chief alleles in the current population of Australian Holsteins, we detected two genes, ARL4 and BMP4 (Fig. 2 A and B), that had been identified previously as candidate genes for QTL (21, 22). Other functional candidates are given in Table 2. (A complete list of genes within selected intervals is presented in SI Appendix, Table S7.)
Fig. 2.
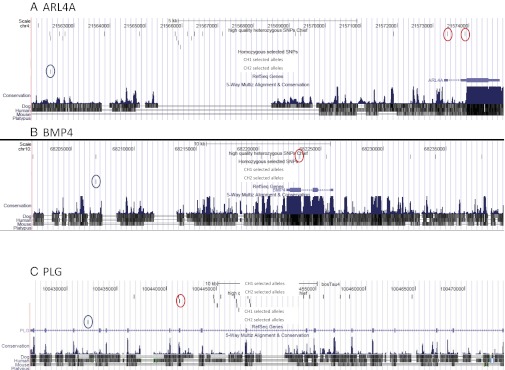
Position of Chief SNPs in candidate genes ARL4A (A), BMP4 (B), and PLG (C). Blue ovals indicate positions of the SNP50 SNPs with the strongest selection signature within Chief alternative haplotypes. Red ovals show positions of exonic SNPs in the adjacent gene. On the basis of reconstruction of allele phases in Chief chromosomes, a prediction can be made for which allele in CH1 or CH2 has been selected in each case.
Table 2.
Genes with SNPs in selected regions of Chief chromosomes with functions related to milk production and disease resistance
| No. SNPs |
||||||
| Gene* | Chromosome | Start | End | Nonsynonymous | 5′UTR | 3′UTR |
| ITGA6 | 2 | 24966035 | 25051812 | 1 | 0 | 0 |
| PLA2G2F | 2 | 137036850 | 137050098 | 1 | 2 | 6 |
| ARL4A | 4 | 21573320 | 21574858 | 0 | 2 | 0 |
| CSF2RB | 5 | 81005400 | 81026598 | 1 | 0 | 0 |
| SULT1E1 | 6 | 88173894 | 88229946 | 0 | 0 | 4 |
| CXCL10† | 6 | 94129095 | 94131447 | 0 | 0 | 3 |
| SCARB2† | 6 | 94258594 | 94341321 | 1 | 0 | 8 |
| SAR1B | 7 | 45321215 | 45352216 | 0 | 0 | 1 |
| PLG† | 9 | 100426314 | 100473934 | 1 | 0 | 0 |
| CYP19A1 | 10 | 59440433 | 59504627 | 0 | 1 | 8 |
| BMP4 | 10 | 68222238 | 68225971 | 0 | 0 | 0 |
| MYD88 | 22 | 11727055 | 11731271 | 0 | 0 | 1 |
*Only best candidate genes are shown. For complete list of gene mutations in selected regions, see SI Appendix, Table S7.
†These genes are located within intervals where allele frequencies differ in descendants and nondescendants of Chief (see SI Appendix, Table S8).
Discussion
The goals of our study were to reconstruct the haplotypes of two influential ancestors in the pedigrees of the contemporary Holstein-Friesian population and to use this information to identify chromosome segments and polymorphisms in dairy cattle that have been subjected to recent selection. To do so, we sequenced the genomes of two of the most influential dairy sires in history, Walkway Chief Mark and his father Pawnee Farm Arlinda Chief. Mark was born in 1978, sired more than 60,000 daughters by artificial insemination, and had many sons that later became popular sires (23). Chief was born in 1962. His descendants include Mark and many prominent dairy sires bred throughout the world. Each of these bulls accounts for ∼7% of the genomes of the current North American Holstein cow populations (http://aipl.arsusda.gov/). Obtaining the sequence of these two bulls thus provides an important milestone in dairy cattle genetics and serves as the foundation for the detection and utilization of selected haplotypes resulting from more than 50 y of modern dairy cattle breeding.
The Mark and Chief genomes were sequenced to ∼13.5× and ∼7.3× coverage, respectively, using 454 sequencing technology (Table 1). More than 2.5 million high-quality autosomal SNPs were detected (SI Appendix, Table S3); ∼1.2 million and ∼757,000 of these high-quality SNPs were identified in the genomes of Mark and Chief, respectively. Of these SNPs, only 523,191 positions (25.4%) were present in cattle dbSNP. In Mark, there was 98.3% agreement for 11,273 SNP50 SNPs that overlapped with the equivalent bases called by DNA sequencing (SI Appendix, Table S4), whereas in Chief the agreement was 91.3% for 8,801 SNPs. Visual inspection of the discrepancies revealed that the great majority could be explained by allele dropout in Chief caused by lower sequence coverage relative to Mark. With the deeper sequence coverage that now can be obtained for these bulls with other sequencing platforms, the accuracy of SNP calling certainly will improve, resulting in recovery of a greater fraction of the existing polymorphisms.
The genome-wide haplotypes shared by Mark and Chief, as well as the alternative Chief and Mark haplotypes, were reconstructed with an average resolution of one SNP per 2.7 kbp. The first successful genome-wide reconstruction of individual haplotypes was reported by Levy and coworkers (12) but was limited to SNPs present in overlapping Sanger reads or mate pairs. As a consequence, long-range haplotype blocks covered only 58% of human genes. A more complete reconstruction of haplotypes in families became feasible after the introduction of next-generation sequencing techniques. For example, Roach et al. (24) reconstructed individual and paternal haplotypes by resequencing two human siblings and their parents to ∼60× average coverage. De novo mutations were identified, and the haplotype information was used to narrow the number of candidate genes for Miller syndrome and primary ciliary dyskinesia to four genes. Here we report a haplotype reconstruction by resequencing only two first-order relatives. We confirmed the high accuracy of the haplotype reconstruction by genotyping one of the individuals and 92 of his offspring. We then used the reconstructed haplotypes of one sire to identify regions in his genome showing evidence of selection in his descendants and deduced a set of candidate genes for traits that have been under selection in the dairy cattle population for several generations.
In modern human populations, limited patient pedigree information and historical DNA collections and the lack of strong selection limits the ability to trace the frequency of individual haplotype blocks (e.g., those related to disease resistance) over extended periods of time. In contrast, dairy cattle have been under strong selection for lactation and reproduction traits for more than half a century, and retrospective DNA collections are available throughout the world. We had access to the SNP50 genotypes of 1,149 recorded descendants of Chief in the Australian Holstein population, allowing us to trace the frequencies of Chief haplotype blocks and alleles across seven generations. We identified 49 chromosome intervals that have a significantly higher than expected fraction of Chief alleles. In 11 of these regions the frequency of Chief alleles was significantly higher in his descendants than in nondescendants, suggesting selection of Chief haplotypes. We found several regions of the genome that have a significantly higher frequency of homozygous Chief alleles in the whole population. These areas may represent haplotypes that are moving toward fixation in Holsteins as a result of artificial selection.
An analysis of polymorphisms in 49 chromosomal segments showing evidence of selection revealed several interesting candidate genes for economically important traits. For example, the gene encoding ADP ribosylation factor-like 4A (ARL4A) on BTA4 contains a putative quantitative trait nucleotide for milk-production traits (21). A Chief SNP with a strong signal for recent selection was found 69.7 kbp downstream of ARL4 (SI Appendix, Table S7). The Chief haplotypes reconstructed by DNA resequencing show that Chief is heterozygous for two SNPs that are located in the 5′UTR of ARL4A (Fig. 2A). One of these SNPs at BTA4 position 21,573,888 was reported to be associated with milk-production traits in the North American Holstein population (21). We also detected selection of Chief alleles on BTA10 in a region previously shown to be under selection in Holstein cattle (22); (Fig. 2B), with the most significant SNP50 SNP falling within 114 kbp of the gene encoding bone morphogenetic protein (BMP4). This gene has been shown to potentiate growth factor-induced proliferation of cultured mammary epithelial cells (25). Two BMP4 SNPs in Chief that were identified as heterozygous by resequencing and haplotype imputation were located in exon 3 and an intron sequence, respectively, but did not cause an amino acid or splice site change, indicating that another unidentified polymorphism(s) on the haplotype may be responsible for the associated phenotype.
A strong selection signature was detected in the region of SCARB2 on BTA6, a receptor for groups A, B, and C of EV-71, the causative agent of hand, foot, and mouth disease in humans that is related to the picornavirus that causes foot-and-mouth disease in cattle. This gene has 118 SNPs heterozygous in Chief, of which seven are in exons (an Arg-to-Lys substitution in exon 6), and eight are in the 3′UTR. SCARB2 is located in one of the 11 regions in which the frequencies of Chief alleles are overrepresented in his descendants as compared with nondescendants. Another strong candidate gene located in one of these intervals on BTA9 is plasminogen (PLG). PLG deficiency in mice leads to milk stasis and premature mammary gland involution (26). In sheep, plasminogen activity in mammary gland is affected during mastitis (27). This gene has one detected coding mutation in Chief that changes Pro to Thr in exon 12.
Our data demonstrate that chromosomal regions that have undergone recent selection can be identified using a combination of whole-genome resequencing of ancestors, haplotype reconstruction, and high-density genotyping of their descendants. Among Chief’s descendants, continued breeding emphasis on selected haplotypes should have favorable economic consequences. Furthermore, imputing SNP genotypes obtained from haplotype reconstructions shows promise as an efficient strategy for rapid identification of causal mutations controlling simple and complex traits under selection. This strategy, we term “haplotracking,” broadly impacts genomic selection schemes for genetic improvement of livestock species (28) and also can be adapted for resolving mutations affecting complex traits in humans when multigeneration pedigree and phenotype data are available.
Methods
Sequencing Strategy.
A strategy was devised to obtain the maximum power for reconstruction of the shared genome-wide chromosomal haplotypes assuming different levels of sequence depth. A total of 18× genome coverage using 454 Titanium technology was determined to be adequate for haplotype reconstruction (SI Appendix). For the present study, we selected two famous bulls to test our resequencing strategy: Walkway Chief Mark (“Mark”; HOUSAM1773417) and his father Pawnee Farm Arlinda Chief (“Chief”; HOUSAM1427381). A target of 6× genome coverage was selected for Chief and 12× for Mark under the assumption that the shared inherited autosomes and thus the shared alleles of Chief would be sequenced at an average depth of 9× [(0.5 × 6×) + (0.5 × 12×)]. Mark was selected for greater coverage because he was the higher-impact bull, with the greatest number of progeny-tested sons and sons’ DNA samples in the US Dairy Bull DNA Repository (DBDR) (7, 29). Having a large number of sons with DNA in the DBDR collection was important, because it permitted verification of the sequence-based haplotype reconstruction using high-density SNP genotyping.
Whole-Genome Shotgun Sequencing.
DNA was extracted from Mark and Chief and 92 Mark sons in the DBDR collection using a salting-out DNA extraction protocol (29). Sequencing libraries were constructed following the General Library Preparation Method Manual (Roche). Emulsion-based clonal amplification and sequencing were performed on a 454 Genome Sequencer FLX-Titanium system (454 Life Sciences) according to the manufacturer’s instructions. A total of 50 and 87.5 GS-Titanium 70 × 75 picotiter plates were sequenced from Chief and Mark genomic DNA libraries, respectively. Signal processing and base calling were performed using the bundled 454 Data Analysis Software version 2.0.00.
Alignment and Mapping.
Sequence reads from each plate were mapped against bovine genome assembly Btau4.0 using Newbler (15) with the default criteria. All reads with a single match in the bovine genome, read coverage of ≥90%, and read length of ≥50 bp were considered as mapped. To this collection we added reads that had a single match with ≥90% coverage in chromosome sequences (excluding unassigned contigs) and reads that had a best Newbler hit in a chromosome sequence. All sequences that were not mapped using Newbler default criteria were aligned against the Btau4.0 cattle chromosome sequences using BLAT (16) with -ooc, -fastMap options. The mapping criteria described above were applied to the reads aligned by BLAT. A reference-based assembly of the unmapped reads was performed using the MOSAIK assembler (17).
Identification of SNPs.
GigaBayes software, a new version of POLYBAYES (30), was used for the initial call of SNPs from the reference-based, multisequence, combined assemblies of Mark and Chief autosomes. We allowed a minimum of 2× read coverage for the initial identification of putative SNPs. GigaBayes was used to distinguish between the reads originating from the Mark and Chief genomes based on an extension added to PyroBayes software (17). This approach allowed us to control for the allele status of each bull separately for every putative SNP locus. Details of the methods used for allele calling and SNP filtering are given in SI Appendix.
Genotyping.
DNA samples from Mark, Chief, and 92 Mark sons were genotyped for 54,001 SNPs using the Illumina BovineSNP50 (SNP50) BeadChips (Illumina) according to the manufacturer’s instructions. There were 1,672 (3.1%) SNPs that had no chromosomal assignment in Btau4.0 and were thus excluded from the analysis (SI Appendix, Table S4).
Reconstruction of Mark and Chief Haplotypes from Sequence Data.
In silico SNPs heterozygous in one bull but homozygous in the other were used to reconstruct Mark’s and Chief’s haplotypes from the sequence data using the following rules: (i) all SNP loci at which Mark was homozygous and Chief was heterozygous, or vice-versa, were identified; (ii) an allele present in both bulls was assigned to the haplotype inherited by Mark from Chief and also to the second haplotype of the bull homozygous at this SNP locus; (iii) an allele present only in the bull heterozygous for the SNP locus was assigned to the second haplotype of this bull; (iv) steps ii–iv were repeated for all SNP loci that fit criteria i. As a result, we identified alleles that were inherited by Mark from Chief. Together they constitute Mark’s paternally inherited haplotype on a chromosome-by-chromosome, whole-genome basis. In addition, we imputed Mark’s maternally inherited haplotype by the difference from the Mark–Chief shared chromosomal haplotypes and Chief’s alternative (i.e., not inherited by Mark) chromosomal haplotypes.
Reconstruction of Mark and Chief Haplotypes from Genotyping Data.
Genotypes of 1,183 Holstein Friesian cattle, including Chief and Mark, had their haplotypes reconstructed using the ChromoPhase algorithm (19). The software makes use of the long chromosome segments shared by (closely or distantly) related animals (31). Haplotypes were reconstructed by transferring information between animals with matching chromosome segments. Before using ChromoPhase on the Chief multigeneration data, we tested how well Mark’s haplotypes were tracked using SNP50 genotypes of a separate sample from 92 of his sons. The ChromoPhase-based segregation of alleles on all autosomes was determined, and putative recombinants were identified (SI Appendix, Fig. S8).
Identification of IBD Segments.
Chief’s haplotypes were tracked in our sample of Holstein cattle by identifying stretches of alleles that were in common for consecutive loci between Chief and a descendant. The length of common chromosome segments needed to exceed 50 loci and have less than 15% missing alleles in the corresponding Chief segment to ensure the segment was IBD. Chief’s haplotypes were assigned unique labels: CH1, CH2, and homozygous. When a common chromosome segment was identified, this haplotype label was transferred to alleles in the descendant’s matching segment. Pedigree analysis identified Chief 1,149 descendants in our sample. Proportions of alleles that Chief had in common with descendants and nondescendants were calculated at each locus by dividing the counts of labels by the total allele count in his descendants. This process allowed us to identify chromosome segments where Chief shared more than the expected proportion of alleles in his descendents. Furthermore, it allows identification of loci where one Chief chromosome is significantly more represented than the other in his descendants, indicating selective pressures.
Signatures of Selection.
Peaks in proportions could be caused by the selection of favorable Chief alleles or by genetic drift. We used a simulation study to differentiate peaks caused by drift from peaks caused by selection. Chiefs’ alleles on the SNP50 array were sampled at random and “gene dropped” (32) through his descendants in the Australian Holstein pedigree, assuming random mating and no selection. The parental source of alleles was randomly assigned, and the allele frequency distribution then was calculated in Chief’s descendants. This process was replicated 1,000 times, allowing reconstruction of a distribution for the allele frequencies under the null hypothesis of no selection. A P value of 0.01 at the genome-wide level (e.g., accounting for the multiple testing across all of the SNP50 SNPs) was used.
Allele frequencies could be similar in both Chief descendants and nondescendants. A second test fitted a linear model to differentiate peaks where Chief descendants differed significantly from nondescendants. See SI Appendix for details.
Detection of Genes and SNPs in Selected Regions.
We merged all selected Chief SNPs located <1 Mbp apart into 49 regions and extracted annotation for cattle RefSeq genes (University of California, Santa Cruz Genome Browser, Btau4.0) located within the selected regions. The ANNOVAR program (33) then was used to distinguish between SNPs located in gene and intergenic intervals of the cattle genome. The program reports SNPs located within introns and exons of annotated genes as well as within 5′ and 3′ UTRs and SNPs upstream and downstream from gene positions. For SNPs present within exons, the information is provided if the SNP changed an amino acid.
Supplementary Material
Acknowledgments
The authors thank Mr. Gene McCoy for assistance with obtaining samples for DNA sequencing. Funding for this research was provided in part by the US Department of Agriculture Cooperative State Research Education and Extension Service, Livestock Genome Sequencing Initiative Grants 538 AG2009-34480-19875 and 538 AG 58-1265-0-031.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequences reported in this paper have been submitted to the National Center for Biotechnology Information (NCBI) dbSNP Short Genetic Variations database, http://www.ncbi.nlm.nih.gov/SNP/snp_viewBatch.cgi?sbid=1055441.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1114546109/-/DCSupplemental.
References
- 1.Goddard ME, Hayes BJ. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet. 2009;10:381–391. doi: 10.1038/nrg2575. [DOI] [PubMed] [Google Scholar]
- 2.Ron M, et al. Combining mouse mammary gland gene expression and comparative mapping for the identification of candidate genes for QTL of milk production traits in cattle. BMC Genomics. 2007;8:183. doi: 10.1186/1471-2164-8-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oltenacu PA, Algers B. Selection for increased production and the welfare of dairy cows: Are new breeding goals needed? Ambio. 2005;34:311–315. [PubMed] [Google Scholar]
- 4.Matukumalli LK, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE. 2009;4:e5350. doi: 10.1371/journal.pone.0005350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elsik CG, et al. Bovine Genome Sequencing and Analysis Consortium The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science. 2009;324:522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norman HD, Wright JR, Hubbard SM, Miller RH, Hutchison JL. Reproductive status of Holstein and Jersey cows in the United States. J Dairy Sci. 2009;92:3517–3528. doi: 10.3168/jds.2008-1768. [DOI] [PubMed] [Google Scholar]
- 7.Da Y, et al. 5th World Congress on Genetics Applied to Livestock Production. (Guelph, Canada), Vol 21, 1994. The dairy bull DNA repository: A resource for mapping quantitative trait loci; pp. 229–232. [Google Scholar]
- 8.Kim YS, Stephan W. Allele frequency changes in artificial selection experiments: Statistical power and precision of QTL mapping. Genet Res. 1999;73(2):177–184. [Google Scholar]
- 9.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibbs RA, et al. Bovine HapMap Consortium Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009;324:528–532. doi: 10.1126/science.1167936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frazer KA, et al. International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vergara CI, et al. A Six-SNP haplotype of ADAM33 is associated with asthma in a population of Cartagena, Colombia. Int Arch Allergy Immunol. 2010;152(1):32–40. doi: 10.1159/000260081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lasky-Su J, et al. Genome-wide association scan of quantitative traits for attention deficit hyperactivity disorder identifies novel associations and confirms candidate gene associations. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1345–1354. doi: 10.1002/ajmg.b.30867. [DOI] [PubMed] [Google Scholar]
- 15.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinlan AR, Stewart DA, Strömberg MP, Marth GT. Pyrobayes: An improved base caller for SNP discovery in pyrosequences. Nat Methods. 2008;5:179–181. doi: 10.1038/nmeth.1172. [DOI] [PubMed] [Google Scholar]
- 18.Liu GE, et al. Analysis of recent segmental duplications in the bovine genome. BMC Genomics. 2009;10:571. doi: 10.1186/1471-2164-10-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daetwyler HD, Wiggans GR, Hayes BJ, Woolliams JA, Goddard ME. Imputation of missing genotypes from sparse to high density using long-range phasing. Genetics. 2011;189:317–327. doi: 10.1534/genetics.111.128082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu ZL, Fritz ER, Reecy JM. AnimalQTLdb: A livestock QTL database tool set for positional QTL information mining and beyond. Nucleic Acids Res. 2007;35(Database issue):D604–D609. doi: 10.1093/nar/gkl946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rincón G, et al. Fine mapping and association analysis of a quantitative trait locus for milk production traits on Bos taurus autosome 4. J Dairy Sci. 2009;92:758–764. doi: 10.3168/jds.2008-1395. [DOI] [PubMed] [Google Scholar]
- 22.Qanbari S, et al. A genome-wide scan for signatures of recent selection in Holstein cattle. Anim Genet. 2010;41:377–389. doi: 10.1111/j.1365-2052.2009.02016.x. [DOI] [PubMed] [Google Scholar]
- 23.Vierhout CN, Cassell BG, Pearson RE. Comparisons of cows and herds in two progeny testing programs and two corresponding states. J Dairy Sci. 1999;82:822–828. doi: 10.3168/jds.S0022-0302(99)75302-6. [DOI] [PubMed] [Google Scholar]
- 24.Roach JC, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montesano R, Sarközi R, Schramek H. Bone morphogenetic protein-4 strongly potentiates growth factor-induced proliferation of mammary epithelial cells. Biochem Biophys Res Commun. 2008;374:164–168. doi: 10.1016/j.bbrc.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Green KA, Nielsen BS, Castellino FJ, Rømer J, Lund LR. Lack of plasminogen leads to milk stasis and premature mammary gland involution during lactation. Dev Biol. 2006;299:164–175. doi: 10.1016/j.ydbio.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 27.Leitner G, et al. Changes in milk composition as affected by subclinical mastitis in sheep. J Dairy Sci. 2004;87:46–52. doi: 10.3168/jds.S0022-0302(04)73140-9. [DOI] [PubMed] [Google Scholar]
- 28.Drögemüller C, et al. Identification of the bovine Arachnomelia mutation by massively parallel sequencing implicates sulfite oxidase (SUOX) in bone development. PLoS Genet. 2010;6:e1001079. doi: 10.1371/journal.pgen.1001079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heyen DW, et al. A genome scan for QTL influencing milk production and health traits in dairy cattle. Physiol Genomics. 1999;1:165–175. doi: 10.1152/physiolgenomics.1999.1.3.165. [DOI] [PubMed] [Google Scholar]
- 30.Marth GT, et al. A general approach to single-nucleotide polymorphism discovery. Nat Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- 31.Kong A, et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat Genet. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacCluer JW, Vandeburg JL, Read B, Ryder OA. Pedigree analysis by computer simulation. Zoo Biol. 1986;5(2):149–160. [Google Scholar]
- 33.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.