

Abstract
Hydrophobicity is thought to underlie self-assembly in biological systems. However, the protein surface comprises hydrophobic and hydrophilic patches, and understanding the impact of such a chemical heterogeneity on protein self-assembly in water is of fundamental interest. Here, we report structural and thermodynamic investigations on the dimer formation of full-length amyloid-β proteins in water associated with Alzheimer’s disease. Spontaneous dimerization process—from the individual diffusive regime at large separations, through the approach stage in which two proteins come close to each other, to the structural adjustment stage toward compact dimer formation—was captured in full atomic detail via unguided, explicit-water molecular dynamics simulations. The integral-equation theory of liquids was then applied to simulated protein structures to analyze hydration thermodynamic properties and the water-mediated interaction between proteins. We demonstrate that hydrophilic residues play a key role in initiating the dimerization process. A long-range hydration force of enthalpic origin acting on the hydrophilic residues provides the major thermodynamic force that drives two proteins to approach from a large separation to a contact distance. After two proteins make atomic contacts, the nature of the water-mediated interaction switches from a long-range enthalpic attraction to a short-range entropic one. The latter acts both on the hydrophobic and hydrophilic residues. Along with the direct protein–protein interactions that lead to the formation of intermonomer hydrogen bonds and van der Waals contacts, the water-mediated attraction of entropic origin brings about structural adjustment of constituent monomer proteins toward the formation of a compact dimer structure.
Keywords: solvation thermodynamics, dehydration, protein interface
Water-mediated interaction such as hydrophobic interaction plays a key role in protein folding and protein self-assembly (1–3), yet elucidating its physical and chemical basis still remains a challenge. Recent theoretical advances in understanding the hydrophobic effect have elucidated that the hydrophobic interaction between small apolar groups is markedly different from that between large hydrophobic solutes, with the cross-over length-scale being of the order of 10 Å (see, e.g., refs. 4 and 5 for review). However, most of previous studies employed solutes such as cylinders, plates, or hydrophobic chains, and a direct applicability of findings therefrom to proteins is not obvious. A major obstacle here is the fact that protein surfaces are both topologically and chemically heterogeneous (6, 7): Protein surfaces are irregular in shape and hydrophobic residues are laced with hydrophilic ones. The main focus of the present work is to contribute to the understanding of the impact of chemical heterogeneity on protein self-assembly in water via the study of solvation for a realistic system of protein and water.
Self-assembly of large hydrophobic solutes is thought to be driven by a drying transition of interfacial water (4, 5). It was demonstrated by simulation that, when two large hydrophobic plates are brought together to a certain critical distance, interfacial water exhibits a drying transition (8). Such a dewetting transition gives rise to a strong driving force for hydrophobic collapse as confirmed by theory and subsequent simulations (9–11). Possible relevance of the dewetting-induced collapse to protein self-assembly has also been examined (12–14). (See ref. 15 for a critical view on the potential role of the dewetting-induced self-assembly, refs. 16 and 17 on the “lubrication” scenario in the absence of the dewetting transition, and ref. 18 on the possible role of water confined in hydrophobic pores.) The existence of the dewetting transition was found to be sensitive to the details of protein–water interactions (12–14). In particular, the surface chemical heterogeneity is expected to have a profound effect. Indeed, recent works on model hydrophilic plates have addressed a related issue and shown that the presence of hydrophilic group provokes wetting against dewetting behavior (19–21). This line of study was extended to a protein with flattened surface topology, where a high sensitivity of the dewetting to the local hydrophobicity/hydrophilicity of the planar protein interface was demonstrated (22).
Here, we expand these pioneering works on the nature of the protein self-assembly in water, and this is done in twofold. Firstly, we deal with a realistic protein whose surface is both topologically and chemically heterogeneous. Secondly, a whole dimerization process of two proteins is examined from the individual diffusive regime at large separations, through the approach stage in which two proteins come close to each other, to the structural adjustment stage toward compact dimer formation. Thereby, we address the role of hydrophobic and hydrophilic residues in each stage of the self-assembly process. To this end, we present structural and thermodynamic investigations on the dimer formation of full-length, 42-residue amyloid-β (Aβ42) proteins in water. Aggregation of Aβ proteins is linked to Alzheimer’s disease (23). Aβ dimer is of particular interest because it is the smallest neurotoxic Aβ oligomeric species as revealed by a recent clinical study (24). So far, computational studies have been carried out for the dimer formation of Aβ fragments (14, 25, 26) and for the Aβ dimerization in a continuum solvent (27, 28) due to the heavy computational load. Here, a spontaneous Aβ42 dimerization process was captured in full atomic detail via an unbiased, explicit-water molecular dynamics (MD) simulations starting from two isolated Aβ42 monomers. The simulated dimer structure was validated through the collision cross-section measured by the ion-mobility mass spectrometry (29). We then performed solvation thermodynamic and potential of mean force analyses by applying the integral-equation theory of liquids (30, 31) to simulated protein conformations. The component analysis of the hydration thermodynamic properties (32, 33) was further conducted to elucidate how hydrophobic and hydrophilic patches on the protein surface manifest themselves in the thermodynamic driving force for the Aβ42 self-assembly in water.
Results
Structural Characteristics of the Dimerization Process.
We carried out Aβ42 dimerization simulations at the temperature T = 300 K and the pressure P = 1 bar under neutral pH (see Materials and Methods and SI Text). The initial monomer structure was taken from our previous study (34), whose collision cross-section is computed to be 752 Å2 that is in fair agreement with the experimental value 702 Å2 (29) considering the size and complexity of Aβ42 protein. Two monomers were initially placed at 45 Å apart from each other with a random orientation, and no artificial attraction force was employed between them. The dimerization process was monitored via the center-of-mass (COM) distance between two monomers, the number of intermonomer heavy atom contacts, and the collision cross-section (Fig. 1 A–C). Three independent dimerization simulations were performed with different random initial relative orientations and velocities. All three trajectories exhibit a similar Aβ42 dimerization process, except the time scales for the dimerization events vary (see Figs. S1–S4). In the following, we present the results based on the single representative trajectory.
Fig. 1.

Structural characteristics of the Aβ42 dimerization process. (A) COM distance between two Aβ42 monomers, (B) the number of intermonomer heavy atom contacts (the heavy atom contact is counted when the distance between two heavy atoms is less than 5.4 Å), and (C) the collision cross-section as a function of time. Vertical dashed lines refer to 32 and 47 ns separating the diffusive regime (0 to 32 ns), the approach stage (32 to 47 ns, colored by light yellow), and the structural adjustment stage (47 to 100 ns, colored by light orange). (D) Representative Aβ42 dimer conformations during the dimerization process. Each monomer structure is color-coded according to the sequence, ranging from blue to red at N and C termini, respectively.
The Aβ42 dimerization occurs via two stages—the approach and structural adjustment stages—that stem out from diffusive dynamics (Fig. 1). Up to around 32 ns, two monomers exhibit individual diffusive motions. The collision cross-section stays around 1,500 Å2 in this time regime that is simply a double of that for monomer (752 Å2). A transient contact is formed at around 18 ns, but it readily dissociates into two diffusing monomers. After 32 ns, a significant decrease in the COM distance is observed up to 47 ns, and we refer to this time regime as the approach stage. A large drop in the collision cross-section also occurs here that indicates that a considerable structural overlap between two monomers starts to be developed. The approach stage is not monotonous and involves relative reorientational dynamics of two monomers. The second contact that is formed at 33 ns dissociates at 42 ns because relative orientation of two monomers in this contact does not allow the development of sufficient intermonomer contacts to stabilize the dimer structure. Two monomers start to contact again at 45 ns with a different relative orientation. The number of intermonomer heavy atom contacts further increases after 47 ns, and two monomers do not dissociate till the end of our 100 ns simulation. The time regime between 47 and 100 ns is the structural adjustment stage because the most distinctive feature here is the conformational adjustment of two monomers to form favorable interactions between them. Such conformational adjustment leads to a more compact dimer structure that is reflected in the further decrease in the collision cross-section during the structural adjustment stage. The computed collision cross-section (1,287 Å2) of the final Aβ42 dimer structure is in good agreement with the experimental value (1,256 Å2) (29), and a similar level of agreement is achieved from other trajectories (Figs. S1 and S3).
Thermodynamic Features of the Dimerization Process.
The two-stage nature of the Aβ42 dimerization process has its thermodynamic counterpart. We computed the total protein internal energy 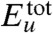 (the sum of intramonomer and intermonomer energies) directly from the force field used for the simulations. Solvation free energy Δμtot and its enthalpy (Δhtot) and entropy (TΔstot) components along the simulation trajectory were calculated using the integral-equation theory of liquids (30, 31) (see SI Text). The approach stage of the dimerization (32 to 47 ns) is characterized by the increase in the protein internal energy and by the decrease in the solvation free energy (Fig. 2
A and B). The decrease in the solvation free energy is dominated by its enthalpy component (Fig. 2C). On the other hand, just the opposite changes are observed in the structural adjustment stage (47 to 100 ns). The decrease in the protein internal energy here reflects the energetic stabilization of the dimer structure. These thermodynamic features are reproduced in other independent trajectories (Figs. S2 and S4).
(the sum of intramonomer and intermonomer energies) directly from the force field used for the simulations. Solvation free energy Δμtot and its enthalpy (Δhtot) and entropy (TΔstot) components along the simulation trajectory were calculated using the integral-equation theory of liquids (30, 31) (see SI Text). The approach stage of the dimerization (32 to 47 ns) is characterized by the increase in the protein internal energy and by the decrease in the solvation free energy (Fig. 2
A and B). The decrease in the solvation free energy is dominated by its enthalpy component (Fig. 2C). On the other hand, just the opposite changes are observed in the structural adjustment stage (47 to 100 ns). The decrease in the protein internal energy here reflects the energetic stabilization of the dimer structure. These thermodynamic features are reproduced in other independent trajectories (Figs. S2 and S4).
Fig. 2.

Thermodynamics of the Aβ42 dimerization process. (A) Total protein internal energy 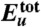 , (B) solvation free energy Δμtot, and (C) solvation enthalpy Δhtot and solvation entropy -TΔstot for dimer conformation along the simulation trajectory. In these and the following panels, the initial values are set to zero, and vertical dashed lines refer to 32 and 47 ns indicating the approach stage (32 to 47 ns, colored by light yellow) and the structural adjustment stage (47 to 100 ns, colored by light orange). By combining
, (B) solvation free energy Δμtot, and (C) solvation enthalpy Δhtot and solvation entropy -TΔstot for dimer conformation along the simulation trajectory. In these and the following panels, the initial values are set to zero, and vertical dashed lines refer to 32 and 47 ns indicating the approach stage (32 to 47 ns, colored by light yellow) and the structural adjustment stage (47 to 100 ns, colored by light orange). By combining 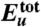 and Δμtot, we obtain a free energy
and Δμtot, we obtain a free energy  shown in D whose average over protein conformations yields the Gibbs free energy up to the protein configurational entropy (35). Its enthalpy component (
shown in D whose average over protein conformations yields the Gibbs free energy up to the protein configurational entropy (35). Its enthalpy component (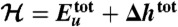 ) and entropy component (
) and entropy component (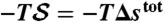 ) are presented in E and F, respectively. (Because the protein configurational entropy is not taken into account in
) are presented in E and F, respectively. (Because the protein configurational entropy is not taken into account in 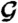 , its entropy component is solely given by the solvation entropy.) Red horizontal bars in D–F represent averages over each 5-ns time interval.
, its entropy component is solely given by the solvation entropy.) Red horizontal bars in D–F represent averages over each 5-ns time interval.
Enthalpy and entropy components of the Gibbs free energy reveal the thermodynamic driving force for the dimerization process. By combining the protein internal energy and the solvation free energy, we obtain a free energy  whose average over protein conformations yields the Gibbs free energy up to the protein configurational entropy (35). (The contribution from the pressure–volume term was found to be negligible, in which case Gibbs and Helmholtz free energies can be used interchangeably.) Because the protein configurational entropy would decrease upon dimerization, the driving force for the dimerization must come from
whose average over protein conformations yields the Gibbs free energy up to the protein configurational entropy (35). (The contribution from the pressure–volume term was found to be negligible, in which case Gibbs and Helmholtz free energies can be used interchangeably.) Because the protein configurational entropy would decrease upon dimerization, the driving force for the dimerization must come from  . In fact, the free energy
. In fact, the free energy 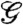 decreases as the dimerization proceeds (Fig. 2D and Table 1). We find that the approach stage is mainly driven by its enthalpy component
decreases as the dimerization proceeds (Fig. 2D and Table 1). We find that the approach stage is mainly driven by its enthalpy component  (Fig. 2E and Table 1). Because the solvation enthalpy decreases while the protein internal energy increases in this stage, it is the solvation enthalpy that initiates the approach stage of the dimerization. On the other hand, the enthalpy
(Fig. 2E and Table 1). Because the solvation enthalpy decreases while the protein internal energy increases in this stage, it is the solvation enthalpy that initiates the approach stage of the dimerization. On the other hand, the enthalpy 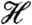 in the structural adjustment stage increases because the increase in the solvation enthalpy is found to prevail over the decrease in the protein internal energy. It is therefore a favorable increase in the solvation entropy (i.e., decrease in -TΔstot) that primarily drives the structural adjustment stage (Fig. 2F and Table 1).
in the structural adjustment stage increases because the increase in the solvation enthalpy is found to prevail over the decrease in the protein internal energy. It is therefore a favorable increase in the solvation entropy (i.e., decrease in -TΔstot) that primarily drives the structural adjustment stage (Fig. 2F and Table 1).
Table 1.
Thermodynamic changes in kcal/mol during the approach stage and the structural adjustment stage of the Aβ42 dimerization process
| Approach stage | Adjustment stage | |
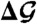 |
−22.0 | −9.4 |
 |
−30.7 | +14.7 |
 |
+8.6 | −24.1 |
Impact of Chemical Heterogeneity on the Dimerization Process.
Not only the surface of each Aβ42 monomer but also the contact interface of Aβ42 dimer are composed of hydrophobic and hydrophilic patches (Fig. 3). To quantify the impact of such a chemical heterogeneity on the driving force for the dimerization, we partitioned (see SI Text) the solvation enthalpy as well as the solvation entropy into the contributions from hydrophobic and hydrophilic residues (Fig. 4). Because the electrostatic interaction is much stronger than the van der Waals interaction, the solvation enthalpy change is dominated by the hydrophilic-residue contribution (Fig. 4A). On the other hand, the solvation entropy change is dominated by the nonelectrostatic component (Fig. 4B). Hence, both the hydrophobic and hydrophilic residues contribute to the solvation entropy change to a similar extent.
Fig. 3.

Chemical heterogeneity of the Aβ42 dimer interface. (A) The Aβ42 dimer structure at 100 ns, with hydrophobic residues colored by gray and hydrophilic residues by red, is first separated so that each monomer structure can be seen, and then each monomer is rotated so that the interface area is facing the reader. We used the hydrophobicity scales by Kyte and Doolittle (36) according to which A, F, L, M, I, and V are considered hydrophobic amino acids whereas D, E, H, K, N, Q, R, S, and G are considered hydrophilic. (B) Conceptual figure on how the contact area is calculated based on the solvent accessible surface area (SASA) of dimer and monomers. (C) Total contact area as a function of time and its components from hydrophobic-hydrophobic residue contacts, hydrophobic-hydrophilic-residue contacts, and hydrophilic-hydrophilic-residue contacts.
Fig. 4.

Component analysis of the solvation enthalpy and the solvation entropy. The solvation enthalpy Δhtot (A) and the solvation entropy TΔstot (B) are partitioned into nonelectrostatic and electrostatic components (Upper) and into hydrophobic- and hydrophilic-residue components (Lower). In all the panels, the initial values are set to zero, and vertical dashed lines refer to 32 and 47 ns indicating the approach stage (32 to 47 ns, colored by light yellow) and the structural adjustment stage (47 to 100 ns, colored by light orange). In the partitioning of TΔstot, horizontal bars representing averages over each 5-ns time interval are presented for clarity: original data before taking those averages are presented in Fig. S6. Although the solvation enthalpy change is dominated by the electrostatic component, the nonelectrostatic component prevails in the solvation entropy change. As a result, the hydrophilic-residue component dominates the solvation enthalpy change, whereas both the hydrophobic and hydrophilic residues contribute to the solvation entropy change to a similar extent.
A key concept to understand the changes in these thermodynamic quantities is the hydration/dehydration. When an amino acid residue gets more hydrated, a favorable residue–water interaction is formed at the expense of a residue–residue interaction. This leads to the gain (i.e., decrease) in the solvation enthalpy and the loss (increase) in the protein internal energy. This also explains why these quantities exhibit an antiphase correlation (Fig. 2 A and C). Just the opposite changes occur in the solvation enthalpy (increase) and the protein internal energy (decrease) when an amino acid residue is dehydrated. Solvation entropy change is also affected by the hydration status. The hydration is a kind of “trapping” of surrounding water molecules, and this leads to the decrease in the solvation entropy. On the other hand, the solvation entropy increases upon dehydration because trapped water molecules are “liberated” to the bulk.
The results shown in Fig. 4 consistently indicate that hydrophilic residues get more hydrated in the approach stage of the dimerization, whereas both hydrophobic and hydrophilic residues are dehydrated in the subsequent structural adjustment stage. These behaviors can be confirmed by the number of water molecules surrounding hydrophobic and hydrophilic residues directly obtained from the simulations (Fig. S5). Thus, it is those hydrophilic residues being more hydrated that enthalpically drive the approach stage of the Aβ42 dimerization process. On the other hand, the dehydration of both hydrophobic and hydrophilic residues entropically drives the formation of a compact dimer structure in the subsequent structural adjustment stage.
Nature of the Water-Mediated Attraction Between Proteins.
To investigate the nature of the water-mediated interaction between Aβ42 proteins, we consider the solvent contribution to the potential of mean force (PMF). The PMF is the reversible work required to bring two solutes from an infinite separation to some distance R apart, and its derivative with respect to R determines the average force acting between them (37). For proteins, the PMF depends also on the relative orientation as well as on the protein conformation, but such dependences shall not be resolved here to ease the analysis. In this case, the solvent contribution to the PMF is given by (37)
Here Δμtot(r1,r2; R), simply denoted as Δμtot so far for brevity, represents the solvation free energy when two monomers having configurations r1 and r2 are separated by a COM distance R. Δμm(ri) (i = 1,2) denotes the solvation free energy for an isolated monomer. The quantity δΔμ(R) accounts for the water-mediated interaction between two monomers including the hydrophobic interaction (37). δΔμ(R) comprises its enthalpy [δΔh(R)] and entropy [-TδΔs(R)] components. Unlike Δμtot, which is concerned with the hydration of whole protein surfaces, δΔμ is associated with that in the interfacial region. (Notice a similarity between the definition of δΔμ and that of the contact area explained in Fig. 3B).
Let us consider the time intervals between 32 and 35 ns and between 44 and 47 ns where a large drop in the intermonomer COM distance is observed and two monomers start to contact each other (Fig. 5A). As mentioned above, it is the decrease in the solvation enthalpy that mainly drives two monomers to approach each other. Fig. 5B confirms that the enthalpic component δΔh(R) of the water-mediated interaction is in fact attractive when two monomers are approaching from a large separation. The long-range nature of this attractive interaction reflects the electrostatic origin of the solvation enthalpy change. Because the solvation enthalpy change is dominated by the hydrophilic-residue contribution, such a long-range water-mediated force acts on the hydrophilic residues. This demonstrates a key role of the hydrophilic residues in initiating the dimerization.
Fig. 5.

Solvent contribution to the potential of mean force between two Aβ42 proteins. (A) We focus on the time intervals between 32 and 35 ns (Left) and between 44 and 47 ns (Right) where a large drop in the intermonomer COM distance is observed and two monomers start to contact each other. In all the panels, data points referring to the time regimes before and after two monomers make atomic contacts are distinguished by blue and red colors, respectively. The COM distance at which the contacts start differs in Left and Right because it depends also on the relative orientation of two approaching monomers (see Fig. 1D). (B,C) Parametric plots for the solvation enthalpy component δΔh(R) and the solvation entropy component -TδΔs(R) of the potential of mean force versus the COM distance R. The attraction (repulsion) is expressed by a negative (positive) change in these functions as the COM distance R is decreased. These functions exhibit a multivalued feature because they are projected onto the R axis without resolving, e.g., the relative-orientation dependence of two monomers. Both δΔh(R) and -TδΔs(R) for large R are attractive before two monomers make atomic contacts (blue data points), but the magnitude of the former is much stronger than that of the latter [notice different ordinate scales for δΔh(R) and -TδΔs(R)]. After two monomers make atomic contacts (red data points), water molecules in the interfacial region have to be dehydrated. Such a dehydration penalty shows up as a repulsive barrier in δΔh(R) for small R. On the other hand, the dehydration leads to a favorable increase in the solvation entropy, which results in the attractive portion in -TδΔs(R) for small R.
The attractive water-mediated force switches from a long-range enthalpic force to a short-range entropic one after two monomers make atomic contacts. Water molecules in the interfacial region have to be dehydrated when the intermonomer contacts are formed. The dehydration leads to an increase in the solvation enthalpy and shows up as a repulsive barrier in δΔh(R) for small R (Fig. 5B). On the other hand, the solvation entropy increases upon dehydration, and such a favorable change results in the attractive portion in the entropy component -TδΔs(R) of the water-mediated interaction (Fig. 5C). Due to the nonelectrostatic origin of the solvation entropy change, this attractive force is of short range. Such a short-range entropic force acts both on the hydrophobic and hydrophilic residues because they contribute to the solvation entropy change to a similar extent.
When two proteins make atomic contacts, direct protein–protein interactions come into play. The formation of intermonomer hydrogen bonds and van der Waals contacts energetically stabilizes the contact dimer structure (Fig. 6 A and B). Together with the entropic water-mediated attraction just mentioned (Fig. 6C), these favorable changes in the protein internal energy and solvation entropy overcome an unfavorable increase in the solvation enthalpy due to the dehydration and bring about structural adjustment of constituent monomer proteins toward the formation of a compact dimer structure (Fig. 6D).
Fig. 6.

Direct protein–protein interactions and the water-mediated attraction responsible for the compact dimer formation. (A) Presence (indicated by vertical bars) of intermonomer hydrogen bonds, (B) the intermonomer van der Waals interaction energy, and (C) the solvation entropy component -TδΔs of the PMF plotted as a function of time. In these panels, time regimes where intermonomer heavy atom contacts are present (see Fig. 1B) are colored by light green. In A, A(X)-B(Y) indicates that atom X in residue A of monomer 1 makes a hydrogen bond with atom Y in residue B of monomer 2. (D) Aβ42 dimer structures at 47, 53, 76, and 100 ns with surrounding water molecules. Two monomers are colored by yellow and cyan, respectively, and those residues that are to make intermonomer heavy atom contacts at 100 ns are shown in sphere representation. Water molecules are drawn in stick representation (O: red, H: white). Direct protein–protein interactions lead to the formation of intermonomer hydrogen bonds and van der Waals contacts. Concomitant dehydration of interfacial water gives rise to a water-mediated attraction of entropic origin quantified by -TδΔs. These favorable changes in the protein internal energy and the solvation entropy prompt the formation of compact dimer structure.
Discussion
The nature of the water-mediated interaction depends not only on the solute size but also on the surface chemical character. Here, we investigate the impact of the surface chemical heterogeneity (hydrophobic and hydrophilic) on the protein self-assembly in water via structural and thermodynamic analyses of the Aβ42 dimerization process. Because order parameters characterizing the Aβ42 dimerization pathway are not known a priori and protein reorientational and conformational fluctuations can significantly affect the features of protein self-assembly, a use of the thermodynamic perturbation technique is not appropriate, which has been conventionally adopted in the study of nanoscale plates as a function of the intersolute separation under prespecified relative orientation (see, e.g., ref. 8). Unbiased dimerization simulations—from the initial approach stage out of large separations to the structural adjustment stage toward compact dimer formation—are therefore carried out in full atomic detail. These are explicit-water MD simulations in which such a spontaneous Aβ42 dimerization process is captured starting from two isolated monomers. Having those simulated protein conformations along the dimerization pathway allows us to analyze the hydration thermodynamic properties and the water-mediated interaction with the help of the liquid integral-equation theory.
We demonstrate a crucial role of hydrophilic residues in initiating the dimerization process. A long-range hydration force of electrostatic and enthalpic origin acting on the hydrophilic residues is the major thermodynamic force that drives two proteins to approach from a large separation to a contact distance. This is in contrast to the traditional view that the self-assembly in water is initiated by the hydrophobic collapse. The importance of the water-mediated interaction between the hydrophilic residues has also been argued for the protofilament formation (38). Protein conformational fluctuations play an important role here because they alter the hydration status of the surface hydrophilic residues (32). In fact, the major thermodynamic driving force—the decrease in the solvation enthalpy—is provided by those hydrophilic residues being more hydrated during the approach of two monomers. The approach stage also involves protein reorientational dynamics in pursuit of the relative orientation of two monomers that allows the development of sufficient intermonomer contacts in the subsequent structural adjustment stage (Fig. 1D).
After two proteins make atomic contacts, the nature of the water-mediated attraction changes from a long-range enthalpic interaction to a short-range entropic one. This short-range water-mediated attraction acts both on the hydrophobic and hydrophilic residues. Along with the direct protein–protein interactions leading to the formation of intermonomer hydrogen bonds and van der Waals contacts, it induces the structural adjustment of constituent monomers toward the formation of a compact dimer structure. We observe the dewetting transition neither in the approach stage nor in the structural adjustment stage in our Aβ42 dimerization simulations: The water expulsion concomitantly occurs as the intermonomer contacts are formed. In the absence of a dewetting transition, a lubrication scenario was proposed in which water facilitates the formation of a compact protein structure rather than provides a driving force for self-assembly (16, 17). Our result differs from this scenario in that the “escape” of interfacial water toward the bulk in quest of entropy is thermodynamically driving the compact structure formation (Table 1). This is consistent with the observations in the previous related studies that the intermonomer hydrogen bondings provide only a limited contribution to the stability of the Aβ42 dimer (27, 28) and that the water-mediated attraction originating from the dehydration is more important to the stabilization of the dimer structure than the electrostatic interactions (25). (See SI Text for the structural comparison with the previous dimer studies).
Due to the heavy computational load to handle the dimerization of full-length Aβ42 proteins in explicit water, the simulation length of each trajectory is limited to 100 ns. This might be insufficient to sample potentially more stable Aβ42 dimer conformations. Furthermore, we did not observe the formation of the D23–K28 salt bridge in our simulations that is thought to play an important role in the self-association of Aβ proteins (39, 40). We note in this connection that the dimerization of amyloidogenic proteins has been suggested to be kinetically controlled involving metastable conformations (41). Whereas our limited simulations of having few rather than a more complete ensemble of trajectories do not allow us to analyze the structural relaxation time of the Aβ42 dimer, it should be much longer than microseconds estimated for short peptides (41). In light of the fast (sub-100 ns) association observed in our simulations, we expect that the Aβ42 dimerization is also kinetically controlled. Nevertheless, it makes sense to perform the solvation thermodynamic analysis as we presented here because the water relaxation occurs on the time scale of picoseconds, i.e., there are enough times for such an analysis to be applied to elucidate the nature of the water-mediated interaction during the Aβ42 dimerization process.
In summary, we demonstrate the impact of the surface chemical heterogeneity on the protein self-assembly in water. A long-range water-mediated attraction of enthalpic origin acting on the hydrophilic residues plays a key role in facilitating the protein dimerization. Subsequently, a short-range water-mediated attraction of entropic origin exerted on the hydrophobic as well as hydrophilic residues, along with the direct protein–protein interaction, imparts the stability of a resulting compact dimer structure.
Materials and Methods
The explicit-water Aβ42 dimerization simulations were carried out for 100 ns at 300 K and 1 bar under neutral pH with SANDER module of the AMBER9 program package using the ff99 force field (42). Solvation thermodynamic and potential of mean force analyses were then performed by applying the liquid integral-equation theory (30, 31) to simulated protein conformations. The component analysis of the hydration thermodynamic properties was further conducted using the method developed in ref. 32. Further details are provided in SI Text.
Supplementary Material
ACKNOWLEDGMENTS.
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (KRF-2008-313-C00404, 20090065791, and 2011-0012096). The authors would like to acknowledge the support from Korea Institute of Science and Technology supercomputing center.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1120646109/-/DCSupplemental.
References
- 1.Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 2.Tanford C. The hydrophobic effect and the organization of living matter. Science. 1978;200:1012–1018. doi: 10.1126/science.653353. [DOI] [PubMed] [Google Scholar]
- 3.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 4.Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 5.Berne BJ, Weeks JD, Zhou R. Dewetting and hydrophobic interaction in physical and biological systems. Annu Rev Phys Chem. 2009;60:85–103. doi: 10.1146/annurev.physchem.58.032806.104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng YK, Rossky PJ. Surface topography dependence of biomolecular hydrophobic hydration. Nature. 1998;392:696–699. doi: 10.1038/33653. [DOI] [PubMed] [Google Scholar]
- 7.Granick S, Bae SC. A curious antipathy for water. Science. 2008;322:1477–1478. doi: 10.1126/science.1167219. [DOI] [PubMed] [Google Scholar]
- 8.Wallqvist A, Berne BJ. Computer simulation of hydrophobic hydration forces on stacked plates at short range. J Phys Chem. 1995;99:2893–2899. [Google Scholar]
- 9.Lum K, Chandler D, Weeks JD. Hydrophobicity at small and large length scales. J Phys Chem B. 1999;103:4570–4577. [Google Scholar]
- 10.Huang DM, Chandler D. Temperature and length scale dependence of hydrophobic effects and their possible implications for protein folding. Proc Natl Acad Sci USA. 2000;97:8324–8327. doi: 10.1073/pnas.120176397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang X, Margulis CJ, Berne BJ. Dewetting-induced collapse of hydrophobic particles. Proc Natl Acad Sci USA. 2003;100:11953–11958. doi: 10.1073/pnas.1934837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou R, Huang X, Margulis CJ, Berne BJ. Hydrophobic collapse in multidomain protein folding. Science. 2004;305:1605–1609. doi: 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- 13.Liu P, Huang X, Zhou R, Berne BJ. Observation of a dewetting transition in the collapse of the melittin tetramer. Nature. 2005;437:159–162. doi: 10.1038/nature03926. [DOI] [PubMed] [Google Scholar]
- 14.Krone MG, et al. Role of water in mediating the assembly of Alzheimer amyloid-β Aβ 16–22 protofilaments. J Am Chem Soc. 2008;130:11066–11072. doi: 10.1021/ja8017303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ball P. Water as an active constituent in cell biology. Chem Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- 16.Cheung MS, Garcia AE, Onuchic JN. Protein folding mediated by solvation: Water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci USA. 2002;99:685–690. doi: 10.1073/pnas.022387699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shea J-E, Onuchic JN, Brooks CL., III Probing the folding free energy landscape of the src-SH3 protein domain. Proc Natl Acad Sci USA. 2002;99:16064–16068. doi: 10.1073/pnas.242293099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasaiah JC, Garde S, Hummer G. Water in nonpolar confinement: From nanotubes to proteins and beyond. Annu Rev Phys Chem. 2008;59:713–740. doi: 10.1146/annurev.physchem.59.032607.093815. [DOI] [PubMed] [Google Scholar]
- 19.Giovambattista N, Rossky PJ, Debenedetti PG. Effect of pressure on the phase behavior and structure of water confined between nanoscale hydrophobic and hydrophilic plates. Phys Rev E. 2006;73:041604. doi: 10.1103/PhysRevE.73.041604. [DOI] [PubMed] [Google Scholar]
- 20.Giovambattista N, Debenedetti PG, Rossky PJ. Hydration behavior under confinement by nanoscale surfaces with patterned hydrophobicity and hydrophilicity. J Phys Chem C. 2007;111:1323–1332. [Google Scholar]
- 21.Hua L, Zangi R, Berne BJ. Hydrophobic interactions and dewetting between plates with hydrophobic and hydrophilic domains. J Phys Chem C. 2009;113:5244–5253. [Google Scholar]
- 22.Giovambattista N, Lopez CF, Rossky PJ, Debenedetti PG. Hydrophobicity of protein surfaces: Separating geometry from chemistry. Proc Natl Acad Sci USA. 2008;105:2274–2279. doi: 10.1073/pnas.0708088105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardy JA, Higgins GA. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 24.Shankar GM, et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tarus B, Straub JE, Thirumalai D. Probing the initial stage of aggregation of the Aβ10–35-protein: Assessing the propensity for peptide dimerization. J Mol Biol. 2005;345:1141–1156. doi: 10.1016/j.jmb.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 26.Gnanakaran S, Nussinov R, Garcia AE. Atomic-level description of amyloid β-dimer formation. J Am Chem Soc. 2006;128:2158–2159. doi: 10.1021/ja0548337. [DOI] [PubMed] [Google Scholar]
- 27.Urbanc B, et al. Molecular dynamics simulation of amyloid β dimer formation. Biophys J. 2004;87:2310–2321. doi: 10.1529/biophysj.104.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitternacht S, Staneva I, Härd T, Irbäck A. Monte Carlo study of the formation and conformational properties of dimers of Aβ42 variants. J Mol Biol. 2011;410:357–367. doi: 10.1016/j.jmb.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein SL, et al. Amyloid β-protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovalenko A. In: Molecular Theory of Solvation. Hirata F, editor. Dordrecht, The Netherlands: Kluwer Academic; 2003. pp. 164–275. [Google Scholar]
- 31.Imai T, Harano Y, Kinoshita M, Kovalenko A, Hirata F. A theoretical analysis on hydration thermodynamics of proteins. J Chem Phys. 2006;125:024911. doi: 10.1063/1.2213980. [DOI] [PubMed] [Google Scholar]
- 32.Chong S-H, Ham S. Atomic decomposition of the protein solvation free energy and its application to amyloid-beta protein in water. J Chem Phys. 2011;135:034506. doi: 10.1063/1.3610550. [DOI] [PubMed] [Google Scholar]
- 33.Chong S-H, Lee C, Kang G, Park M, Ham S. Structural and thermodynamic investigations on the aggregation and folding of acylphosphatase by molecular dynamics simulations and solvation free energy analysis. J Am Chem Soc. 2011;133:7075–7083. doi: 10.1021/ja1116233. [DOI] [PubMed] [Google Scholar]
- 34.Lee C, Ham S. Characterizing amyloid-beta protein misfolding from molecular dynamics simulations with explicit water. J Comput Chem. 2011;32:349–355. doi: 10.1002/jcc.21628. [DOI] [PubMed] [Google Scholar]
- 35.Chong S-H, Ham S. Configurational entropy of protein: A combined approach based on molecular simulation and integral-equation theory of liquids. Chem Phys Lett. 2011;504:225–229. [Google Scholar]
- 36.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 37.Ben-Naim A. Hydrophobic Interactions. New York: Plenum; 1980. [Google Scholar]
- 38.Reddy G, Straub JE, Thirumalai D. Dry amyloid fibril assembly in a yeast prion peptide is mediated by long-lived structures containing water wires. Proc Natl Acad Sci USA. 2010;107:21459–21464. doi: 10.1073/pnas.1008616107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. A β40-Lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry. 2005;44:6003–6014. doi: 10.1021/bi0474867. [DOI] [PubMed] [Google Scholar]
- 40.Tarus B, Straub JE, Thirumalai D. Dynamics of Asp23-Lys28 salt-bridge formation in A β10–35 monomers. J Am Chem Soc. 2006;128:16159–16168. doi: 10.1021/ja064872y. [DOI] [PubMed] [Google Scholar]
- 41.Hwang W, Zhang S, Kamm RD, Karplus M. Kinetic control of dimer structure formation in amyloid fibrillogenesis. Proc Natl Acad Sci USA. 2004;101:12916–12921. doi: 10.1073/pnas.0402634101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Case DA, et al. AMBER9. San Francisco: University of California; 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.