

Abstract
More than 50 y of research have provided great insight into the physiology, metabolism, and molecular biology of Salmonella enterica serovar Typhimurium (S. Typhimurium), but important gaps in our knowledge remain. It is clear that a precise choreography of gene expression is required for Salmonella infection, but basic genetic information such as the global locations of transcription start sites (TSSs) has been lacking. We combined three RNA-sequencing techniques and two sequencing platforms to generate a robust picture of transcription in S. Typhimurium. Differential RNA sequencing identified 1,873 TSSs on the chromosome of S. Typhimurium SL1344 and 13% of these TSSs initiated antisense transcripts. Unique findings include the TSSs of the virulence regulators phoP, slyA, and invF. Chromatin immunoprecipitation revealed that RNA polymerase was bound to 70% of the TSSs, and two-thirds of these TSSs were associated with σ70 (including phoP, slyA, and invF) from which we identified the −10 and −35 motifs of σ70-dependent S. Typhimurium gene promoters. Overall, we corrected the location of important genes and discovered 18 times more promoters than identified previously. S. Typhimurium expresses 140 small regulatory RNAs (sRNAs) at early stationary phase, including 60 newly identified sRNAs. Almost half of the experimentally verified sRNAs were found to be unique to the Salmonella genus, and <20% were found throughout the Enterobacteriaceae. This description of the transcriptional map of SL1344 advances our understanding of S. Typhimurium, arguably the most important bacterial infection model.
Keywords: transcriptional mapping, noncoding RNA, posttranscriptional regulation, pathogenicity, genome sequence
Large numbers of human deaths are caused by Salmonella bacteria, particularly in developing countries. Typhoidal serovars kill ∼244,000 people (1), and nontyphoidal serovars kill ∼155,000 people each year (2). The number of cases of human Salmonellosis in the United States remains at 17.6 per 100,000 people, a rate that is as high today as it was a decade ago (3). Indeed, half of the recent outbreaks of food-borne disease in England and Wales were caused by Salmonella enterica, more than any other pathogen (4). The S. enterica species is divided into >2,300 serovars that can be distinguished on the basis of surface-exposed lipopolysaccharide and flagellin molecules (5). One serovar, Salmonella Typhimurium, causes a considerable level of human disease in developed nations, and variants of S. Typhimurium have arisen in Africa that cause a highly invasive form of nontyphoidal Salmonellosis (6, 7).
After ingestion by a mammalian host, S. Typhimurium progresses through the diverse environments of the gastrointestinal tract and subsequently crosses the intestinal epithelial barrier. Its ability to persist within macrophages as well as the gall bladder makes it a formidable pathogen that causes both acute and chronic infections (8). The ease of genetic manipulation coupled with a detailed understanding of core metabolism has made S. Typhimurium the preeminent model for studying host–pathogen interactions and intracellular survival (9). Unfortunately, reliance upon an Escherichia coli archetype and the paucity of well-annotated genome sequences of virulent S. Typhimurium strains have limited the analysis of regulatory functions in relation to S. Typhimurium infection. The majority of gene regulatory studies have focused on the Salmonella pathogenicity islands (SPI)1 and SPI2, but it has become clear from transcriptomic analyses that additional global changes in metabolic and physiological processes are required for adaptation to host environments (10). To gain insight into host–pathogen interactions we must characterize the genetic regulatory programs that allow S. Typhimurium to cause infection. Despite a decade of intensive research and the beginning of systems-level analysis (11), we still have many unanswered questions about the global transcriptional networks of S. Typhimurium. For example, where are gene promoters located? Is antisense transcription widespread? What is the complement of small regulatory RNAs expressed by S. Typhimurium? To answer these questions, we defined the global transcriptional map of the virulent S. Typhimurium strain SL1344.
Until recently, transcriptomic analysis of S. Typhimurium has relied upon DNA microarray-based technology (12). Now, RNA sequencing (RNA-seq) has become the ideal technique for visualizing transcription at the genomic level (13–15). As well as allowing comparative gene expression, RNA-seq can also identify novel transcripts at the single-nucleotide level. Individual −10 and −35 promoter motifs can be found by characterizing the first nucleotide of a transcript, termed the transcriptional start site (TSS). Recently, a novel differential RNA-sequencing (dRNA-seq) approach was developed to discover TSSs at a genome-wide scale (16). It uses the 5′-monophosphate–dependent terminator exonuclease (TEX) that specifically degrades 5′-monophosphorylated RNA species such as processed RNA including mature rRNA and tRNA, whereas 5′-triphosphorylated RNA species (primary transcripts) are protected and remain intact. This approach results in an enrichment of primary transcripts, allowing the TSSs to be identified by comparison of the TEX-treated with untreated libraries.
We used a combination of chromatin immunoprecipitation coupled with microarray hybridization (ChIP-chip), RNA-seq, dRNA-seq, and Hfq coimmunoprecipitation coupled with RNA-seq (Hfq-coIP-seq) to generate a robust and comprehensive picture of the transcriptional organization of the genome of S. enterica. Key insights include the identification of the 832 σ70-associated promoters in S. Typhimurium, as well as the discovery of 60 small RNAs.
Results and Discussion
The SL1344 Genome.
S. Typhimurium strain SL1344 has played an important role in the analysis of Salmonella infection, starting with its use as a virulent Salmonella strain for vaccine research. The ancestral strain ST4/74 was originally isolated from the bowel of a calf with Salmonellosis (17) and used by Bruce Stocker to generate a histidine auxotroph named SL1344 (18).
Here, we report the complete and annotated genome sequence of S. Typhimurium SL1344 (Fig. S1A and Dataset S1). SL1344 shares a similar GC ratio with other S. enterica serovars, 52.3%, which is significantly higher than that of other enteric species like E. coli (19). The SL1344 genome contains 4,742 protein-coding genes (Dataset S1). A total of 4,530 of these genes are present on the chromosome, and 212 genes are encoded by three plasmids, pSLTSL1344, pCol1B9SL1344 (also known as p2), and pRSF1010SL1344. The plasmid pCol1B9SL1344 is responsible for horizontal gene transfer via conjugation to E. coli during infection of the murine gut (20). The relatively high proportion of regulatory and metabolic genes in S. Typhimurium contributes to the physiological versatility of this robust pathogen (Dataset S1) (21).
Comparative Genomics of SL1344.
To put the gene content of SL1344 into a broader context, we performed an iterative BLAST analysis against 31 sequenced enterobacterial genomes. The annotation of the resulting BLAST Atlas shows the 13 SPIs and five prophages present in the SL1344 genome and their conservation within the chromosomes of six S. Typhimurium strains and 13 other Salmonella serovars (Fig. S1B). The 13 SPI regions are absent from E. coli K12, from three other E. coli pathovars, and from four more disparate members of the Enterobacteriaceae (Shigella, Pectobacterium, Yersinia, and Serratia).
The first S. Typhimurium genome sequence was published in 2001 for the attenuated type strain, LT2 (22). The attenuation of strain LT2 is largely due to suboptimal translation of the RpoS (σ38) sigma factor (23). The SL1344 genome sequence confirmed that the rpoS coding sequence of SL1344 begins with an optimal ATG translational start at location 3,088,055. Comparison of the LT2 and SL1344 genome sequences identified 260 genes that are not present in LT2. The largest difference in gene complement is explained by the absence of the plasmids pCol1B9SL1344 and pRSF1010SL1344 and the phages Gifsy-2 and Fels-2 from LT2 and several other S. Typhimurium strains (Fig. S1B).
Identification of Transcriptional Start Sites Under Infection-Relevant Conditions.
A promoter is defined as a DNA sequence that binds RNA polymerase (RNAP) to initiate the transcription of RNA. To understand the transcriptional control of S. Typhimurium virulence genes that are required for infection we must determine the precise location of promoter regions. This process will allow transcriptional regulatory networks to be assembled and allow the DNA-binding motifs of different transcription factors to be identified. Promoter identification was previously done “one gene at a time,” and up to now promoters have been assigned to only 2% of S. Typhimurium genes (Dataset S2).
We used RNA-seq–based approaches to globally define the TSSs of S. Typhimurium grown to early stationary phase (ESP) (Fig. 1 and Dataset S2). ESP is an infection-relevant growth condition associated with high levels of expression of the SPI1 virulence genes that are responsible for invasion of epithelial cells (24). To ensure that the identified TSSs were robust and reproducible, we used five biological replicates of RNA-seq (including three dRNA-seq replicates) and a combination of 454 and Illumina-based sequence platforms (Figs. 1 and 2A). The identification of small regulatory RNAs was aided by the enrichment of one of the RNA samples for small RNA fragments (<500 nt). We complemented the standard RNA-seq protocol by using the flow cell reverse transcription sequencing (FRT-seq) approach; this method involved the synthesis of cDNA on the sequencing flow cell to improve cDNA library representation (25). The dRNA-seq technique identified examples of processed transcripts, such as the small RNAs ArcZ and RprA, and succeeded in precisely localizing the TSS to a single nucleotide (26, 27). Two FRT-seq sequencing reactions were conducted on one of the biological replicates, one of which was depleted for rRNA (Fig. 1A) (25). The sequencing statistics and the number of biological replicates are shown in Fig. 1B and Dataset S1. We mapped >12 million sequence reads uniquely to the S. Typhimurium SL1344 genome, amounting to 120-fold coverage. A total of ∼3.5 million (23%) of all sequenced reads mapped to the annotated coding sequences (CDS), whereas just ∼200,000 reads (1.75%) mapped antisense to CDS.
Fig. 1.

RNA-seq and ChIP-chip–based strategy to identify promoters, transcribed regions, and small RNAs. (A) The different cDNA libraries that were generated and sequenced in this study. (B) The distribution of mapped reads to different genome locations (IGR, intergenic region). Names of the 11 sequence datasets are defined in Dataset S1. The three libraries marked with TEX were treated with terminator exonuclease (TEX) to enrich the cDNA libraries for primary transcripts that carried a 5′ triphosphate (dRNA-seq). The suffix numbers link the “RNA-seq” (TEX-untreated) libraries with the appropriate TEX-enriched libraries. The RNA-seq* library sample was enriched for small RNA species (mirVana) before cDNA library generation. The FRT-seq_dep sample was depleted of rRNA, and both FRT-seq and FRT-seq_dep were done with RNA from the same biological replicate (Materials and Methods). (C) The workflow that uses the dRNA-seq, RNA-seq, and ChIP-chip data to identify small RNAs, TSSs, promoters, and transcribed regions throughout the chromosome of S. Typhimurium SL1344 (Rif, rifampicin; RNAP, RNA polymerase; TSS, transcriptional start site).
Fig. 2.
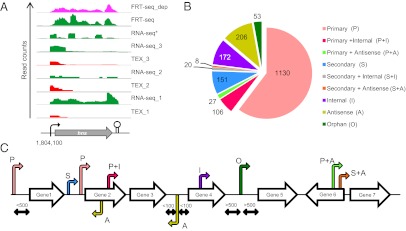
Global identification and categorization of transcriptional start sites (TSSs). (A) Visualization of cDNA sequencing reads obtained from RNA-seq, dRNA-seq, and FRT-seq experiments using the Integrated Genome Browser (IGB) version 6.5.3 (65) for the hns gene. The vertical “read count” scale for the TEX_1, RNA-seq_1, TEX_2, RNA-seq_2, FRT-seq, and FRT-seq_dep libraries is 0–10 sequencing reads and for TEX_3, RNA-seq_3, and RNA-seq* it is 0–100 reads. The TSS for the hns gene was identified at nucleotide 1,804,100 in the SL1344 chromosome. (B) Categorization of 1,873 TSSs identified in this study, according to C. The majority of TSSs were identified as primary starts (1,263/1,873 = 67%). (C) Schematic explanation of TSS categorization, as in ref. 16. The TSS abbreviations refer to the designations shown in B.
The dRNA-seq data often confirmed the TSSs that were already clearly apparent from RNA-seq and FRT-seq, as seen for the hns gene (Fig. 2A). When the location of the start of transcripts was not clear from RNA-seq and FRT-seq, the dRNA-seq became more important. A conservative approach was used to identify the precise nucleotide used for transcriptional initiation: The same “+1” nucleotide of each TSS was identified in at least two biological replicates using dRNA-seq. A total of 1,873 TSSs were classified into eight promoter categories (16) (Fig. 2 B and C). We assigned primary starts to 1,130 protein-coding genes of S. Typhimurium and 87 transcriptional starts were assigned to known or newly identified small RNAs (see below). We observed 206 TSSs for transcripts located antisense to ORFs and 172 internal starts, highlighting the complexity of transcription and gene expression in Salmonella.
Validation of S. Typhimurium TSSs.
The dRNA-seq approach has already been validated in Helicobacter, Synechocystis, and other organisms (16, 28–31), but not in S. Typhimurium. It was important to put our global TSS approach into the context of the wealth of the Salmonella literature. We found publications that described the TSSs of 57 genes. Fig. S2 and Dataset S2 show the overlap between 37 of the published transcriptional start sites that are present within our dRNA-seq dataset. Thirty-one of 37 transcriptional starts lie within ± 2nt of the published start, with 15 starts matching exactly.
To corroborate our approach, we performed a series of 5′-RACE experiments that unambiguously identified TSSs for 10 genes, namely invF, hilD, ompA, osmC, phoP, prgH, slyA, sodB, yfgE, and yibP (Fig. S2). The 5′-RACE data were in complete agreement with the TSSs, confirming that the 1,873 TSSs represent a robust database that describes the transcription of S. Typhimurium genes at ESP.
Reannotation of the SL1344 Genome Sequence.
The availability of experimental evidence describing the location of TSSs of S. Typhimurium led us to examine whether our data could be used to improve the accuracy of SL1344 CDS annotation. We found five examples of TSSs that lay downstream of predicted translational start sites, suggesting that an incorrect translational start had been annotated for cysJ, infC, himA, pps, and prfB. In addition, 17 small ORFs (sORFs) that have been experimentally confirmed in E. coli were found to be conserved in S. Typhimurium and various other Gram-negative bacteria (32–37) (Fig. S3 and Dataset S1). Transcripts of 9 of these sORFs were visible in the RNA-seq data, showing that these coding sequences were expressed during ESP (Dataset S1). The locations of the sORF-encoding genes and the genes with incorrect translational starts were reannotated on the SL1344 genome (Dataset S1).
Transcriptional Activity Across the SL1344 Chromosome.
Bacterial promoters are regions of DNA that bind RNA polymerase holoenzyme (E) and drive transcript initiation. To confirm that the identified TSSs were indeed associated with bacterial promoters, we experimentally defined the transcriptionally active areas of the S. Typhimurium chromosome. RNAP is an abundant protein complex in bacterial cells, with measurements varying between 2,600 and 13,000 molecules per cell (38, 39). We performed a ChIP-chip experiment with a monoclonal antibody that recognized the β-subunit of RNAP. A stringent approach was used to analyze the ChIP-chip data to identify the S. Typhimurium chromosomal regions that showed only reproducible binding of RNAP (Dataset S3). In total, 645 chromosomal regions showed dynamic binding of RNAP that extended across highly expressed operons such as those present in SPI1 (Fig. 3).
Fig. 3.

Integration of the S. Typhimurium transcriptome with genome-wide binding of RNAP and Sigma70 at early stationary phase (ESP). (A) Visualization of binding of the transcriptional machinery (ChIP-chip data: blue, Sigma70; purple, RNAP; orange, RNAP+rif) (Materials and Methods), in the context of the transcriptome, shown with RNA-seq (green) and dRNA-seq (red) datasets using IGB. The TEX+/− RNA-seq libraries of RNA-seq_3 are shown. (B) Detailed view of Salmonella pathogenicity island 1 (SPI1). (Upper) ChIP-chip data are presented as quantitative data in the top lane, with ChIPotle-identified binding sites depicted below each lane as bars. Binding of RNAP or Sigma70 was signified by more than twofold enrichment over input DNA. TSSs identified in SPI1 are depicted as black arrows in the TSS lanes. Most SPI1 genes are actively transcribed during growth at ESP. The TSSs of short (<100 nt) antisense transcripts are located within the sipA, sipD, and sipC genes. (C) Detailed view of the prgI-hilD region of SPI1. Three hilD transcriptional start sites were identified, and one TSS was found for prgH. The ChIP-chip data show that these TSSs are bound by RNAP, RNAP+rif, and Sigma70.
The RNAP-binding regions covered 690,500 bp or 14% of the SL1344 chromosome. We found that 817 of the 1,873 (44%) TSSs were bound by RNAP (Fig. 4A). To locate promoter regions with more precision, we pretreated the bacteria with rifampicin (Rif) before isolation of chromatin for ChIP-chip. Rifampicin is an inhibitor of transcriptional elongation and so confines RNAP to promoter regions (40). The resulting static map of RNAP showed 1,099 smaller binding regions that were largely located upstream of annotated genes (Dataset S3). More than 70% of the TSSs map to a RNAP+Rif binding region (1,318 of 1,873 TSSs, Fig. 3A), significantly increasing the overlap of localization between RNAP and transcriptional start sites. E. coli transcripts are longer lived than RNAP-promoter complexes (41, 42), which might explain the many TSS that do not show RNAP binding in ChIP datasets.
Fig. 4.

Interaction of the transcriptional machinery with transcriptional start sites in S. Typhimurium. (A) The transcriptional start sites bound by RNAP, RNAP+rif, and Sigma70 were identified by integrating the ChIP-chip and TSS data. The bar charts indicate the number (and percentages) of the 1,873 transcriptional start sites that lie within the binding region of the particular factor. (B) The majority (70%) of the identified TSSs are bound by RNAP+rif, of which 63% are also bound by Sigma70, indicating that most TSSs are Sigma70 dependent at early stationary phase.
To define the relative importance of the σ70 (RpoD) sigma factor in the initiation of transcription at ESP, we performed the dynamic RNAP ChIP experiment with an anti-σ70 monoclonal antibody. This method identified 835 regions that were bound by σ70 (Dataset S3). Of the 1,318 TSSs bound by RNAP at promoter sites, 832 (63%) TSSs were also associated with σ70, consistent with σ70 being the major sigma factor of transcription initiation at early stationary phase in S. Typhimurium (Fig. 4B). The fact that the ChIP-chip data show a strong overlap between the locations of bound RNAP and σ70 suggests that the two proteins are predominantly associated as Eσ70 holoenzyme. We note that σ70 is present at higher levels than RNAP, with measurements of between 7,200 and 17,000 molecules per cell (38, 39).
The SPI2 pathogenicity island of S. Typhimurium plays a critical role during the intracellular life of the pathogen. We identified primary and secondary TSSs for the ssrAB transcript that encodes the sensor kinase and response regulator that activate SPI2 transcription (Dataset S2). The ChIP-chip data showed that transcription of ssrAB is driven by σ70, consistent with a recent report that ssrAB expression is independent of σ38 (43). Apart from the ssaB promoter, no other TSSs were identified for the SPI2 secretion system and effector genes, perhaps due to the low level expression of SPI2 genes at the ESP growth condition.
Identification of σ70 Motifs in S. Typhimurium.
The consensus structure of a S. Typhimurium promoter has not been experimentally defined by large-scale sequence comparisons. To test whether the S. Typhimurium σ70 targets the same DNA sequence motifs as those of E. coli, we analyzed the primary TSSs of S. Typhimurium that were overlapped by both RNAP and σ70 in the ChIP-chip datasets (n = 717). We used unbiased motif searching with the Meme and BioProspector algorithms (44, 45) to identify canonical σ70 motifs upstream of the TSSs (Fig. 5A). The same algorithms identified very similar σ70 −10 and −35 motifs in the experimentally determined σ70-binding sites of E. coli (n = 857). S. Typhimurium has a stronger “extended” −10 motif, and this motif contains a G at position −3 within the −10 element. Such extended −10 sequences are common in σ70-driven promoters that lack or have a very weak consensus −35 sequence (46). Our finding is consistent with extended −10 elements playing a more significant role in S. Typhimurium promoter recognition than in E. coli.
Fig. 5.

Similarities and differences between the Sigma70 binding site motifs of S. Typhimurium and E. coli and features of the initiating nucleotide of transcripts and the 5′-UTR of S. Typhimurium at early stationary phase. (A) The 717 transcription start sites bound by both Sigma70 and RNAP in S. Typhimurium (Fig. 4B) were used to identify Sigma70 binding site motifs (Materials and Methods) and are aligned with similar motifs identified in the 857 Sigma70 binding regions in E. coli (46). Base positions are numbered as in Shultzaberger et al. (46). (B) Distribution and frequency of the length of the 5′-UTR of mRNAs. A total of 1,294 primary (red) and secondary (black) start sites were used to visualize the 5′-UTRs of S. Typhimurium at ESP, revealing that >50% of all 5′-UTRs are between 20 nt and 65 nt. (Inset) The initiating nucleotide of transcripts compiled from 1,873 TSSs. The majority of transcripts (84%) possess a purine (50%, A; 34%, G) at their +1 position. The positions −1 and +2 are dominated by pyrimidines.
Initiating Nucleotide of S. Typhimurium Transcription.
The first nucleotide in a transcript acts as a ligand to catalyze open complex formation and transcription initiation by RNAP. Consequently, the availability of this nucleotide in cellular NTP pools directly regulates the rate of transcription initiation (47). A well-characterized example of this regulation occurs at ribosomal RNA promoters that express the most abundant transcripts in the cell encoding the ribosomal translation machinery. Because RNA and protein synthesis are energetically very costly, the production of ribosomes is controlled to efficiently conserve cellular energy. rRNA genes initiate with either ATP or GTP, thus linking rRNA transcription directly to the availability of the primary energy-carrying molecules (ATP and GTP) (48).
Of the 1,873 TSSs mapped in S. Typhimurium, the majority (84%) of transcripts initiate with a purine nucleotide (ATP = 50%, GTP = 34%) (Fig. 5B), suggesting that transcription initiation is regulated by the levels of energy pools under these experimental conditions. Pools of pyrimidine nucleotides are less abundant in bacterial cells than purine nucleotides (49). The preference for pyrimidine nucleotides at the −1 and +2 positions immediately flanking the TSSs could reflect a mechanism to reduce unintentional transcription initiation from these flanking positions.
5′-Untranslated Regions of S. Typhimurium.
A 5′-untranslated region (5′-UTR) is defined as the transcribed nucleotides located between the transcriptional start and the translational start codon in a bacterial mRNA. Some 5′-UTR sequences are required for optimal translation and can also harbor regulatory elements such as riboswitches. Here, we show that the average length of the S. Typhimurium 5′-UTR is between 20 nt and 65 nt long, which is strikingly similar to the length of the 5′-UTR in Helicobacter pylori (16), and might represent an optimal length for efficient translation (Fig. 5B). We found 23 leaderless mRNAs and confirmed the TSSs of two of these candidates by 5′-RACE (yfgE and yibP) (Fig. S2 and Dataset S2). These leaderless mRNAs amount to 1.2% of the transcripts, and they all contain the AUG translational start codon that can also promote ribosome binding (50).
Identification of 60 S. Typhimurium sRNAs.
In recent years, it has become evident that small RNAs (sRNAs) are a ubiquitous class of regulatory elements carrying out important roles in posttranscriptional gene regulation and that many of these sRNAs act as regulators of multiple target genes (51). Small RNAs have now been discovered in different bacteria using microarray or deep sequencing-based transcriptomic techniques, often combined with a coimmunoprecipitation of the RNA chaperone Hfq, computer-based prediction methods, or shotgun cloning of cDNA (24, 52–56).
To reveal the sRNA complement of S. Typhimurium at ESP, we combined the RNA-seq and dRNA-seq analyses with our published Hfq-coIP-seq approach (55). The identity of candidate sRNAs was assigned conservatively (Materials and Methods) and they were generally small (<500 nt) transcripts expressed from intergenic regions or antisense to characterized ORFs. Surprisingly, we found two small RNAs that were expressed from within an ORF, in the same strand as the coding sequence (STnc1290 and STnc1680, Dataset S1).
S. Typhimurium expressed 140 sRNAs at ESP (Dataset S1). These include 60 newly identified sRNAs, of which 29 were confirmed by Northern blot (Fig. 6 and Fig. S4). A representative example, STnc1390, is shown in Fig. 6B. We discovered that the expression of 9 sRNAs was environmentally regulated, being differentially expressed throughout the growth phase and in conditions that induce the expression of SPI1 or SPI2. We determined that STnc1020 was maximally expressed at ESP during growth and STnc1080 was highly up-regulated under SPI2-inducing conditions (Fig. 6A). In addition, some sRNAs (i.e., STnc1120) show multiple bands with varying prominence, suggesting condition-specific processing profiles (compare late stationary phase with SPI1-inducing conditions, Fig. 6A). We anticipate that more sRNAs will be identified in other growth conditions.
Fig. 6.

Identification of small RNAs in S. Typhimurium. (A) Differential gene expression of newly identified small RNAs shown by Northern blot. RNA samples were taken from cells grown in L broth (OD = 0.5, 1, 2, and 2 + 6 h) and from SPI1- and SPI2-inducing conditions. (B) Identification of the unique small RNA STnc1390. (Left) Northern blot of STnc1390; (Right) sequence reads from all RNA-seq libraries used in this study. STnc1390 is located in the intergenic region between genes SL2591 and SL2592.
S. Typhimurium sRNA Conservation Between Enteric Bacteria.
The sRNA complement of S. Typhimurium was used for an evolutionary overview of S. Typhimurium sRNAs within the Enterobacteriaceae. We used a bioinformatic approach to assess the conservation of the 113 S. Typhimurium sRNAs that have been experimentally verified, here and elsewhere (Dataset S1). Sequence identity is shown across the sequenced genomes of 29 enterobacterial strains (Fig. 7 and Fig. S5). The cluster analysis shows that the S. Typhimurium sRNAs comprise six distinct phylogenetic groups. We found 6 sRNAs that are S. Typhimurium specific, including IsrK (57). A further 8 sRNAs are conserved in the serovars Typhimurium, Paratyphi, Newport, Virchow, Saintpaul and Schwarzengrund, including the virulence regulator IsrJ (57). The identification of a total of 48 sRNAs that are Salmonella specific raises the possibility that these sRNAs might play a role in infection and these sRNAs include the SPI1-encoded InvR (24). More than 48% of the 93 sRNAs that are found across the Salmonella genomes are also conserved in three pathovars of E. coli and include many sRNAs that have previously been shown to be conserved between E. coli K12 and S. Typhimurium LT2 (58), including GcvB, OxyS, MicF, and ArcZ (27). Finally, we identified a total of just 20 sRNAs that were conserved in all of the enterobacterial strains that were examined, such as RybB (59). The 60 sRNAs that were discovered in this study showed a varied pattern of evolution, with 7 being confined to S. enterica subspecies I and others being Salmonella specific or conserved in both Salmonella and E. coli. Once the mRNA targets of S. Typhimurium have been identified, it will be interesting to compare the phylogenetic patterns of the targets and the sRNAs.
Fig. 7.

Conservation of S. Typhimurium sRNAs within enteric bacteria. Heat map shows the conservation of S. Typhimurium SL1344 sRNAs in 29 genome sequences of bacteria belonging to the family Enterobacteriaceae. Homology was identified with Exonerate software (Materials and Methods). Columns and rows represent sRNAs and bacterial genomes, respectively. In the heat map, red indicates the highest homology as 95–100% identity, and pink shows 85–95% identity. The three blue colors indicate between 85% and 55% identity, and white shows <55% sequence identity. Colored bars at the bottom indicate six phylogenetic groups of S. Typhimurium sRNAs: black (conserved in Typhimurium), gray (conserved in Typhimurium, Paratyphi, Newport, Virchow, Saintpaul and Schwarzengrund), blue (conserved in all Salmonella enterica subspecies 1 serovars), yellow (conserved in all S. enterica subspecies 1 serovars plus Salmonella arizonae and Salmonella bongori), orange (conserved in all Salmonella and E. coli strains), and green (conserved throughout enteric bacteria).
Minority of S. Typhimurium sRNAs Are Located Within Prophages and Pathogenicity Islands.
To put Fig. 7 into some evolutionary context, we examined the chromosomal location of the sRNAs. Twenty of the 113 sRNAs were located on pathogenicity islands or bacteriophages, as shown in Dataset S1. These include 10 sRNAs (InvR, IsrA, IsrB-1, IsrB-2, IsrC, IsrG, IsrI, IsrJ, IsrK, and IsrL) that were originally identified as island associated (24, 57). The majority of the STnc sRNAs (7/10) were carried on the Gifsy-1, Gifsy-2, and SLP203 prophages. We identified 3 sRNAs that are associated with pathogenicity islands and conserved only within the Salmonella genus: STnc1220 is antisense to the SPI2 ssaK gene, and STnc150 and STnc520 are intergenic within SPI11.
Community Data Resources.
To maximize the impact of the transcriptional map of S. Typhimurium, we have provided direct access to all of the data featured in this paper via an easily searchable online visual interface for the benefit of the broader microbiological community (www.imib-wuerzburg.de/research/salmonella). The identities of orthologs of the 4,742 coding genes of SL1344 also present in the S. Typhimurium strains LT2, 14028, U.K.-1, and D23580 are shown in Dataset S1, to allow researchers to identify genes of interest in these important S. Typhimurium strains. The findings include the locations of all TSSs, all sRNAs, and the positions of reannotated genes and have been included in the genome annotation (Datasets S1 and S2).
Conclusion
The interaction of S. Typhimurium with mammalian cells has been used extensively to understand both bacterial virulence and host cell responses to bacterial infection (60). However, the lack of a fundamental understanding of the structure and function of S. Typhimurium promoters has hampered the identification of the binding sites of key transcription factors involved in the regulation of bacterial virulence gene expression.
The development of high-throughput sequencing techniques to interrogate large populations of RNA molecules has now allowed the visualization of the transcriptional map of the bacterial chromosome. We have defined the most basic element of gene expression in this system, the S. Typhimurium promoter. Unlike previous extrapolation from E. coli data, we have now identified the promoters that are controlled by the predominant σ70 transcription factor. The identification of σ70-dependent and σ70-independent promoter sequences will now allow conserved DNA-binding sites to be characterized and will facilitate the global identification of transcriptional regulators of these genes.
Here, we present a valuable data resource that informs the regulation of the majority of S. Typhimurium genes and operons. Fig. 3B is an example of the value of this approach, showing TSSs that control important virulence genes present in SPI1. As well as the expected primary TSSs that promote expression of key operons, we also report internal transcriptional start sites that allow expression of individual virulence genes and identify a number of antisense transcripts. The dRNA-seq data revealed the TSSs of the SPI1 and SPI2 regulatory genes phoP, slyA, and invF, which were validated by 5′-RACE. The finding that the phoP and invF promoters were bound by both RpoD and RNAP describes the fundamental mechanism that controls the expression of these genes. It is anticipated that in the future many alternate TSSs will be identified at different stages of growth and during the process of infection of the mammalian host.
Significantly less antisense transcription was identified in Salmonella (1.5%) than observed in E. coli (20%) (61). One of these antisense transcripts was complementary to the ssrA gene, which is the master regulator of SPI2. However, we note that the level of bacterial antisense transcription identified by RNA-seq can vary between 3% and 50% (62), raising the possibility that a proportion of antisense sequence reads could reflect the cDNA library preparation protocol used in different studies. Our approach relied upon the addition of a 5′-RNA linker before cDNA synthesis, an approach that was also used for the recent Helicobacter study that identified 27% antisense transcription (19).
The discovery of sRNAs that are expressed at early stationary phase will permit the characterization of the transcriptional network controlled by sRNAs in S. Typhimurium. Nearly half of the experimentally verified sRNAs were uniquely found in the Salmonella genus and relatively few sRNAs were conserved throughout the Enterobacteriaceae. This pattern of sRNA conservation may have significance for the development of transcriptional regulation during evolution. It will be interesting to determine whether the mRNA targets of some of the six phylogenetic groups of sRNAs have been horizontally acquired or are members of the core S. Typhimurium genome.
We anticipate that in the future the detailed understanding of the global impact of important transcription factors, coupled with the mapping of promoters under additional infection-relevant growth conditions, will herald a new era for research on the regulation of gene expression during infection by S. Typhimurium.
Materials and Methods
Bacterial Strains and Growth Conditions.
Bacterial strain S. enterica serovar Typhimurium SL1344 and its parental strain ST4/74 were used throughout the study (18, 63). Nucleotide differences that differentiate these two strains (eight SNPs) are shown in Dataset 1. Liquid growth medium was Lennox (L) (10 g/L Bacto tryptone, 5 g/L Bacto yeast extract, 5 g/L NaCl) or Luria broth (LB) (10 g/L Bacto tryptone, 5 g/L Bacto yeast extract, 10 g/L NaCl) or SPI2-inducing phosphate carbon nitrogen (PCN) medium (pH 5.8, 0.4 mM Pi) (64). All cultures were incubated in 25 mL media in 250-mL flasks at 37 °C and 220 rpm, unless stated otherwise. Samples taken from different conditions were described earlier in detail (55).
Oligonucleotides used in this study are listed in Table S1, and information on S. Typhimurium genome sequence, RNA isolation, cDNA library construction, RNA-seq, dRNA-seq, RNA-seq data analysis, sRNA identification, Northern blot analysis, 5′-RACE, ChIP-chip, identification of consensus motifs, and determination of sRNA conservation is provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Stephen Busby and José Puente for critical appraisal of our data; Fritz Thümmler for cDNA library preparation and sequencing; Profs. Paul Barrow, Gordon Dougan, Tom Humphrey, and Mark Roberts for initiating the sequencing of strain SL1344 at the Wellcome Trust Sanger Institute; Mark Stevens and Mick Watson for sharing the sequence of strain ST4/74 prior to publication; Lira Mamanova for kindly providing aliquots of the chimeric RNA-DNA adapter oligos required for FRT-seq; Tyrrell Conway and Joe Grissom for advice and help with Jbrowse; Cynthia Sharma for help at early stages of this project; the Trinity Centre for High Performance Computing for computational resources and Leanne Hays, Shabarinath Srikumar, and Jane Twohig for their assistance during this project. We also thank Science Foundation Ireland for financial support (Grants 08/IN.1/B2104 and 07/IN.1/B918).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The S. Typhimurium SL1344 genome and plasmid sequences reported in this paper have been deposited in the European Molecular Biology Laboratory database, www.ebi.ac.uk/embl/ (accession nos. FQ312003, HE654724, HE654725, and HE654726) and microarray data have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE35827).
See Author Summary on page 7606 (volume 109, number 20).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201061109/-/DCSupplemental.
References
- 1.Crump JA, Luby SP, Mintz ED. The global burden of typhoid fever. Bull World Health Organ. 2004;82:346–353. [PMC free article] [PubMed] [Google Scholar]
- 2.Majowicz SE, et al. International Collaboration on Enteric Disease ‘Burden of Illness’ Studies The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis. 2010;50:882–889. doi: 10.1086/650733. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention (CDC) Vital signs: Incidence and trends of infection with pathogens transmitted commonly through food—foodborne diseases active surveillance network, 10 U.S. sites, 1996-2010. MMWR Morb Mortal Wkly Rep. 2011;60:749–755. [PubMed] [Google Scholar]
- 4.Gormley FJ, et al. A 17-year review of foodborne outbreaks: Describing the continuing decline in England and Wales (1992-2008) Epidemiol Infect. 2011;139:688–699. doi: 10.1017/S0950268810001858. [DOI] [PubMed] [Google Scholar]
- 5.Lan RT, Reeves PR, Octavia S. Population structure, origins and evolution of major Salmonella enterica clones. Infect Genet Evol. 2009;9:996–1005. doi: 10.1016/j.meegid.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Kingsley RA, et al. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 2009;19:2279–2287. doi: 10.1101/gr.091017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon MA, Graham SM. Invasive salmonellosis in Malawi. J Infect Dev Ctries. 2008;2:438–442. doi: 10.3855/jidc.158. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Escobedo G, Marshall JM, Gunn JS. Chronic and acute infection of the gall bladder by Salmonella Typhi: Understanding the carrier state. Nat Rev Microbiol. 2011;9:9–14. doi: 10.1038/nrmicro2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bäumler AJ, Winter SE, Thiennimitr P, Casadesus J. Intestinal and chronic infections: Salmonella lifestyles in hostile environments. Env Microbiol Rep. 2011;3:508–517. doi: 10.1111/j.1758-2229.2011.00242.x. [DOI] [PubMed] [Google Scholar]
- 10.Hautefort I, et al. During infection of epithelial cells Salmonella enterica serovar Typhimurium undergoes a time-dependent transcriptional adaptation that results in simultaneous expression of three type 3 secretion systems. Cell Microbiol. 2008;10:958–984. doi: 10.1111/j.1462-5822.2007.01099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDermott JE, et al. Technologies and approaches to elucidate and model the virulence program of salmonella. Front Microbiol. 2011;2:121. doi: 10.3389/fmicb.2011.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hébrard M, Kröger C, Sivasankaran SK, Händler K, Hinton JC. The challenge of relating gene expression to the virulence of Salmonella enterica serovar Typhimurium. Curr Opin Biotechnol. 2011;22:200–210. doi: 10.1016/j.copbio.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Ozsolak F, Milos PM. RNA sequencing: Advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croucher NJ, Thomson NR. Studying bacterial transcriptomes using RNA-seq. Curr Opin Microbiol. 2010;13:619–624. doi: 10.1016/j.mib.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorek R, Cossart P. Prokaryotic transcriptomics: A new view on regulation, physiology and pathogenicity. Nat Rev Genet. 2010;11:9–16. doi: 10.1038/nrg2695. [DOI] [PubMed] [Google Scholar]
- 16.Sharma CM, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 17.Rankin JD, Taylor RJ. The estimation of doses of Salmonella typhimurium suitable for the experimental production of disease in calves. Vet Rec. 1966;78:706–707. doi: 10.1136/vr.78.21.706. [DOI] [PubMed] [Google Scholar]
- 18.Hoiseth SK, Stocker BA. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature. 1981;291:238–239. doi: 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]
- 19.Fookes M, et al. Salmonella bongori provides insights into the evolution of the Salmonellae. PLoS Pathog. 2011;7:e1002191. doi: 10.1371/journal.ppat.1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stecher B, et al. Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc Natl Acad Sci USA. 2012;109:1269–1274. doi: 10.1073/pnas.1113246109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becker D, et al. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature. 2006;440:303–307. doi: 10.1038/nature04616. [DOI] [PubMed] [Google Scholar]
- 22.McClelland M, et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 23.Wilmes-Riesenberg MR, Foster JW, Curtiss R., 3rd An altered rpoS allele contributes to the avirulence of Salmonella typhimurium LT2. Infect Immun. 1997;65:203–210. doi: 10.1128/iai.65.1.203-210.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfeiffer V, et al. A small non-coding RNA of the invasion gene island (SPI-1) represses outer membrane protein synthesis from the Salmonella core genome. Mol Microbiol. 2007;66:1174–1191. doi: 10.1111/j.1365-2958.2007.05991.x. [DOI] [PubMed] [Google Scholar]
- 25.Mamanova L, et al. FRT-seq: Amplification-free, strand-specific transcriptome sequencing. Nat Methods. 2010;7:130–132. doi: 10.1038/nmeth.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sittka A, Sharma CM, Rolle K, Vogel J. Deep sequencing of Salmonella RNA associated with heterologous Hfq proteins in vivo reveals small RNAs as a major target class and identifies RNA processing phenotypes. RNA Biol. 2009;6:266–275. doi: 10.4161/rna.6.3.8332. [DOI] [PubMed] [Google Scholar]
- 27.Papenfort K, et al. Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol Microbiol. 2009;74:139–158. doi: 10.1111/j.1365-2958.2009.06857.x. [DOI] [PubMed] [Google Scholar]
- 28.Jäger D, et al. Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci USA. 2009;106:21878–21882. doi: 10.1073/pnas.0909051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitschke J, et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci USA. 2011;108:2124–2129. doi: 10.1073/pnas.1015154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albrecht M, et al. The transcriptional landscape of Chlamydia pneumoniae. Genome Biol. 2011;12:R98. doi: 10.1186/gb-2011-12-10-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong RS, McMurry LM, Levy SB. ‘Intergenic’ blr gene in Escherichia coli encodes a 41-residue membrane protein affecting intrinsic susceptibility to certain inhibitors of peptidoglycan synthesis. Mol Microbiol. 2000;37:364–370. doi: 10.1046/j.1365-2958.2000.01998.x. [DOI] [PubMed] [Google Scholar]
- 33.Fozo EM, et al. Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol Microbiol. 2008;70:1076–1093. doi: 10.1111/j.1365-2958.2008.06394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol Microbiol. 2008;70:1487–1501. doi: 10.1111/j.1365-2958.2008.06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gassel M, Möllenkamp T, Puppe W, Altendorf K. The KdpF subunit is part of the K(+)-translocating Kdp complex of Escherichia coli and is responsible for stabilization of the complex in vitro. J Biol Chem. 1999;274:37901–37907. doi: 10.1074/jbc.274.53.37901. [DOI] [PubMed] [Google Scholar]
- 36.Wadler CS, Vanderpool CK. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc Natl Acad Sci USA. 2007;104:20454–20459. doi: 10.1073/pnas.0708102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alix E, Blanc-Potard AB. Peptide-assisted degradation of the Salmonella MgtC virulence factor. EMBO J. 2008;27:546–557. doi: 10.1038/sj.emboj.7601983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grigorova IL, Phleger NJ, Mutalik VK, Gross CA. Insights into transcriptional regulation and sigma competition from an equilibrium model of RNA polymerase binding to DNA. Proc Natl Acad Sci USA. 2006;103:5332–5337. doi: 10.1073/pnas.0600828103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piper SE, Mitchell JE, Lee DJ, Busby SJ. A global view of Escherichia coli Rsd protein and its interactions. Mol Biosyst. 2009;5:1943–1947. doi: 10.1039/B904955j. [DOI] [PubMed] [Google Scholar]
- 40.Herring CD, et al. Immobilization of Escherichia coli RNA polymerase and location of binding sites by use of chromatin immunoprecipitation and microarrays. J Bacteriol. 2005;187:6166–6174. doi: 10.1128/JB.187.17.6166-6174.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reppas NB, Wade JT, Church GM, Struhl K. The transition between transcriptional initiation and elongation in E. coli is highly variable and often rate limiting. Mol Cell. 2006;24:747–757. doi: 10.1016/j.molcel.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 42.Bernstein JA, Lin PH, Cohen SN, Lin-Chao S. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc Natl Acad Sci USA. 2004;101:2758–2763. doi: 10.1073/pnas.0308747101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cameron AD, Dorman CJ. A fundamental regulatory mechanism operating through OmpR and DNA topology controls expression of Salmonella pathogenicity islands SPI-1 and SPI-2. PLoS Genet. 2012;8(3) doi: 10.1371/journal.pgen.1002615. e1002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bailey TL, Williams N, Misleh C, Li WW. 2006. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34(Web Server issue):W369–W373. [DOI] [PMC free article] [PubMed]
- 45.Liu X, Brutlag DL, Liu JS. BioProspector: dDscovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac Symp Biocomput. 2001;6:127–138. [PubMed] [Google Scholar]
- 46.Shultzaberger RK, Chen Z, Lewis KA, Schneider TD. Anatomy of Escherichia coli sigma70 promoters. Nucleic Acids Res. 2007;35:771–788. doi: 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaal T, Bartlett MS, Ross W, Turnbough CL, Jr, Gourse RL. 1997. Transcription regulation by initiating NTP concentration: rRNA synthesis in bacteria. Science 278(5346):2092–2097. [DOI] [PubMed]
- 48.Murray HD, Schneider DA, Gourse RL. Control of rRNA expression by small molecules is dynamic and nonredundant. Mol Cell. 2003;12:125–134. doi: 10.1016/s1097-2765(03)00266-1. [DOI] [PubMed] [Google Scholar]
- 49.Buckstein MH, He J, Rubin H. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J Bacteriol. 2008;190:718–726. doi: 10.1128/JB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brock JE, Pourshahian S, Giliberti J, Limbach PA, Janssen GR. Ribosomes bind leaderless mRNA in Escherichia coli through recognition of their 5′-terminal AUG. RNA. 2008;14:2159–2169. doi: 10.1261/rna.1089208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papenfort K, Vogel J. Regulatory RNA in bacterial pathogens. Cell Host Microbe. 2010;8:116–127. doi: 10.1016/j.chom.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Vogel J, et al. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res. 2003;31:6435–6443. doi: 10.1093/nar/gkg867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang A, et al. Global analysis of small RNA and mRNA targets of Hfq. Mol Microbiol. 2003;50:1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x. [DOI] [PubMed] [Google Scholar]
- 54.Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001;15:1637–1651. doi: 10.1101/gad.901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sittka A, et al. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sridhar J, et al. sRNAscanner: A computational tool for intergenic small RNA detection in bacterial genomes. PLoS ONE. 2010;5:e11970. doi: 10.1371/journal.pone.0011970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Padalon-Brauch G, et al. Small RNAs encoded within genetic islands of Salmonella typhimurium show host-induced expression and role in virulence. Nucleic Acids Res. 2008;36:1913–1927. doi: 10.1093/nar/gkn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hershberg R, Altuvia S, Margalit H. A survey of small RNA-encoding genes in Escherichia coli. Nucleic Acids Res. 2003;31:1813–1820. doi: 10.1093/nar/gkg297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papenfort K, et al. SigmaE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol Microbiol. 2006;62:1674–1688. doi: 10.1111/j.1365-2958.2006.05524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsolis RM, Xavier MN, Santos RL, Bäumler AJ. How to become a top model: Impact of animal experimentation on human Salmonella disease research. Infect Immun. 2011;79:1806–1814. doi: 10.1128/IAI.01369-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Georg J, Hess WR. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev. 2011;75:286–300. doi: 10.1128/MMBR.00032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lasa I, et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci USA. 2011;108:20172–20177. doi: 10.1073/pnas.1113521108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Richardson EJ, et al. Genome sequences of Salmonella enterica serovar typhimurium, Choleraesuis, Dublin, and Gallinarum strains of well- defined virulence in food-producing animals. J Bacteriol. 2011;193:3162–3163. doi: 10.1128/JB.00394-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Löber S, Jäckel D, Kaiser N, Hensel M. Regulation of Salmonella pathogenicity island 2 genes by independent environmental signals. Int J Med Microbiol. 2006;296:435–447. doi: 10.1016/j.ijmm.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 65.Nicol JW, Helt GA, Blanchard SG, Jr, Raja A, Loraine AE. The Integrated Genome Browser: Free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25:2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
