

Abstract
CD40L is excessively produced in both human and murine lupus and plays a role in lupus pathogenesis. To address how excess CD40L induces autoantibody production, we crossed CD40L-transgenic mice with the anti-DNA H-chain transgenic mouse lines 3H9 and 56R, well-characterized models for studying B-cell tolerance to nuclear antigens. Excess CD40L did not induce autoantibody production in 3H9 mice in which anergy maintains self-tolerance, nor did it perturb central tolerance, including deletion and receptor editing, of anti-DNA B cells in 56R mice. In contrast, CD40L/56R mice restored a large number of marginal zone (MZ) B cells reactive to Sm/ribonucleoprotein (RNP) and produced autoantibody, whereas these B cells were deleted by apoptosis in MZ of 56R mice. Thus, excess CD40L efficiently blocked tolerance of Sm/RNP-reactive MZ B cells, leading to production of anti-Sm/RNP antibody implicated in the pathogenesis of lupus. These results suggest that self-reactive B cells such as anti-Sm/RNP B cells, which somehow escape tolerance in the bone marrow and migrate to MZ, are tolerized by apoptotic deletion in MZ and that a break in this tolerance may play a role in the pathogenesis of lupus.
Keywords: peripheral tolerance, lupus-related autoantibody, marginal zone B cells
Maintenance of B-cell tolerance occurs at various checkpoints throughout B-cell development and maturation and involves multiple mechanisms. Self-reactive immature B cells are subjected to central tolerance mechanisms in the bone marrow, including deletion by apoptosis (clonal deletion), functional inactivation (clonal anergy), or Ig V gene replacement (receptor editing) (1–4). B-cell tolerance can also occur after B cells leave the bone marrow and home to peripheral lymphoid organs. This peripheral tolerance involves mechanisms such as anergy, ignorance, and maturation arrest (5–7). Moreover, how tolerance is maintained in self-reactive B cells, which are anergized in bone marrow and migrate to the peripheral lymphoid tissues, has been extensively studied. These B cells are excluded from the follicle or marginal zone (MZ) and show reduced longevity (2, 8–10) attributable to increased dependence on B cell-activating factor belonging to the tumor necrosis factor family (BAFF) for survival (11, 12). However, it is not fully understood how peripheral tolerance mechanisms are broken in autoimmunity.
B-cell self-tolerance has been addressed using antibody-transgenic (Tg) mice in which most of the B cells are reactive to a particular self-antigen. The anti-DNA H-chain Tg mice 3H9 and 56R are well-characterized models for studying B-cell tolerance to nuclear antigens (1, 10, 13–16), to which autoantibodies are characteristically produced in various autoimmune diseases including systemic lupus erythematosus (SLE). The 56R H chain was generated by introducing an arginine residue to the 3H9 VH region to increase affinity of the transgene-encoded antibodies for DNA. When paired with almost any of the mouse endogenous Ig L chains, both the 3H9 and the 56R H chains form antibodies that bind DNA. In these mice, DNA-reactive B cells are regulated by one or more of several central tolerance mechanisms including deletion, receptor editing, and anergy. High-avidity anti-DNA B cells produced in 56R Tg mice are tolerized by central deletion and receptor editing (13, 14, 17), whereas, low-avidity anti-DNA B cells produced in 3H9 Tg mice are either arrested developmentally or rendered anergic (10).
CD40L (CD154), the ligand for CD40, is excessively produced in patients with SLE and in mouse models of SLE (18) and appears to play a role in the development of the autoimmune disease because treatment with blocking antibodies to CD40L markedly reduced the disease activity in both mouse and human SLE (18). Previously, we demonstrated that CD40L Tg mice in which excess CD40 signaling is generated in B cells through an autocrine pathway (19) by constitutive CD40L expression in B cells spontaneously develop lupus-like disease (20), as is the case for CD40L Tg mice that overexpress CD40L in T cells (21). To address the impact of excess CD40 signaling on B-cell tolerance to nuclear antigens, we crossed CD40L Tg mice with 3H9 Tg mice and with 56R Tg mice. Here, we demonstrate that CD40 signaling specifically breaks tolerance of 56R B cells reactive to RNA-related antigens implicated in pathogenesis of lupus (22, 23). These B cells appear in the MZ in the spleen, undergo apoptosis, and are rapidly cleared by phagocytes. Excess CD40L blocks the MZ deletion, leading to generation of self-reactive MZ B cells and autoantibody production. Our finding strongly suggests that deletion of self-reactive MZ B cells serves a crucial function in self-tolerance and implicates abrogation of this deletion in the pathogenesis of SLE.
Results
CD40 Signaling Induces Autoantibody Production in 56R Mice.
To address how CD40 signaling perturbs B-cell tolerance, we crossed CD40L Tg mice with each of the anti-DNA μH-chain Tg mice 3H9 and 56R. When we measured the concentrations of IgM anti-DNA antibodies in sera from 8- to 15-wk-old F1 mice, anti-DNA antibodies were not detected in the F1 mice carrying either 3H9 or 56R alone (Fig. 1A), in agreement with previous findings (14, 15). F1 mice expressing both 3H9 and CD40L did not produce detectable anti-DNA antibodies, indicating that B-cell tolerance is maintained in 3H9 mice despite excess CD40L expression. In contrast, F1 mice expressing both 56R and CD40L showed marked anti-DNA antibody production. The mean total serum IgM concentration in the double Tg mice was comparable to the mean in WT mice, indicating that this autoantibody production is likely the result of abrogated B-cell tolerance and not of a generalized polyclonal B-cell activation.
Fig. 1.

Excess CD40L induces autoantibody production in 56R mice without perturbing clonal deletion in bone marrow. Anti-DNA antibody H chain-Tg mice 3H9 and 56R mice on the BALB/c background were each crossed with CD40L Tg mice on the C57BL/6 background to generate F1 mice. (A) Sera were obtained from 8- to 15-wk-old F1 mice, and the titers of IgM anti-DNA antibodies (Left) and total serum IgM (Right) were measured by ELISA. Numbers of mice that were analyzed are indicated in parentheses. The titer of anti-DNA antibodies was determined by ELISA relative to a standard curve of DNA binding of anti-DNA monoclonal antibody BW28-20-1 derived from NZB/W F1 mice (IgM; a gift from Hirose, Juntendo University, Tokyo, Japan). Horizontal bars represent mean values. NS, not significant. (B) Bone marrow cells were obtained from 8- to 15-wk-old F1 mice and stained for B220 and IgM expression. Cells in the lymphocyte gate were analyzed by flow cytometry as described previously (50), and percentages of pro/pre B cells, immature B cells, transitional B cells, and mature B cells in lymphocyte-gated cells were calculated. Representative data of three experiments are shown.
Flow cytometric analysis of bone marrow lymphoid cells demonstrated a marked reduction of immature B cells in 56R mice compared with WT (Fig. 1B) as described previously (13). The percentage of immature B cells in CD40L/56R bone marrow was similar to that in 56R mice, suggesting that self-reactive B cells are deleted at the immature B-cell stage in CD40L/56R mice as effectively as in the 56R mice. The percentage of mature recirculating B cells was increased in CD40L/56R mice compared with 56R mice probably because of increase in the number of mature B cells in the peripheral lymphoid organs such as spleen. Thus, the perturbation of B-cell tolerance in CD40L/56R occurs despite intact clonal deletion at the immature B-cell stage.
CD40 Signaling Perturbs Self-Tolerance of Vκ38C+anti-Sm/ Ribonucleoprotein Antibody-Producing Cells in 56R Mice.
To address how CD40L perturbs self-tolerance in 56R mice, we generated hybridoma panels from LPS-stimulated spleen cells obtained from 56R and CD40L/56R mice. We first assayed for presence of the 56R H-chain gene by PCR and found that 60% of 56R hybridomas retained the 56R H-chain gene, in agreement with the previous finding in 56R mice (14). In contrast, the 56R H chain gene was retained in 82% of double Tg hybridomas, suggesting less stringent B-cell selection in double Tg mice. We then analyzed 56R H-chain+ hybridomas for anti-DNA antibody production by ELISA. The percentage of anti-DNA antibody-producing hybridomas was much higher in the CD40L/56R hybridoma panel than in the 56R panel, especially in those from 33-wk-old mice (Fig. 2A), in agreement with spontaneous autoantibody production in CD40L/56R mice (Fig. 1A).
Fig. 2.

Excess CD40L perturbs self-tolerance of anti-Sm/RNP Vκ38C+ B cells in 56R mice. (A–C) Hybridomas were generated from LPS-stimulated spleen cells obtained from 56R and CD40L/56R mice at 8 and 33 wk of age. 56R H chain+ hybridomas were analyzed for expression of Ig κ and λ L chain and reactivity to dsDNA by ELISA and for Vκ gene segment use by PCR (A). Total numbers of 56R H chain+ hybridomas are indicated by the numeral in the center of each circle diagram. Frequencies of hybridomas using Vκ21D and Vκ38C are indicated by the inner circle shading. Frequencies of anti-DNA hybridomas are indicated by the outer circle shading. Detailed results of VL gene use are shown in Table S1. Antibodies (3 μg/mL) from hybridoma supernatants using the indicated Vκ and Vλ gene segments obtained from 8-wk-old mice were assayed by ELISA for reactivity to dsDNA, ssDNA, histone, and Sm/RNP (B). Dots represent individual hybridomas. Numbers of hybridomas that were analyzed are indicated in parentheses. Out of 52 Vκ21D+ hybridomas, 44 hybridomas that do not react to any of these antigens are not shown. R462 is shown by red dots. Supernatant from the Vκ38C+ hybridoma D627 derived from CD40L/56R mice was examined for ANA staining pattern (C). (D) Anti-Sm/RNP antibody production was examined in sera from 16- to 24-wk-old WT (C57BL/6 × BALB/c) F1, 56R, CD40L, and CD40L/56R mice by ELISA. The antibody titer was determined relative to a standard curve generated from pooled MRL/lpr sera. The numbers in parentheses indicate the numbers of mice that were analyzed.
Next, we analyzed use of κ and λ L chains by ELISA and Vκ gene segment use by PCR in the 56R and CD40L/56R hybridoma panels. It was previously demonstrated that B cells in 56R mice use only a restricted set of Vκ segments called editors such as Vκ20, Vκ21D and Vκ38C (17). In the hybridoma panel from 56R mice, roughly 70% of the hybridomas used the Vκ21D gene segment, whereas Vκ38C was used in only 5% of hybridomas (Fig. 2A and Table S1), in agreement with the previous findings (17). In contrast, 13% of CD40L/56R hybridomas from the 8-wk-old mouse fusion expressed Vκ38C, and this fraction increased to 33% in the 33-wk-old mouse fusion. Vκ38C+ but not Vκ21D+ hybridomas showed reactivity to DNA, and 60% of the DNA-reactive hybridomas from CD40L/56R mice used Vκ38C, suggesting that Vκ38C+hybridomas are responsible for increased self-reactivity of the CD40L/56R hybridoma panel.
To characterize the Vκ38C+ hybridomas, we measured reactivity of antibodies from the hybridoma supernatants that contain more than 3 μg/mL IgM from 8-wk-old 56R and CD40L/56R mice to DNA- and RNA-related antigens. One hybridoma antibody (R462) that uses a yet unknown Vκ shows a strong reactivity to both DNA and histone (Fig. 2B). Reactivities to these antigens were weaker in most of the Vκ38C+hybridomas, suggesting that Vκ38C+hybridomas secrete low affinity anti-DNA antibody. Because antibodies to RNA-related antigens are involved in development of lupus (22, 23), we tested hybridoma supernatants for reactivity to Sm/ ribonucleoprotein (RNP). Surprisingly, antibodies from Vκ38C+clones reacted to Sm/RNP more strongly than those using any other VL. Accordingly, the Vκ38C+ antibody D627 stained cell nuclei in a speckled pattern (Fig. 2C) characteristic of autoantibodies to RNA-related antigens but not in the homogenous pattern characteristic of anti-DNA antibody. We tested four other Vκ38C+ hybridoma supernatants and all showed the same anti-nuclear antibody (ANA) staining pattern (Fig. S1A). Moreover, ELISA using components of Sm/RNP demonstrated that Vκ38C+ antibodies strongly react to the glycine-arginine-rich C-terminal part (residues 83–119) of SmD1 (Fig. S2), the major epitope recognized by anti-Sm antibodies from SLE patients (24, 25). Thus, Vκ38C+ antibodies show antigen specificity similar to anti-Sm antibodies in SLE, although 56R/Vκ38C+are potentially reactive to other self-antigens (26).
We then examined Jκ gene segment use in hybridoma panels from 8-wk and 33-wk-old 56R and CD40L/56R mice. Although Jκ1 is the predominant Jκ used in the normal B-cell repertoire, hybridomas from 56R mice were shown to use distal Jκs at higher than normal frequency as a result of receptor editing (1, 4). When we analyzed our hybridoma panels, hybridomas from the CD40L/56R fusion demonstrated a Jκ use distribution similar to the 56R fusion (Table 1). Vκ21D-expressing hybridomas used mostly Jκ2, and those expressing other Vκs used mostly Jκ4 and Jκ5 regardless of whether the hybridomas were generated from 56R or CD40L/56R mice. Thus, hybridomas from CD40L/56R mice used distal Jκs, as is the case for 56R hybridomas, suggesting that receptor editing is not perturbed in CD40L/56R mice.
Table 1.
Jκ use of hybridomas from 56R and CD40L/56R mice
| 56R |
CD40L/56R |
|||||||||
| Clones | Jκ1 | Jκ2 | Jκ4 | Jκ5 | Clones | Jκ1 | Jκ2 | Jκ4 | Jκ5 | |
| Vκ20 | 6 | 0 | 0 | 4 | 2 | 18 | 0 | 9 | 6 | 3 |
| Vκ21D | 41 | 0 | 35 | 4 | 2 | 33 | 1 | 25 | 6 | 1 |
| Vκ38C | 3 | 0 | 0 | 1 | 2 | 30 | 8 | 4 | 7 | 11 |
| Others | 29 | 12 | 2 | 3 | 12 | 24 | 1 | 1 | 7 | 15 |
| Total | 79 | 12 | 37 | 12 | 18 | 105 | 10 | 39 | 26 | 30 |
Data represent number of VκJκ rearrangements. Others, all other light chains.
Vκ38C+ Sm/RNP-Reactive B Cells Are Negatively Selected in Spleen.
To address how CD40 signaling perturbs tolerance of Sm/RNP-reactive B cells, we generated monoclonal anti-idiotype antibodies to recognize each of the L chain Vκs Vκ38C or Vκ21D when paired with the 56R H chain (Table S2) and examined Vκ21D+ and Vκ38C+ B cells in spleen from 16- to 20-wk-old 56R and CD40L/56R mice. Flow cytometric analysis revealed that the number of marginal zone (MZ) B cells was increased and that of follicular (FO) B cells was decreased in 56R spleen compared with WT spleen (Fig. 3A), in agreement with previous findings (27, 28). The number of MZ B cells in CD40L/56R spleen was comparable to that in 56R spleen, whereas the number of FO B cells was increased to the normal level in CD40L/56R spleen, in agreement with our previous finding that the number of FO B cells but not MZ B cells is increased in CD40L-Tg mice (19). Both Vκ21D+ and Vκ38C+ B cells were accumulated in type 2 transitional (T2) and MZ B cells but not in FO B cells (Fig. 3B). On average, 40% of T2 cells were Vκ38C+ in both 56R and CD40L/56R spleen. The percentage of Vκ38C+ cells in MZ B cells was reduced compared with that in T2 cells in 56R mice but not in CD40L/56R mice. In contrast, the percentage of Vκ21D+cells was not changed when B cells matured from T2 cells to MZ B cells. Thus, Vκ38C+ cells, but not Vκ21D+ cells, are deleted in 56R mice when they mature from T2 cells to MZ B cells, and this deletion is reversed in CD40L/56R mice. To confirm this finding, we examined histology of the 56R and CD40L/56R spleen. A large number of Vκ21D+ cells were found mostly in the MZ of both 56R and CD40L/56R spleen (Fig. 3C). Whereas only a few Vκ38C+ B cells were found in 56R spleen, CD40L/56R spleen showed a large number of Vκ38C+ B cells mostly located in MZ, again supporting the notion that excess CD40L perturbs deletion of Vκ38C+ B cells in 56R mice.
Fig. 3.
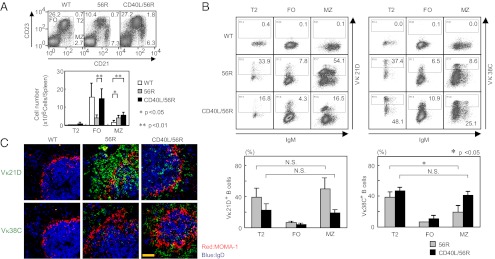
Accumulation and deletion of Sm/RNP-reactive Vκ38C+ B cells in splenic MZ in 56R mice. (A and B) Flow cytometric analysis of spleen cells from 16- to 20-wk-old WT (C57BL/6 × BALB/c) F1, 56R, and CD40L/56R mice. Cells were stained with allophycocyanin (APC)-conjugated rat anti-mouse CD21 and PE-conjugated rat anti-mouse CD23 antibody, and lymphoid gated cells were examined by flow cytometry (A). Percentages of T2, FO, and MZ B cells are indicated. Numbers of T2, FO, and MZ B cells were calculated for each spleen (Lower graph). Means ± SD of three mice are shown. Alternatively, cells were stained with Alexa Fluor 488-conjugated anti-56R/Vκ21D or anti-56R/Vκ38C anti-idiotype antibody together with APC-conjugated rat anti-mouse CD21, PE-conjugated rat anti-mouse CD23, biotinylated goat anti-mouse IgM antibodies, and PerCP-conjugated streptavidin, and lymphoid gated cells were examined by flow cytometry (B). IgM vs. 56R/Vκ21D or 56R/Vκ38C staining is displayed in T2, FO, and MZ B cells. Percentages of 56R/Vκ21D+ and 56R/Vκ38C+ cells in T2, FO, and MZ B cells were calculated. Means ± SD of three mice are shown (Lower graphs). (C) Immunohistological analysis of spleen from WT (C57BL/6 × BALB/c) F1, 56R, and CD40L/56R mice. Sections were stained for MOMA-1 (red), IgD (blue), and either 56R/Vκ21D or 56R/Vκ38C (green) and analyzed by confocal microscopy. [Scale bar (yellow line): 50 μm.] Representative data of three experiments are shown. N.S., not significant.
In contrast, only a small number of Vκ38C+cells and/or Vκ21D+ cells were found in other lymphoid organs such as lymph nodes, Peyer patches and peritoneal cells in 56R and CD40L/56R mice by both flow cytometric (Fig. S3A) and immunohistological analysis (Fig. S3B) probably because of the lack of T2 and MZ B cells in these organs.
Apoptosis of Sm/RNP-Reactive MZ B Cells in 56R Mice.
As CD40 signaling blocks B-cell apoptosis (29, 30), excess CD40L may reverse apoptosis of anti-Sm/RNP B cells in CD40L/56R mice. However, staining of 56R spleen sections from untreated 56R mice with anti-active caspase 3 did not show many apoptotic cells (Fig. 4 A and B, second row). We, thus, hypothesized that anti-Sm/RNP B cells undergo apoptosis but must be rapidly removed by macrophages such as MZ macrophages. To address this possibility, we depleted macrophages in 56R mice by treatment with clodronate liposome (31). We used 56R mice on the BALB/c background because the basal level of Vκ38C+B cells is low in these mice compared with those on the (BALB/c × C57BL/6) F1 background (32). By this treatment, MOMA-1+ marginal metallophilic macrophages were efficiently depleted (Fig. 4B, fourth row), and a large number of Vκ38C+ B cells appeared in the spleen MZ (Fig. 4 A and B, fourth row). Moreover, some of these Vκ38C+ B cells contained active caspase-3 (Fig. 4 A and B); whereas staining of the same section with anti-human IgG1 antibody (IgG2b, κ), a matched control antibody for anti-Vκ38C did not show any staining (Fig. 4A). Nonspecific staining of apoptotic cells by anti-Vκ38C antibody was further excluded by staining with anti-λ L chain antibody (Fig. 4B). In this analysis, λ+ B cells did not contain active caspase-3. Apoptotic Vκ38C+ B cells were also restored by treatment with the recombinant MFG-E8 D89E protein (Fig. 4 A and B, third row), the dominant negative form of MFG-E8 involved in phagocytosis of apoptotic cells (33). Thus, inhibition of phagocytosis restored apoptotic Vκ38C+ B cells in MZ, suggesting that these self-reactive B cells undergo apoptosis and are efficiently phagocytosed in MZ. Moreover, flow cytometry revealed increase of Vκ38C+ MZ B cells, but not T2 cells, by treatment with clodronate liposome (Fig. 4C), suggesting that Vκ38C+B cells are deleted at the MZ B-cell stage.
Fig. 4.
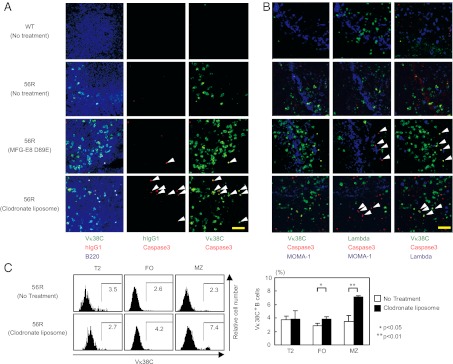
Anti-Sm/RNP Vκ38C+ B cells undergo apoptotic deletion in MZ in 56R mice. Clodronate liposome was injected i.v. to 16- to 24-wk-old 56R mice on BALB/c background at days 0, 8, 16, and 21 and mice were killed at day 23. Alternatively, MFG-E8 D89E was injected i.v. to 14- to 17-wk-old 56R mice on the BALB/c background at days 0, 3, and 6 and killed at day 7. (A and B) Spleen sections were stained for active caspase-3 (A and B), the 56R/Vκ38C idiotype (A and B), B220 (A), λ L chain (B), and MOMA-1 (B). Sections were stained with anti-human IgG1 antibody as an isotype-matched control antibody to the anti-56R/Vκ38C antibody (A). (Left, Center, and Right) The same microscopic fields viewed under different filters. Spleen sections from untreated non-Tg BALB/c mice and untreated 56R mice were examined as controls. Cells expressing both 56R/Vκ38C and active caspase-3 are indicated by arrowheads. [Scale bar (yellow line): 50 μm.] Representative data of three experiments are shown. (C) Spleen cells were stained for CD21, CD23, IgM, and 56R/Vκ38C and analyzed by flow cytometry as in Fig. 3B. 56R/Vκ38C staining is displayed in T2, FO, and MZ B cells (Left). Percentages of 56R/Vκ38C+ cells in T2, FO, and MZ B cells were calculated. Means ± SD of three mice are shown (Right).
Discussion
Because the 56R H chain can bind DNA when paired with nearly all known endogenous Ig L chains (17), 56R H chain Tg mice have been widely used as a model for examining the immunological tolerance of anti-DNA B cells (1, 10, 13–16). We demonstrate here that excess CD40L breaks self-tolerance in 56R mice. Hybridoma analysis showed that Vκ38C+ B cells are the dominant self-reactive B cells activated in CD40L/56R mice. Thus, in the presence of excess CD40L, tolerance of Vκ38C+B cells is preferentially perturbed whereas the remaining B-cell repertoire is subjected to tolerance. This is consistent with our observation that excess CD40L did not affect either receptor editing or reduction of the immature B-cell population that appears to be a consequence of clonal deletion. Hence, self-reactive B cells other than 56R/Vκ38C+ B cells appear to be tolerized, even in the presence of excess CD40L, by central tolerance mechanisms such as clonal deletion and receptor editing. 56R/Vκ38C+ B cells display other unique features. First, their binding specificity is similar to that of anti-Sm antibodies from SLE patients. Second, a sizable number of 56R/Vκ38C+ B cells are present in the T2 population, even in the absence of excess CD40L, and preferentially mature to MZ B cells but not FO B cells. Accumulation of 56R/Vκ38C+ B cells in MZ B cells is in agreement with previous findings (28, 34) and is consistent with the findings that many autoantibody Tg mice show expansion of the MZ B-cell population (27, 35–37). Third, 56R/Vκ38C+ B cells are deleted when they mature from T2 cells to MZ B cells, and this deletion is reversed by excess CD40L. Depletion of macrophages restores apoptotic 56R/Vκ38C+ MZ B cells, suggesting that 56R/Vκ38C+ MZ B cells undergo apoptosis and are removed by macrophages. Thus, a limited fraction of the self-reactive B-cell repertoire, including anti-Sm/RNP B cells, may somehow escape central tolerance, preferentially differentiate into MZ B cells probably because of self-reactivity, and then become tolerized by apoptotic deletion. Excess CD40L appears to specifically perturb this peripheral tolerance mechanism leading to production of anti-Sm/RNP antibody.
Why 56R/Vκ38C+ self-reactive B cells escape central tolerance and mature to peripheral B cells in 56R mice is not clear. Sm/RNP might be less available than DNA in the bone marrow. Alternatively, Sm/RNP might allow maturation of immature bone marrow B cells by interacting with TLR7, an innate receptor for RNA-related antigens essential for production of anti-Sm antibody (22, 23), although TLR7 may also regulate activation of anti-Sm/RNP B cells at the later stage of B-cell differentiation (38). 56R/Vκ38C+ self-reactive B cells preferentially differentiate to MZ B cells probably because of their self-reactivity. Because LPS activates MZ B cells more efficiently than FO B cells (39), 56R/Vκ38C+ hybridomas could overrepresent in the hybridoma panels generated from LPS-stimulated spleen B cells. However, FO B cells may not contribute to autoantibody production as most of the FO B cells in 56R mice produce endogenous H chains (34) and, therefore, appear non-self-reactive. Thus, 56R/Vκ38C+ B cells are the dominant self-reactive B cells in CD40L/56R mice. This conclusion is also supported by the finding that ANA test of sera from CD40L/56R mice show the speckled pattern (Fig. S1B), which is the same as the ANA pattern of 56R/Vκ38C+ antibody.
Self-tolerance in the periphery has been demonstrated in low-avidity, anergic, self-reactive B cells using Tg mice such as 3H9 mice and double Tg mice expressing both anti-HEL antibody and soluble HEL antigen (2, 8–10). In these mice, low-avidity self-reactive B cells are anergized in the bone marrow and migrate to peripheral lymphoid organs, where anergic B cells are excluded from the follicles and MZ and are located at the red pulp and the boundary between the T-cell zone and the follicle (T/B boundary). These anergic self-reactive B cells undergo apoptosis at the T/B boundary (40) because of increased dependence on BAFF (11, 12). In contrast, 56R/Vκ38C+ B cells are located in the MZ (Fig. 3C) and are not found at the T/B boundary (Fig. S4). Thus, 56R/Vκ38C+ cells migrate to and undergo apoptosis in a different location than do anergic B cells. Although apoptosis of anergic B cells is detected at the T/B boundary without inhibition of phagocytosis (40), inhibition of phagocytosis is necessary to detect apoptosis of 56R/Vκ38C+ cells in MZ. The MZ is rich in phagocytes, including metallophilic macrophages and MZ macrophages (41), both of which may immediately phagocytose apoptotic B cells. As a consequence, apoptotic cells are not usually found in the MZ without treatment that inhibits phagocytosis such as clodronate or the mutant form of MFG-E8. Because dead cells generate various immunostimulatory molecules called damage-associated molecular patterns (DAMPs) (42), efficient clearance of apoptotic cells in MZ suggests that the MZ is a suitable location for deletion of self-reactive B cells. CD40L augments survival of both anergic B cells (30) and 56R/Vκ38C+ cells. However, excess CD40L induces autoantibody production in 56R mice but not in 3H9 mice in which low-avidity self-reactive B cells are tolerized by anergy. This suggests that CD40L may fully reverse tolerance of 56R/Vκ38C+ cells but not anergy, although CD40L augments survival of anergic B cells. Thus, anti-Sm/RNP 56R/Vκ38C+ B cells appear to be distinct from anergic B cells in both localization and sensitivity to survival signals such as CD40L for breaking tolerance.
Breakdown of tolerance of anti-Sm B cells appears to be crucial in SLE because its pathogenesis involves antibodies to RNA-related antigens rather than anti-DNA antibodies (23). Previous studies using anti-Sm antibody-Tg mice demonstrated that tolerance of anti-Sm B cells involves mechanisms such as anergy, developmental arrest, and ignorance (7, 22, 36, 43). Whether anti-Sm MZ B cells are deleted by apoptosis in these models needs to be addressed. Nonetheless, our results demonstrate that MZ deletion is one of the crucial tolerance mechanisms for anti-Sm/RNP B cells, suggesting that a defect in such might be involved in pathogenesis of lupus. Tolerance of MZ anti-Sm/RNP B cells but not of anergic B cells is fully reversed by excess CD40L. Excess CD40L was shown in human and murine lupus (18) and appears to play a role in pathogenesis (18), supporting the involvement of the defect in MZ deletion in lupus. Further studies on this tolerance mechanism may elucidate the pathogenesis of autoimmune diseases including SLE, leading to development of new therapies.
Materials and Methods
Standard procedures and methods such as animal handling, flow cytometry, ELISA, and immunohistochemistry, as well as procedures for the generation of anti-idiotype antibodies, are described in SI Materials and Methods.
Hybridoma Analysis.
Spleen cells were obtained from 8- and 33-wk-old female 56R and CD40L/56R mice and were stimulated with 2.5 μg/mL LPS (E. coli 0111:B4) (Sigma). LPS-stimulated spleen cells were fused with X63.Ag8.653 myeloma cells and distributed in culture plates under limiting dilution conditions (500–2,500 cells/well). After confirming that the wells contained only one colony under microscopy, we used hybrids for further study. Genomic DNA was extracted, and the presence or absence of 56R H chain gene was determined by PCR (Table S3) (15). Expression of Ig κ and λ L chain was determined by a sandwich ELISA using goat anti-mouse Ig κ and goat anti-mouse Ig λ antibody (Southern Biotechnology). Vκ use in 56R H-chain-containing hybrids was analyzed by PCR using primers listed in Table S3 as described previously (15, 17, 44–48).
Treatment of Mice with Clodronate Liposomes and MFG-E8 D89E.
Clodronate liposomes were prepared as described previously (31). Details are provided in SI Materials and Methods. The MFG-E8 D89E mutant (a gift from S. Nagata, Kyoto University, Kyoto, Japan) was described previously (33). Mice were injected i.v. with 500 μg clodronate liposome in 100 μL of PBS or 0.4 μg of MFG-E8 D89E (49) in 200 μL of PBS.
Supplementary Material
Acknowledgments
We thank Dr. N. Toyama-Sorimachi (International Medical Center of Japan) and Dr. T. Kina (Kyoto University) for cell lines; Dr. S. Nagata (Kyoto University), Dr. S. Hirose (Juntendo University), Dr. K. Yamamoto (University of Tokyo), Dr. T. Hachiya [Medical and Biological Laboratories Co., Ltd (MBL)], and Drs. Y. Sekine and Y. Sasaki (Tokyo Medical and Dental University) for reagents; Dr. S. Shimizu (Tokyo Medical and Dental University) for a reagent and helpful discussion; Dr. Y. Hitomi (Duke University) for help with statistical analysis; Dr. Y. Aiba and Dr. M. Sumita for initial work of this study; and Ms. Y. Miyamoto and Ms. M. Fujimoto for technical assistance. This work was supported, in part, by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Japan Society for the Promotion of Science.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1204509109/-/DCSupplemental.
References
- 1.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: An approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodnow CC, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 3.Nemazee DA, Bürki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 4.Radic MZ, Erikson J, Litwin S, Weigert M. B lymphocytes may escape tolerance by revising their antigen receptors. J Exp Med. 1993;177:1165–1173. doi: 10.1084/jem.177.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodnow CC, Crosbie J, Jorgensen H, Brink RA, Basten A. Induction of self-tolerance in mature peripheral B lymphocytes. Nature. 1989;342:385–391. doi: 10.1038/342385a0. [DOI] [PubMed] [Google Scholar]
- 6.Hannum LG, Ni D, Haberman AM, Weigert MG, Shlomchik MJ. A disease-related rheumatoid factor autoantibody is not tolerized in a normal mouse: Implications for the origins of autoantibodies in autoimmune disease. J Exp Med. 1996;184:1269–1278. doi: 10.1084/jem.184.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santulli-Marotto S, Retter MW, Gee R, Mamula MJ, Clarke SH. Autoreactive B cell regulation: Peripheral induction of developmental arrest by lupus-associated autoantigens. Immunity. 1998;8:209–219. doi: 10.1016/s1074-7613(00)80473-2. [DOI] [PubMed] [Google Scholar]
- 8.Cyster JG, Goodnow CC. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity. 1995;3:691–701. doi: 10.1016/1074-7613(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 9.Mandik-Nayak L, Bui A, Noorchashm H, Eaton A, Erikson J. Regulation of anti-double-stranded DNA B cells in nonautoimmune mice: Localization to the T-B interface of the splenic follicle. J Exp Med. 1997;186:1257–1267. doi: 10.1084/jem.186.8.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noorchashm H, et al. Characterization of anergic anti-DNA B cells: B cell anergy is a T cell-independent and potentially reversible process. Int Immunol. 1999;11:765–776. doi: 10.1093/intimm/11.5.765. [DOI] [PubMed] [Google Scholar]
- 11.Lesley R, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 12.Thien M, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–798. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Chen C, et al. The site and stage of anti-DNA B-cell deletion. Nature. 1995;373:252–255. doi: 10.1038/373252a0. [DOI] [PubMed] [Google Scholar]
- 14.Chen C, et al. Deletion and editing of B cells that express antibodies to DNA. J Immunol. 1994;152:1970–1982. [PubMed] [Google Scholar]
- 15.Erikson J, et al. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 1991;349:331–334. doi: 10.1038/349331a0. [DOI] [PubMed] [Google Scholar]
- 16.Fukuyama H, Nimmerjahn F, Ravetch JV. The inhibitory Fcgamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nat Immunol. 2005;6:99–106. doi: 10.1038/ni1151. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Jiang Y, Prak EL, Radic M, Weigert M. Editors and editing of anti-DNA receptors. Immunity. 2001;15:947–957. doi: 10.1016/s1074-7613(01)00251-5. [DOI] [PubMed] [Google Scholar]
- 18.Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: The dark side of a great activator. Semin Immunol. 2009;21:293–300. doi: 10.1016/j.smim.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kishi Y, et al. Augmented antibody response with premature germinal center regression in CD40L transgenic mice. J Immunol. 2010;185:211–219. doi: 10.4049/jimmunol.0901694. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi T, et al. Cutting Edge: Ectopic expression of CD40 ligand on B cells induces lupus-like autoimmune disease. J Immunol. 2002;168:9–12. doi: 10.4049/jimmunol.168.1.9. [DOI] [PubMed] [Google Scholar]
- 21.Clegg CH, et al. Thymus dysfunction and chronic inflammatory disease in gp39 transgenic mice. Int Immunol. 1997;9:1111–1122. doi: 10.1093/intimm/9.8.1111. [DOI] [PubMed] [Google Scholar]
- 22.Berland R, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. 2006;25:429–440. doi: 10.1016/j.immuni.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 23.Christensen SR, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Brahms H, et al. The C-terminal RG dipeptide repeats of the spliceosomal Sm proteins D1 and D3 contain symmetrical dimethylarginines, which form a major B-cell epitope for anti-Sm autoantibodies. J Biol Chem. 2000;275:17122–17129. doi: 10.1074/jbc.M000300200. [DOI] [PubMed] [Google Scholar]
- 25.Riemekasten G, et al. A novel epitope on the C-terminus of SmD1 is recognized by the majority of sera from patients with systemic lupus erythematosus. J Clin Invest. 1998;102:754–763. doi: 10.1172/JCI2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witsch EJ, Cao H, Fukuyama H, Weigert M. Light chain editing generates polyreactive antibodies in chronic graft-versus-host reaction. J Exp Med. 2006;203:1761–1772. doi: 10.1084/jem.20060075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Li H, Weigert M. Autoreactive B cells in the marginal zone that express dual receptors. J Exp Med. 2002;195:181–188. doi: 10.1084/jem.20011453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekiguchi DR, et al. Development and selection of edited B cells in B6.56R mice. J Immunol. 2006;176:6879–6887. doi: 10.4049/jimmunol.176.11.6879. [DOI] [PubMed] [Google Scholar]
- 29.Tsubata T, Wu J, Honjo T. B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature. 1993;364:645–648. doi: 10.1038/364645a0. [DOI] [PubMed] [Google Scholar]
- 30.Lesley R, Kelly LM, Xu Y, Cyster JG. Naive CD4 T cells constitutively express CD40L and augment autoreactive B cell survival. Proc Natl Acad Sci USA. 2006;103:10717–10722. doi: 10.1073/pnas.0601539103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 32.Witsch EJ, Bettelheim E. Allelic and isotypic light chain inclusion in peripheral B cells from anti-DNA antibody transgenic C57BL/6 and BALB/c mice. J Immunol. 2008;180:3708–3718. doi: 10.4049/jimmunol.180.6.3708. [DOI] [PubMed] [Google Scholar]
- 33.Hanayama R, et al. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 34.Khan SN, et al. Editing and escape from editing in anti-DNA B cells. Proc Natl Acad Sci USA. 2008;105:3861–3866. doi: 10.1073/pnas.0800025105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandik-Nayak L, Racz J, Sleckman BP, Allen PM. Autoreactive marginal zone B cells are spontaneously activated but lymph node B cells require T cell help. J Exp Med. 2006;203:1985–1998. doi: 10.1084/jem.20060701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian Y, Wang H, Clarke SH. Impaired clearance of apoptotic cells induces the activation of autoreactive anti-Sm marginal zone and B-1 B cells. J Immunol. 2004;172:625–635. doi: 10.4049/jimmunol.172.1.625. [DOI] [PubMed] [Google Scholar]
- 37.Thorn M, Lewis RH, Mumbey-Wafula A, Kantrowitz S, Spatz LA. BAFF overexpression promotes anti-dsDNA B-cell maturation and antibody secretion. Cell Immunol. 2010;261:9–22. doi: 10.1016/j.cellimm.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lau CM, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliver AM, Martin F, Gartland GL, Carter RH, Kearney JF. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. Eur J Immunol. 1997;27:2366–2374. doi: 10.1002/eji.1830270935. [DOI] [PubMed] [Google Scholar]
- 40.Ekland EH, Forster R, Lipp M, Cyster JG. Requirements for follicular exclusion and competitive elimination of autoantigen-binding B cells. J Immunol. 2004;172:4700–4708. doi: 10.4049/jimmunol.172.8.4700. [DOI] [PubMed] [Google Scholar]
- 41.Pillai S, Cariappa A, Moran ST. Marginal zone B cells. Annu Rev Immunol. 2005;23:161–196. doi: 10.1146/annurev.immunol.23.021704.115728. [DOI] [PubMed] [Google Scholar]
- 42.Bianchi ME. DAMPs, PAMPs and alarmins: All we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 43.Borrero M, Clarke SH. Low-affinity anti-Smith antigen B cells are regulated by anergy as opposed to developmental arrest or differentiation to B-1. J Immunol. 2002;168:13–21. doi: 10.4049/jimmunol.168.1.13. [DOI] [PubMed] [Google Scholar]
- 44.Huse WD, et al. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science. 1989;246:1275–1281. doi: 10.1126/science.2531466. [DOI] [PubMed] [Google Scholar]
- 45.Prak EL, Trounstine M, Huszar D, Weigert M. Light chain editing in kappa-deficient animals: A potential mechanism of B cell tolerance. J Exp Med. 1994;180:1805–1815. doi: 10.1084/jem.180.5.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramsden DA, Paige CJ, Wu GE. Kappa light chain rearrangement in mouse fetal liver. J Immunol. 1994;153:1150–1160. [PubMed] [Google Scholar]
- 47.Retter MW, Nemazee D. Receptor editing occurs frequently during normal B cell development. J Exp Med. 1998;188:1231–1238. doi: 10.1084/jem.188.7.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schlissel MS, Baltimore D. Activation of immunoglobulin kappa gene rearrangement correlates with induction of germline kappa gene transcription. Cell. 1989;58:1001–1007. doi: 10.1016/0092-8674(89)90951-3. [DOI] [PubMed] [Google Scholar]
- 49.Asano K, et al. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J Exp Med. 2004;200:459–467. doi: 10.1084/jem.20040342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carsetti R, Köhler G, Lamers MC. Transitional B cells are the target of negative selection in the B cell compartment. J Exp Med. 1995;181:2129–2140. doi: 10.1084/jem.181.6.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.