

Abstract
Fibroblastic reticular cells (FRCs) are lymphoid stromal cells essential to T-cell migration and survival. Although FRCs are targets of multiple viral infections, little is known about their role during infection due to the cells’ scarcity and difficulty in isolating in vivo. To initiate studies of interactions among FRCs, viruses, and immune cells, we isolated and immortalized CD45−gp38+CD35−CD31−CD44+VCAM1+ cell lines from C57BL/6 mice designated as immortalized FRC. Using these cloned cell lines, we have established that FRCs express the major histocompatibility complex (MHC) II molecule, a factor necessary for stimulation of CD4+ T cells thought to be expressed primarily by antigen-presenting cells, along with other T-cell stimulatory ligands in an IFN-γ–dependent manner. In this environment, lymphocytic choriomeningitis virus (LCMV)-infected iFRCs activated naive LCMV-specific CD4+ and CD8+ T cells while limiting expansion of effector LCMV-specific T cells. Thus, FRCs effectively presented antigen along with activating signals during viral infection using both MHC I and MHC II molecules, illustrating a previously undescribed interaction with CD4+ T cells and indicating a unique role for FRCs.
Keywords: reticular network, T cell activation, secondary lymphoid stroma
Clearance of chronic viral infection is dependent upon effective anti-viral CD4+ and CD8+ T-cell responses (1–3) that are generated within secondary lymphoid organs (SLOs), including lymph nodes, spleen, thymus, Peyer’s patches, and tonsils. Anti-viral CD4+ T cells are necessary for anti-viral CD8+ T and B-cell responses and for noncytolytic-mediated viral clearance, whereas CD8+ T-cell responses are primarily necessary for cytolytic killing of infected cells. Within SLOs, mature naive T lymphocytes interact with antigen-presenting cells (APCs), encounter soluble signals and antigens, and undergo activation. All of these functions are facilitated and supported by fibroblastic reticular cells (FRCs), a stromal cell found only within SLOs, which form the reticular network (4, 5). The reticular network is essential to the immune response, as mice that lack lymphoid architecture have impaired immune responses (6–8). Although impairment of the reticular network adversely affects immunity, because of the low numbers of FRCs (<0.5% of total cells in SLOs), their role and activity during an anti-viral immune response is poorly understood.
Nevertheless, the FRC network is deeply involved in T-cell function because the network’s specialized microarchitecture regulates the structure and efficient functioning of SLOs by compartmentalizing T-cell zones and creating a three-dimensional roadway system for the migration and interaction of T lymphocytes and APCs (9, 10). FRCs control migration through SLOs by expressing chemokines CCL19 and CCL21, which bind CCR7 expressed on T cells and APCs (7). In addition to structure, FRCs provide important regulatory signals that control lymphocyte survival. FRCs secrete IL-7 (11, 12), a crucial survival signal for naive T cells. Abrogation of FRC–T-cell interactions results in T-cell loss (12). Furthermore, FRCs may contribute to the maintenance of peripheral CD8+ T-cell tolerance as they are able to present peripheral endogenous antigens (13, 14) and, further, may induce deletion of CD8+ T cells specific to these antigens (15).
Although our understanding of this important cell type has grown over the last several years, very little is known about how FRCs respond during an inflammatory event such as viral infection. The majority of studies describing the function of FRCs have done so under steady-state conditions. However, FRCs are located within SLOs where the main function is immune surveillance and activation. FRCs are the target of at least several viruses, including the lymphocytic choriomeningitis virus (LCMV), Ebola, Lassa, Marburg, and simian immunodeficiency virus, as well as the parasitic eukaryotes Plasmodium yoelli and Leishmania major (16), indicating both their importance during infection and the need to study their cellular, molecular, and signaling actions.
LCMV is a prototypic noncytolytic virus that has been used with great success to examine host–viral interactions. During acute infection with LCMV, FRCs contribute to coordination of T-cell trafficking by transiently down-regulating CCL21 at 3 d post infection, thereby abrogating movement into the T-cell zone (17). FRCs also appear to negatively regulate activated anti-viral T cells. During murine infection with the clone 13 (CL-13) strain of LCMV, which causes a persistent infection characterized by T-cell exhaustion, FRCs within the spleens and lymph nodes are infected and express the programmed death ligand-1 (PD-L1) (18), which likely prevents clearance of infected cells and contributes to T-cell exhaustion (19). Together, these studies suggest that FRCs play an integral role during anti-viral immune responses.
On the basis of the essential role of FRCs under homeostatic conditions and mounting evidence that FRCs are targeted by several viruses and are important during anti-viral T-cell responses, questioning these cells’ influence on activation and maintenance of T-cell responses is important for understanding viral pathogenesis and persistence. However, their relative rarity and the presence of other stromal cell types within SLOs has hindered analysis of specific FRC function during viral infection in vivo. Additionally, sorting enough FRCs to use ex vivo is stressful on the cells and requires an excessively large number of mice as the cells’ source. Thus, to enable better characterization of this important cell type and test its interaction with viruses and its role in anti-viral T-cell responses, we generated immortalized FRC cell (iFRC) lines from the spleens of C57BL/6 mice and tested their interactions with LCMV-specific T cells. We demonstrate that not only do these cells express important T-cell stimulatory ligands, including MHC II when stimulated with IFN-γ, but also that LCMV-infected FRCs are able to stimulate the proliferation of naive CD8+ and CD4+ LCMV-specific T cells while, on the other hand, inhibiting proliferation of activated T cells.
Results
Isolation of Immortalized C57BL/6 FRC Cell Lines.
Because FRCs make up <0.5% of lymph node (11) and <0.1% of spleen cells, we generated three iFRC lines—iFRC-T, iFRC-TN, and iFRC2—from the spleens of C57BL/6 mice for our biological and biochemical studies. For that purpose, splenic stromal cells were cultured from splenocytes isolated from the spleens of C57BL/6 mice digested with collagenase D. Adherent and nonadherent cells were separated and adherent cells were passaged to obtain a culture enriched with stromal cells, which were then immortalized with a doxycycline-regulated lentiviral expression vector encoding both the large and the small SV40 T antigen. We performed this procedure on three separate occasions. Two of the resulting immortalized stromal cultures (SC-T and SC-TN) were highly enriched in nonhematopoietic (CD45−) cells that bore the phenotype of FRCs (gp38+CD31−CD35−) whereas the third culture was less enriched with only 14% CD45− cells (Fig. 1A). Subsequently, the immortalized stromal cultures were sorted to isolate FRCs, and these cells were cloned to derive the final lines that are each CD45−gp38+CD31−CD35−CD44+VCAM1+ (Fig. 1B), markers that are indicative of FRCs in vivo (4, 11, 14).
Fig. 1.

Phenotype of iFRC cell lines cloned from immortalized stromal cultures derived from spleens of C57BL/6 mice. (A) Expression of FRC markers in cultures enriched for splenic stromal cells and then immortalized with lentiviral vector encoding the large and small SV40 T antigen. These cultures were sorted and cloned to obtain the iFRC lines. (B) iFRCs cloned from the three immortalized stromal cultures express markers typical of FRCs in vivo (CD45−gp38+CD31−CD35−CD44+VCAM1+) as assessed by flow cytometry.
All three cell lines display traits that are characteristic of FRCs. Morphologically, they are nebulous, spreading cells with large cytoplasmic processes (Fig. 2A). In vivo, signals from other lymphoid cells are important in defining and maintaining FRC architecture (20). We tested whether iFRCs were responsive to coculture with lymphocytes by incubating them with nonadherent splenic lymphocytes. Cocultured iFRCs exhibited different growth and morphology from that when grown alone, appearing round with thin, long processes rather than the fanning processes (Fig. 2A), which demonstrates their responsiveness to lymphocyte-associated factors. A key role for FRCs is providing survival signals to naive T cells via the expression of IL-7 (11, 12) and CCL19 (11); the latter is also important in APC and T-cell migration (11, 21, 22). All three iFRC lines were positive for expression of il7 and ccl19 mRNA (Fig. 2B). When incubated with naive CD3+ T cells in vitro over a 4-d period, iFRCs enhanced survival of T cells (15–59%) in comparison with T cells alone (2–5%) (Fig. 2C). The iFRC-T and iFRC-2 lines maintained T-cell viability better than the iFRC-TN line. Collectively, these results suggest that the isolated cells are indeed of an FRC phenotype.
Fig. 2.
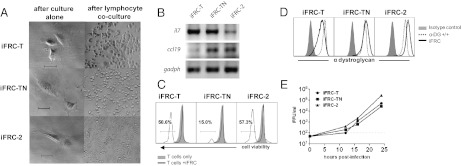
Phenotype of iFRC cell lines and their susceptibility to LCMV infection. (A) Cellular morphology of immortalized FRC lines cultured in the presence and absence of lymphocytes. (Scale bar, 50 μm.) (B) iFRCs express mRNA for T-cell survival factors il7 and ccl19. (C) Coculture of iFRCs and T cells results in enhanced survival of T cells after 4 d as indicated by live/dead cell viability staining. (D) iFRCs express the LCMV receptor, α-dystroglycan, and (E) are susceptible to LCMV infection. iFRCs were infected at an MOI of 0.1. Viral titers are expressed as focus forming units (FFU) per milliliter of media.
iFRCs Produce Infectious LCMV Virus.
During infection of C57BL/6 mice, FRCs are infected by a persistent strain of LCMV, CL-13. The cellular receptor for CL-13 (as well as other Old World arenaviruses) is α-dystroglycan (23), the host receptor for laminin (24). To further validate the iFRCs and to investigate FRC susceptibility, we analyzed iFRC expression of α-dystroglycan. By flow cytometry, all three iFRC clones express high levels of functionally glycosylated α-dystroglycan (Fig. 2D), the form of the receptor necessary for laminin and LCMV binding. iFRC-2 expressed the highest levels, and iFRC-T expressed the lowest levels of the three lines. In vitro, this coincided with susceptibility to LCMV infection as all iFRC lines, when infected with CL-13 (MOI 0.1), produced high titers of infectious virus by 24 h post infection (Fig. 2E) with iFRC-2 producing the highest titers. Considering that iFRCs were reliably infected with CL-13 in vitro, consistent with FRCs’ susceptibility in vivo, our results establish the validity of analyzing virally infected FRC interactions with T cells in this experimental system.
iFRCs Express Factors Necessary for Activation of Naive T Cells.
Activation of T cells requires two interactions: (i) engagement of the T-cell receptor (TCR) by MHC class I (MHC I) for CD8 T cells or MHC class II (MHC II) for CD4 T cells and (ii) engagement of CD28 by CD80 or CD86. To investigate FRC expression of T-cell ligands and changes in expression during inflammation, we investigated iFRC expression of important T-cell stimulatory molecules in the presence of type I and type II IFN. Untreated iFRCs exhibited expression of CD80, with the highest expression in iFRC-T, and little to no expression of CD86 (Fig. 3A). Expression of neither molecule was altered by IFN-α or IFN-β treatment (Fig. S1). Comparatively, IFN-γ treatment up-regulated CD80 highly on iFRC-T and modestly on iFRC-TN but had no effect on CD86 expression. When we examined MHC expression, MHC I and, surprisingly, MHC II were highly up-regulated after IFN-γ treatment (Fig. 3A). MHC II was up-regulated in a dose-dependent manner (Fig. S2A). Analysis of FRCs in vivo (CD45−gp38+CD31−CD35−) also demonstrated MHC II expression in both the spleen and the lymph node (Fig. 3B). To test if MHC II expression was increased with the presence of IFN-γ, FRCs from naive mice were compared with those from mice infected with LCMV Armstrong 53b, which causes acute viral infection with a strong cytotoxic T lymphocyte response accompanied by IFN-γ but does not infect FRCs (18). FRCs from mice 5 d after infection exhibited significantly higher fluorescent intensities than FRCs during homeostasis (Fig. 3 B and C). Because T cells interact so closely with FRCs, MHC II expression strongly indicates the ability to present antigen to CD4+ T cells. Together, these data show that FRCs express the factors necessary for activation of naive CD4+ and CD8+ T cells, suggesting the possibility that FRCs may be able to fulfill a role thought to be restricted to APCs.
Fig. 3.

iFRCs express T-cell stimulatory ligands in response to IFN-γ. (A) Expression of factors necessary for naive T-cell activation in the absence and presence of IFN-γ. MHC II was up-regulated after IFN stimulation. (B) Analysis of FRCs isolated from spleen and lymph nodes of C57BL/6 mice demonstrated MHC II expression. (C) Analysis of mean fluorescent intensity in splenic FRCs indicates that MHC II was elevated during viral infection when IFN-γ is also elevated. (D) Analysis of suppressive ligands in the iFRC–T-cell line shows constitutive expression of galectin-9 (TIM-3 ligand) and up-regulation of PD-L1 when stimulated with IFN-β and IFN-γ. (E) Effect of LCMV infection on expression in the presence and absence of IFN-γ. iFRC-T was infected with LCMV (MOI 1) and then, 24 h later, after infection was established, treated with IFN-γ.
FRCs have also been shown to repress T-cell responses. During chronic LCMV infection, FRCs up-regulate PD-L1, the ligand for PD-1 on activated T cells that down-regulates their effector activity (25). Furthermore, concomitant signaling through PD-1 and the T-cell Ig mucin-3 (TIM-3) cooperatively suppresses effector T-cell activity (26). Examination of PD-L1 and galectin-9 (TIM-3 ligand) revealed constitutive expression of galectin-9 whereas PD-L1 expression was up-regulated by treatment with IFN-β or IFN-γ but not with IFN-α (Fig. 3D) in a dose-dependent manner (Fig. S2B). This result is consistent with other studies describing PD-L1 up-regulation in the presence of IFN-γ (26), further affirming the FRC phenotype of these cell lines.
LCMV Inhibits IFN-γ–Dependent FRC Cell-Surface Expression.
FRCs are a target of viral infection, including during chronic infection with LCMV CL-13. In dendritic cells (DCs), the classical APC, CL-13 causes down-regulation of T-cell stimulatory molecules to hinder T-cell activation (27, 28). To investigate whether viral infection influenced FRC expression of T-cell ligands, we examined their expression during LCMV infection in the presence and absence of IFN-γ. iFRC-T was infected with LCMV [multiplicity of infection (MOI 1)] and, 24 h later, after infection was established, treated with IFN-γ. LCMV infection alone did not alter expression patterns of T-cell ligands compared with uninfected iFRCs (Fig. S3), indicating that infection alone is insufficient to alter the FRC expression pattern. However, in the presence of IFN-γ, infection inhibited up-regulation of MHC I and MHC II, but had minimal effects on PD-L1 and CD80 expression (Fig. 3E). Together, these data indicate that LCMV infection impacts presentation of antigen by FRCs during inflammatory conditions and that in vivo up-regulation of PD-L1 during CL-13 infection is not due to virus but likely to IFN-γ, IFN-β, and/or other signaling factors.
FRCs Activate Naive LCMV-Specific T Cells but Limit Expansion of Activated LCMV-Specific T Cells.
FRCs display peripheral tissue antigens to CD8+ T cells (13, 29); however, there is no published data regarding their ability to present antigen from infectious virus. Furthermore, because MHC II has not previously been described on FRCs, there has been no study of their ability to present antigen to CD4+ T cells. We investigated whether it was possible for FRCs to display and prime naive T cells to foreign antigens by testing the ability of peptide-loaded or LCMV-infected iFRCs to stimulate LCMV-specific CD4+ and CD8+ T cells. Because the iFRC-T line was the most responsive to IFN-γ treatment, we focused on this line for these experiments. To test CD8+ T-cell priming, iFRCs either were incubated with an LCMV CD8+ T-cell epitope (gp33–41) or infected with LCMV CL-13 (MOI 3) for 24 h to achieve 100% infection and then incubated with carboxyfluorescein succinimidyl ester (CFSE)-stained naive LCMV-specific transgenic TCR CD8+ T cells (P14) for 4 d. Splenic DCs were used as positive controls. To ensure that any observed proliferation was due to antigen stimulation by iFRC-T, P14 cells were sorted to obtain the CD44lo population to eliminate cells with a “memory” phenotype. In the presence of peptide-pulsed iFRC-T cells, P14 cells exhibited successive generations of proliferation (Fig. 4A) and demonstrated production of IFN-γ (Fig. 4B). Similarly, LCMV-infected iFRC-Ts were able to stimulate proliferation (Fig. 3A). Control DCs isolated from spleens of C57BL/6 mice also successfully initiated proliferation with higher induction of IFN-γ than iFRC-T when loaded with gp33–41 peptide (Fig. 3 A and B). The DCs did not appear to induce as many P14 cells to proliferate, but this is likely due to general access rather than antigen-presenting ability. DCs were used at an approximate DC:T-cell ratio of 1:20 and iFRCs were used at ∼1:40, but the iFRCs were very large (>50 μm) and continued to divide, whereas DCs were relatively small (<10 μm) and did not divide in vitro.
Fig. 4.

iFRCs present viral antigen and successfully activate LCMV-specific T cells. (A) Proliferation of CFSE-stained P14 CD8+ T cells when stimulated by GP33–41 peptide presented by iFRC-T cells (Upper panels) or viral antigen presented by LCMV-infected iFRC-T cells (Lower panels). Similarly treated DCs are shown as controls. (B) P14 cells stimulated by iFRC-T–presenting GP33–41 peptide are able to express IFN-γ. (C) (Upper panels) Proliferation of CFSE-stained Smarta CD4+ T cells when stimulated by GP61–80 peptide presented by iFRC-T cells in the absence and presence of IFN-γ to up-regulate T-cell ligands. (Lower panels) Smarta proliferation when antigen is presented by LCMV-infected iFRC-T cells. Similarly treated DCs are shown as controls. (D) Expression of CD25 and CD44, markers of T-cell activation, on Smarta CD4+ T cells activated by LCMV-infected iFRCs. (E) Peptide-loaded iFRC-T cells limit proliferation of P14 CD8+ T cells and Smarta CD4+ T cells isolated from mice challenged 5 d before coculture with LCMV Armstrong 53b. Data are representative of three independent experiments.
In a set of similar experiments, LCMV-specific CD4+ T cells were examined for activation by iFRC-T cells incubated with LCMV CD4+ T-cell epitope (gp61–77) or LCMV CL-13 (MOI 3) 24 h before coculturing with CFSE-stained naive LCMV-specific transgenic TCR CD4+ T cells (Smarta). Smarta cells incubated with iFRC-T successfully initiated CD4+ T-cell proliferation when treated with IFN-γ (Fig. 4C) whereas Smarta cells alone with peptide and IFN-γ did not proliferate. Conversely, DCs loaded with peptide instigated Smarta cell proliferation in the absence of IFN-γ but very little in the presence of IFN-γ. This is very likely due to the inhibitory effects of IFN-γ on T-cell proliferation (30, 31). Hence, it is interesting that IFN-γ–treated iFRCs were able to overcome IFN-γ–dependent inhibition of T-cell proliferation. When we tested the ability of iFRC-T to present viral antigen from infectious virus on MHC II, untreated infected iFRCs did not significantly stimulate Smarta cells (Fig. 4C). However, treatment with IFN-γ at the time of infection to up-regulate MHC II expression without preventing infection permitted iFRC-T cells to stimulate Smarta cell proliferation, whereas infected dendritic cells (which express little MHC II) and Smarta-only controls treated in the same manner failed to do so (Fig. 4C). Smarta cells stimulated by infected iFRCs expressed CD44 and CD25, markers for T-cell activation (Fig. 4D). These data demonstrate that iFRCs are able to present intracellular antigens on MHC II to naive CD4+ T cells.
Because FRCs may have one or more mechanism(s) of T-cell suppression, including PD-L1, we tested the effect of iFRCs on activated virus-specific T cells. To activate T cells, we primed mice with virus 5 d in advance before harvesting and isolating the LCMV-specific T cells. Peptide-loaded iFRC-T cells and LCMV-specific T cells were coincubated for 4 d without the addition of IFN-γ. iFRCs limited the expansion of activated P14 cells, resulting in only five generations of proliferation whereas incubation with DCs resulted in six generations (Fig. 4E). Activated Smarta cells were further repressed and did not demonstrate any significant proliferation. There were no significant differences between IFN-γ–treated and untreated wells for either P14 or Smarta cells (Fig. S4); however, activated T cells produce IFN-γ and thus would supply IFN-γ in untreated wells.
Discussion
Viral infection induces anti-viral T-cell responses that play a key role in the clearance of the virus, and orchestration of these T-cell responses is determined at least in part by FRCs. In this study, we developed immortalized cell lines from splenic FRCs of C57BL/6 mice to better illustrate the role of FRCs in viral infection, which to date has been poorly defined. These cells proved to be valuable for use with a variety of transgenic reagents, the majority of which are available only on the H-2b C57BL/6 background. Previous FRC lines generated from long-term culture of lymph node stroma of BALB/c mice (20) cannot be used in this manner due to MHC mismatch. Furthermore, there has been little study of FRCs within the spleen, the SLO that sees the greatest number of T cells per day, making it an important organ in assessing T-cell responses.
Using these cell lines, we found that FRCs are capable of presenting antigen from infectious virus not only to CD8+ T cells but also to CD4+ T cells. This was a unique find, given the paradigm that CD4+ T-cell activation was previously thought to be restricted to APCs because MHC II is expressed only on these cells. Earlier studies showed that FRCs can present peripheral tissue antigens to CD8+ T cells (13) but did not examine CD4+ T cells. Indeed, we show that FRCs express MHC II, both in vitro and in vivo, in an IFN-γ–dependent manner, along with CD80, another factor necessary for T-cell activation. Our study is a unique description of MHC II on non-APC cells within SLOs. Epithelial cells in the lung have been reported to express MHC II (32); however, unlike lung epithelium, FRCs in SLOs regularly interact with naive T cells, as well as with effector T cells in the case of SLO infection, suggesting that FRCs are capable of multiple types of interactions. Interestingly, virally infected iFRCs induced more proliferation than peptide-loaded iFRCs. This is likely due to a constant supply of antigen due to viral replication, whereas peptide was only supplied 1 d before the addition of T cells. After 5 d, any remaining peptide was likely degraded. Additional study is necessary to identify the pathways by which infected FRCs load viral antigen onto MHC II. In DCs, autophagy has been shown to play a role in MHC II presentation of endogenous viral antigens (33, 34), making this pathway a likely candidate for presentation of viral antigens in FRCs.
Our study of the iFRC cell lines revealed not only up-regulation of MHC II in an IFN-γ–dependent manner but also up-regulation of additional cell-surface ligands relevant to T-cell activity. Both type I and type II IFN are released during viral infection; however, type I IFN, which is more ubiquitously produced, did not cause alterations in cell-surface expression of these ligands. Conversely, IFN-γ is released solely by immune cells and primarily by T cells, suggesting that FRCs are programmed to be responsive to T cells and APCs and that IFN-γ is an important communication factor between FRCs and these cells. Although IFN-γ (and IFN-β) also up-regulated PD-L1 on iFRCs, naive T cells do not express significant levels of PD-1 until after activation (35), which likely allows naive T cells to avoid PD-1/PD-L1–mediated suppression. In our experiments, iFRCs activated LCMV-specific T cells in vitro despite IFN-γ–dependent up-regulation of MHC and PD-L1. Together, our data suggest that FRCs may help to augment the initiation of the T-cell response and that FRCs have different functions depending on the activation state of the T-cell and the immune response. Furthermore, these results illustrate that the study of antigen presentation should not be focused solely on DCs. Our data propose a model in which FRCs support activation of naive T cells during the initiation of the anti-viral response and after the T-cell response is underway, FRCs then serve to modulate the resultant response by dampening or enhancing the T-cell activity.
In conclusion, our study highlights a potential role for nonhematopoietic FRCs in SLOs by influencing not just CD8+ but also CD4+ T-cell responses. Further investigation of this rare cell type is necessary to better characterize the role of MHC II expression in vivo by FRCs in SLOs during the immune response. With the availability of iFRCs, an improved understanding of FRC function, biochemistry, cell biology, and regulation during immune responses is now possible and should shed light on new therapeutic methods to modulate immune responses during vaccination and disease.
Materials and Methods
Establishment of Cell Lines.
C57BL/6 mice were acquired from the Rodent Breeding Colony at Scripps Research Institute. The use of mice was approved by the Scripps Research Institute Animal Care Committee. Spleens were harvested from naive C57BL/6 mice, digested for 30 min with collagenase D (1 mg/mL) and Dnase I (100 μg/mL) in RPMI with 5% FBS, and processed into single-cell suspension through a 100-μm cell strainer (BD Biosciences). Erythrocytes were lysed using 0.83% ammonium chloride, and the remaining cells were plated at 5E6 cells per milliliter of media (RPMI, 10% FBS, penicillin/streptomycin, l-glutamine, Hepes, 55 μM β-mercaptoethanol). Cells were incubated overnight, and the next day nonadherent cells were removed by washing the flask four times with PBS. The media was replaced, and the adherent cells were incubated for another 2 d in culture after which they were dissociated using 5 mM EDTA and then plated in a 24-well plate. Cells were monitored daily for 1–2 wk. When the cultures demonstrated proliferation, they were transduced with a doxycycline-regulated lentiviral expression vector (36) encoding both the large and the small SV40 T antigen. The virus encodes the reverse Tet transactivator rtTA3 linked by the foot-and-mouth disease virus (FMDV) cleavage factor 2A peptide sequence to the Zeocin resistance gene, which in turn is fused to the TAg coding sequence by a TaV-2a peptide sequence. The self-processing 2a peptide sequences ensure expression of three distinct proteins from a single mRNA expressed under control of the pTight tet-reponsive promoter. Cells were incubated with vector overnight, after which vector was removed and the cells were maintained in media with 1 μg/mL doxycycline. Once the culture was established, immortalized cells were harvested; stained with fluorescently conjugated anti-CD45, anti-gp38, anti-CD35, and anti-CD31 antibodies; and sorted for CD45−gp38+CD35−CD31− VCAM1+ cells on a FACSAria cell sorter (Becton-Dickinson). These cells were then cloned and restained with fluorescently conjugated anti-CD45, anti-gp38, anti-CD35, anti-CD31, anti-CD44, and anti-VCAM1 antibodies and analyzed on an LSR II flow cytometer (Becton-Dickinson). This process was performed independently three times to generate three separate iFRC cell lines.
Characterization of iFRC Expression.
To characterize cell-surface expression of the iFRC lines, the cell lines were harvested with 5 mM EDTA/PBS. To analyze α-dystroglycan expression, cells were stained with IIH6 antibody (kindly provided by Kevin Campbell, University of Iowa, Iowa City, IA) followed by a secondary anti-mouse IgM antibody conjugated to Alexa Fluor 488 (Invitrogen). For experiments in which cells were treated with IFN, iFRCs were incubated with 100 U/mL of IFN-α, IFN-β, or IFN-γ for 24 h before harvest. To analyze T-cell stimulatory ligands, cells were stained with fluorescently conjugated anti-MHC II (I-A/I-E), anti-MHC I (H-2Db), anti-CD80, anti-CD86, anti-PD-L1, and anti-CD275 antibodies (eBioscience) and analyzed by flow cytometry.
Coincubation with Splenocytes.
iFRCs were cultured in the absence of doxycycline for 4 d and then plated in eight-well slides at 200 cells/well. Lymphocytes were harvested from the spleens of C57BL/6 mice, processed into single-cell suspension, and subjected to lysis of red blood cells. To remove stromal cells that were adherent, the remaining lymphocytes were plated in T175 flasks (5 × 107 cells/flask) and incubated overnight. The next day, nonadherent cells were removed, counted, and added to the iFRC cultures in a total of 200 μL. The cocultures were incubated for 7 d, with 200 μL of fresh media added at day 4. At day 7, nonadherent cells were vigorously washed off with PBS four times, and iFRCs were photographed on an Axiovision S100 inverted microscope fitted with an Axiovision camera (Zeiss).
For assessment of cell viability, iFRCs were cultured as above and then plated at 5 × 104 cells per well in a 24-well plate. To obtain naive T cells, splenocytes were isolated as above and T cells were isolated using the Easysep CD90+ positive selection kit (Stemcell) and then stained with CFSE using the CellTrace CFSE Cell Proliferation Kit (Invitrogen). Subsequently, 1–2 × 106 cells were added to wells with iFRCs. Cells were incubated at 37 °C for 4 d, and then T cells were harvested and stained with the Violet LIVE/DEAD Fixable Dead Cell Stain Kit (Invitrogen) and fluorescently conjugated anti-CD3 antibody. Staining was assessed by flow cytometry.
RNA Isolation and RT-PCR.
iFRCs were cultured in the absence of doxycycline for 4 d, and then RNA was extracted using the Rneasy plus mini kit (Qiagen). First-strand cDNA synthesis (SuperScript III; Invitrogen) with oligo(dT)12–18 primer (Invitrogen) was performed according to the manufacturer’s instructions. Primers previously described for il7 (37) and ccl19 (11) were used to detect mRNA of interest, and the housekeeping gene gadph was amplified as a control.
Characterization of MHC II on in Vivo FRCs.
Spleens and inguinal lymph nodes were harvested from either naive C57BL/6 mice or 5 d after i.v. infection with LCMV Armstrong [2 × 106 plaque forming units (pfu)] (n = 5). Spleens were processed as described above. Inguinal lymph nodes were teased open and digested in collagenase D/Dnase I (1 mg/mL) for 1 h with repeated agitation every 15 min as previously described (11). Lymph node cells were pooled by treatment, whereas spleens were treated individually. The isolated cells were stained with anti-CD45, anti-CD31, anti-CD35, anti-gp38, and anti-MHC II (I-A/I-E) and analyzed by flow cytometry. MHC II analysis was performed on CD45−gp38+CD31−CD35− cells.
T-Cell Proliferation.
iFRCs were cultured in the absence of doxycycline for 4 d and then plated at 2.5–5 × 104 cells per well in a 24-well plate. As a control, DCs were isolated from spleens of C57BL/6 mice after collagenase D treatment using the Easysep CD11c+ positive selection kit (Stemcell) according to the manufacturer’s instructions. This kit resulted in >80% purity of DCs (CD45+CD11chi CD19−CD90−). The isolated DCs were plated at 1 × 105 cells per well. The next day, iFRCs and DCs were treated with MHC I-restricted LCMV GP33–41 peptide (1 μg/mL; immunodominant H2-Db restricted CD8 T-cell epitope), MHC II-restricted LCMV GP61–80 peptide (2 μg/mL; immunodominant I-Ab restricted CD4 T-cell epitope), or LCMV CL-13 (MOI 3). IFN-γ (50 U/mL) was added to specified wells after adding the initial treatment. The following day, spleens were collected from transgenic mice with LCMV-specific CD4+ T cells (Smarta; recognizes I-Ab–restricted GP61–80 epitope) or CD8+ T cells (P14; recognizes H2-Db–restricted GP33–41 epitope) that were either naive or had been challenged 5 d previously with LCMV Armstrong (2 × 105 pfu, intraperitoneally) and processed as described above. CD4+ T cells or CD8+ T cells were enriched using the Easysep CD4+ or CD8+ T-cell–negative selection kit (Stemcell). Naive T cells were stained and sorted on a Moflo XDP flow cytometer (CD3+ CD4+ CD44lo or CD3+ CD8+ CD44lo). Both naive and activated T cells were stained with CFSE (as above), and 1–2 × 106 cells were added to wells with iFRCs. After incubation at 37 °C for 4 d, the T cells were stained with APC anti-CD4 or CD8 and Violet LIVE/DEAD Fixable Dead Cell Stain Kit (Invitrogen) and assessed by flow cyometry. To examine T-cell activation status, CD4+ T cells were also stained with fluorescently conjugated anti-CD44 and anti-CD25. For analysis of IFN-γ, T cells were incubated with brefeldin-A (2 μg/mL) for 5 h before intracellular staining with PE-Cy7 anti–IFN-γ antibody.
Supplementary Material
Acknowledgments
Funding was provided by National Institutes of Health Grant A019484 and Training Grant HL007195 (to C.T.N.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205850109/-/DCSupplemental.
References
- 1.Tishon A, Eddleston M, de la Torre JC, Oldstone MB. Cytotoxic T lymphocytes cleanse viral gene products from individually infected neurons and lymphocytes in mice persistently infected with lymphocytic choriomeningitis virus. Virology. 1993;197:463–467. doi: 10.1006/viro.1993.1613. [DOI] [PubMed] [Google Scholar]
- 2.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger DP, Homann D, Oldstone MB. Defining parameters for successful immunocytotherapy of persistent viral infection. Virology. 2000;266:257–263. doi: 10.1006/viro.1999.0074. [DOI] [PubMed] [Google Scholar]
- 4.Balogh P, Fisi V, Szakal AK. Fibroblastic reticular cells of the peripheral lymphoid organs: Unique features of a ubiquitous cell type. Mol Immunol. 2008;46:1–7. doi: 10.1016/j.molimm.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol. 2009;9:618–629. doi: 10.1038/nri2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger DP, et al. Lymphotoxin-beta-deficient mice show defective antiviral immunity. Virology. 1999;260:136–147. doi: 10.1006/viro.1999.9811. [DOI] [PubMed] [Google Scholar]
- 7.Gunn MD, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller S, et al. Role of an intact splenic microarchitecture in early lymphocytic choriomeningitis virus production. J Virol. 2002;76:2375–2383. doi: 10.1128/jvi.76.5.2375-2383.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katakai T, et al. A novel reticular stromal structure in lymph node cortex: An immuno-platform for interactions among dendritic cells, T cells and B cells. Int Immunol. 2004;16:1133–1142. doi: 10.1093/intimm/dxh113. [DOI] [PubMed] [Google Scholar]
- 10.Kaldjian EP, Gretz JE, Anderson AO, Shi Y, Shaw S. Spatial and molecular organization of lymph node T cell cortex: A labyrinthine cavity bounded by an epithelium-like monolayer of fibroblastic reticular cells anchored to basement membrane-like extracellular matrix. Int Immunol. 2001;13:1243–1253. doi: 10.1093/intimm/13.10.1243. [DOI] [PubMed] [Google Scholar]
- 11.Link A, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8:1255–1265. doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- 12.Zeng M, et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest. 2011;121:998–1008. doi: 10.1172/JCI45157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fletcher AL, et al. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J Exp Med. 2010;207:689–697. doi: 10.1084/jem.20092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turley SJ, Fletcher AL, Elpek KG. The stromal and haematopoietic antigen-presenting cells that reside in secondary lymphoid organs. Nat Rev Immunol. 2010;10:813–825. doi: 10.1038/nri2886. [DOI] [PubMed] [Google Scholar]
- 15.Nichols LA, et al. Deletional self-tolerance to a melanocyte/melanoma antigen derived from tyrosinase is mediated by a radio-resistant cell in peripheral and mesenteric lymph nodes. J Immunol. 2007;179:993–1003. doi: 10.4049/jimmunol.179.2.993. [DOI] [PubMed] [Google Scholar]
- 16.Steele KE, Anderson AO, Mohamadzadeh M. Fibroblastic reticular cells and their role in viral hemorrhagic fevers. Expert Rev Anti Infect Ther. 2009;7:423–435. doi: 10.1586/eri.09.13. [DOI] [PubMed] [Google Scholar]
- 17.Mueller SN, et al. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science. 2007;317:670–674. doi: 10.1126/science.1144830. [DOI] [PubMed] [Google Scholar]
- 18.Mueller SN, et al. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci USA. 2007;104:15430–15435. doi: 10.1073/pnas.0702579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller SN, et al. PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J Clin Invest. 2010;120:2508–2515. doi: 10.1172/JCI40040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katakai T, Hara T, Sugai M, Gonda H, Shimizu A. Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J Exp Med. 2004;200:783–795. doi: 10.1084/jem.20040254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansson M, et al. Dendritic cells express CCR7 and migrate in response to CCL19 (MIP-3beta) after exposure to Helicobacter pylori. Microbes Infect. 2006;8(3):841–850. doi: 10.1016/j.micinf.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Jang MH, et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J Immunol. 2006;176:803–810. doi: 10.4049/jimmunol.176.2.803. [DOI] [PubMed] [Google Scholar]
- 23.Cao W, et al. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science. 1998;282:2079–2081. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- 24.Oldstone MB, Campbell KP. Decoding arenavirus pathogenesis: Essential roles for alpha-dystroglycan-virus interactions and the immune response. Virology. 2011;411(2):170–179. doi: 10.1016/j.virol.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 26.Jin HT, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA. 2010;107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sevilla N, Kunz S, McGavern D, Oldstone MB. Infection of dendritic cells by lymphocytic choriomeningitis virus. Curr Top Microbiol Immunol. 2003;276:125–144. doi: 10.1007/978-3-662-06508-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J Clin Invest. 2004;113:737–745. doi: 10.1172/JCI20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fletcher AL, Malhotra D, Turley SJ. Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol. 2011;32:12–18. doi: 10.1016/j.it.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rennick D, Jackson J, Moulds C, Lee F, Yang G. IL-3 and stromal cell-derived factor synergistically stimulate the growth of pre-B cell lines cloned from long-term lymphoid bone marrow cultures. J Immunol. 1989;142:161–166. [PubMed] [Google Scholar]
- 31.Hugo P, Gorczynski RM, Oth D, Potworowski EF. Interactions between lymphoid cells and a thymic stromal cell line in vitro. Adv Exp Med Biol. 1988;237:357–362. doi: 10.1007/978-1-4684-5535-9_53. [DOI] [PubMed] [Google Scholar]
- 32.Kreisel D, et al. Cutting edge: MHC class II expression by pulmonary nonhematopoietic cells plays a critical role in controlling local inflammatory responses. J Immunol. 2010;185:3809–3813. doi: 10.4049/jimmunol.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J Virol. 2009;83:12164–12171. doi: 10.1128/JVI.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paludan C, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 35.Blackburn SD, et al. Tissue-specific differences in PD-1 and PD-L1 expression during chronic viral infection: implications for CD8 T-cell exhaustion. J Virol. 2010;84:2078–2089. doi: 10.1128/JVI.01579-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markusic D, Oude-Elferink R, Das AT, Berkhout B, Seppen J. Comparison of single regulated lentiviral vectors with rtTA expression driven by an autoregulatory loop or a constitutive promoter. Nucleic Acids Res. 2005;33:e63. doi: 10.1093/nar/gni062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang H, Madison B, Gumucio DL, Teitelbaum DH. Specific overexpression of IL-7 in the intestinal mucosa: The role in intestinal intraepithelial lymphocyte development. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1421–G1430. doi: 10.1152/ajpgi.00060.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.