

Abstract
The mechanisms underlying hypermethylation of tumor-suppressor gene promoters in cancer is not well understood. Here, we report that lysine acetylation of the oncogenic transcription factor STAT3 is elevated in tumors. We also show that genetically altering STAT3 at Lys685 reduces tumor growth, which is accompanied by demethylation and reactivation of several tumor-suppressor genes. Moreover, mutating STAT3 at Lys685 disrupts DNA methyltransferase 1–STAT3 interactions in cultured tumor cells and in tumors. These observations are confirmed by treatment with an acetylation inhibitor, resveratrol. Furthermore, reduction of acetylated STAT3 in triple-negative breast cancer cells leads to demethylation and activation of the estrogen receptor-α gene, sensitizing the tumor cells to antiestrogens. Our results also demonstrate a correlation between STAT3 acetylation and methylation of estrogen receptor-α in melanoma, which predicts melanoma progression. Taken together, these results suggest a role of STAT3 acetylation in regulating CpG island methylation, which may partially explain aberrant gene silencing in cancer. These findings also provide a rationale for targeting acetylated STAT3 for chemoprevention and cancer therapy.
The importance of phosphorylation in regulating STAT3 functions has been a focus since the discovery of this transcription factor. However, STAT3 is also acetylated (1, 2). Converting Lys685 to Arg has been shown to disrupt STAT3 dimerization, thereby abrogating STAT3 DNA-binding and transcriptional activation of oncogenes in response to cytokine stimulation (1, 2). However, the role of STAT3 acetylation in human cancer has not been determined. Although STAT3 is a well-known transcriptional activator for many genes (3, 4), recently it has also been reported to inhibit gene expression (5–9). Although the underlying mechanisms remain to be further explored, STAT3 has been shown to increase CpG island methylation of certain tumor-suppressor genes through regulating expression and interacting with DNA methyltransferase 1 (DNMT1) (5–7). DNMT1 is primarily involved in the maintenance of methylation (10–12), but it also is required for aberrant CpG methylation in human cancer cells (13, 14).
In addition to many tumor-suppressor genes, the estrogen receptor-α (ERα) gene is also frequently methylated and silenced in several types of cancer, including breast cancer, colorectal cancer, lung cancer, and melanoma (15–19). ERα silencing via CpG island methylation precludes the use of effective antiestrogen therapeutics in cancer patients (15, 16, 20). In this study, using both a genetic approach and an inhibitor of acetylation, resveratrol, we identify a role of STAT3 acetylation in cancer CpG methylation and in silencing key tumor-suppressor and therapeutic target genes. These findings suggest the importance of acetylated STAT3 as a target for chemoprevention and cancer therapy.
Results
STAT3 Acetylation Affects Tumor Growth, DNA Methylation, and Tumor-Suppressor Gene Silencing.
We first noticed strikingly increased STAT3 acetylation in melanoma tissues, compared with normal skin specimens (Fig. 1A). Similar immunohistochemical (IHC) analyses of human colon cancer tissues also showed that STAT3 acetylation was elevated in malignant areas compared with normal tissue areas (Fig. S1A). Furthermore, STAT3 acetylation was found to be increased in tumor vs. matching normal tissues from triple-negative breast cancer (TNBC) patients (Fig. S1B).
Fig. 1.

The effects of K685R acetylation defective mutant of STAT3 on tumor growth, expression and promoter DNA methylation of tumor suppressor genes. (A) IHC analyses of normal human skin vs. melanoma tissue sections by staining with antibodies against acetylated STAT3 at Lys685. (Scale bars, 100 μm.) (B) Growth curve of A2058 tumors overexpressing either STAT3 wild-type or acetylation mutant (KR) (n = 6); ***P < 0.0001. (C) Real-time RT-PCR analysis of tumor-suppressor gene mRNA expression levels in A2058 tumors (n = 3). (D) Gene-specific promoter methylation analysis by methyl-dependent immunoprecipitation of A2058 tumor cells transfected with either STAT3 WT or KR plasmid expression vectors. Preimmune serum (PIS) was used as nonspecific antibody control. Ordinant shows enrichment over input; shown are means ± SD.
To address whether STAT3 acetylation affects tumor growth, we overexpressed in A2058 human melanoma cells a STAT3 mutant (K685R) in which Lys685 of STAT3 was converted to Arg. In mice, growth of A2058 melanoma cells expressing the STAT3 K685R mutant was significantly slower than that of A2058 tumor cells with the wild-type STAT3 gene (Fig. 1B). Analysis of the A2058 tumors harvested from the mice showed that mutating Lys685 of STAT3 up-regulated expression of several key tumor-suppressor genes in human cancers, including p53 and PTPN6 (SHP-1), the expression of which is known to be inhibited by STAT3 (Fig. 1C) (7, 9). The mutation affecting STAT3 acetylation also increased Ptpn6 and Cdkn2a expression in mouse embryonic fibroblasts (MEFs) to similar levels as those expressing control vectors (Fig. S1C). However, expression of the Rb tumor-suppressor gene in MEF cells was not affected by mutating STAT3 at acetylation site (Fig. S1C). STAT3 acetylation also contributed to Dnmt1 expression in MEF cells (Fig. S1C). STAT3 K685R expressing A2058 tumors harvested from the mice displayed a reduction of CpG island methylation in several key tumor suppressor gene promoters (Fig. 1D).
Loss of Endogenous STAT3 Acetylation Results in Reactivation and Demethylation of Tumor-Suppressor Genes.
To assess more physiologically the role of STAT3 acetylation in cancer cells, we used a homologous recombination-mediated knock-in strategy to specifically mutate the Lys685 site of the endogenous STAT3 in the human colon cancer cell line HCT116. Western blotting analysis, after immunoprecipitation with either preimmune serum or STAT3 antibodies, confirmed that the Lys685 mutation had little effect on STAT3 phosphorylation (Fig. S2). We then assayed for and found the reactivation of a number of tumor-suppressor genes, the silencing of which is important for colon cancer development and progression (Fig. 2A) (13, 21–25). Several of these reactivated tumor-suppressor genes, such as CDKN2A, DLEC1, and STAT1, are known to be frequently hypermethylated in cancer (13, 21, 25, 26). Reactivation of these tumor-suppressor genes by mutation of endogenous STAT3 acetylation at Lys685 was associated with demethylation of their promoters (Fig. 2B). STAT3 has been shown to facilitate DNMT1 binding to the PTPN6 promoter in a T-cell lymphoma cell line (7). To test whether acetylation was crucial for STAT3 and DNMT1 binding to promoters of the tumor-suppressor genes, we performed ChIP assays in the HCT116 parental (wild-type) cancer cell line and its variant with an endogenous Lys685 mutation (KR), which were treated with tumor-conditioned medium (TCM) to further activate STAT3, thereby facilitating detection of STAT3-DNMT1 binding to the promoters. As shown in Fig. 2C, both STAT3 and DNMT1 bound to the tumor-suppressor gene promoters, and the mutation on STAT3 K685R abrogated this binding.
Fig. 2.
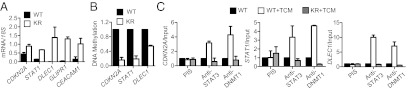
Mutating endogenous STAT3 at K685 results in up-regulation and promoter demethylation of tumor-suppressor genes and abrogates DNMT1 recruitment to their promoters. (A) Levels of mRNA of tumor-suppressor genes in HCT116 tumor cells with endogenous STAT3 wild-type or K685R acetylation mutant (KR). (B) Methylation levels of the indicated tumor-suppressor gene promoters in HCT116 tumor cells with WT STAT3 or KR mutant. The relative amount of methylated DNA is normalized to input DNA and compared with methylation levels in parental HCT116 tumor cells (STAT3 WT), which is designated as 1. (C) ChIP analysis to detect the recruitment of STAT3 and DNMT1 to gene promoters; means ± SD. HCT116 cells were treated with TCM in all cases.
STAT3 Acetylation at Lys685 Mediates STAT3–DNMT1 Interaction.
To further investigate how acetylation of STAT3 mediates DNA methylation, we assessed whether the STAT3 K685R mutation affects STAT3 interaction with DNMT1. We detected acetylated STAT3 in the same protein complex with DNMT1 in A2058 melanoma tumor cells overexpressing a wild-type STAT3 gene fused to YFP (Fig. S3A). In contrast, when Lys685R-mutated STAT3 was expressed in the same tumor cells, the interaction between STAT3 and DNMT1 was reduced. MCF7 cells do not display elevated STAT3 activity in vitro, but we found that overexpressing p300 and STAT3 led to not only enhanced STAT3 acetylation, but also increased interaction between STAT3 and DNMT1 (Fig. S3B). However, with mutation at Lys685, STAT3 no longer efficiently interacted with DNMT1. Conversely, treating tumor cells with a histone deacetylase (HDAC) inhibitor, Trichostatin, led to an increase in STAT3 acetylation and STAT3-DNMT1 interaction (Fig. S3B). We also assessed acetylation-dependent STAT3-DNMT1 interaction during tumor growth. Results from these experiments indicated that STAT3 was in the same complex with DNMT1 in A2058 tumors with wild-type STAT3, but this complex was disrupted when STAT3 K685R mutant was expressed in the tumors (Fig. 3A). We next examined the effects of inhibiting acetylation on the STAT3-DNMT1 interaction. Treating the TNBC cell line MDA-MB468 with the acetyltransferase inhibitor anacardic acid led to a reduction of acetylated STAT3 and coimmunoprecipitated DNMT1 (Fig. S3C). Resveratrol is a HDAC activator known to inhibit STAT3 acetylation (27, 28). Treating mice bearing A2058 human melanoma tumors with resveratrol led to tumor growth inhibition, accompanied by disruption of STAT3-DNMT1 complex formation and demethylation of several tumor-suppressor gene promoters (Fig. 3 B–D). To determine whether acetylation of STAT3 might facilitate STAT3-DNMT1 interaction in tumors from patients, we analyzed several types of tumor specimens. The results showed that acetylated STAT3 colocalized well with DNMT1 in different types of human cancer cells (Fig. S3D).
Fig. 3.
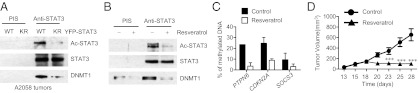
STAT3 acetylation affects its interaction with DNMT1. (A) Western blotting following immunoprecipitation with STAT3 antibodies detecting interaction of acetylated STAT3 with DNMT1 in growing A2058 tumors expressing either STAT3 wild-type or acetylation mutant (KR). (B) Effects of resveratrol on acetylated STAT3 as well as its interaction with DNMT1 in A2058 tumors analyzed by Western blotting. (C) DNA methylation analysis of the PTPN6, CDKN2A, and SOCS3 promoters upon abrogating STAT3 acetylation in tumors (the mean and range for two experiments with pooled tumors is shown). (D) A2058 tumor growth measured at the indicated time points. Postintratumoral injection of either solvent control or 50 μM of resveratrol. n = 8; ***P < 0.0001.
Blocking STAT3 Acetylation Reactivates the ERα Gene and Sensitizes TNBC Cells to Therapy.
One of the main reasons that many breast cancer patients do not benefit from front-line antiestrogen therapy is lack of expression of the ERα gene in tumor cells because of DNA methylation (19, 20). Because acetylated STAT3 is highly elevated in TNBC tissues (Fig. S1B), we tested whether inhibiting STAT3 acetylation could reactivate ERα expression in MDA-MB468 TNBC cells. Treating the TNBC cells with resveratrol increased ERα gene expression at both the RNA and protein levels, which was associated with a decrease in STAT3 acetylation (Fig. 4A and Fig. S4A), as well as a decrease in methylation of the ERα gene promoter in these cell lines (Fig. 4B and Fig. S4B). We next determined whether treating the TNBC cells with resveratrol could sensitize them to tamoxifen-induced cell death. In vitro combination treatment with resveratrol and tamoxifen significantly reduced tumor cell viability more than either drug alone (Fig. 4C and Fig. S4C). Importantly, although tamoxifen itself did not cause significant growth inhibition of the TNBC tumors in mice, treatment with resveratrol in combination with tamoxifen effectively blocked breast cancer growth (Fig. 4D).
Fig. 4.
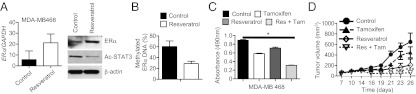
Resveratrol reduces STAT3 acetylation, restores ERα expression and sensitizes TNBC cells to antiestrogen therapy. (A) RNA expression levels of ERα (n = 3) (Left) or protein expression levels of ERα and acetylated STAT3 (9) in MDA-MB468 cells treated with either solvent control or resveratrol. (B) Quantification of ERα promoter DNA methylation levels in control and resveratrol-treated MDA-MB468 cells; means ± SD. (C) Cell proliferation assay to measure effects of treatments with tamoxifen or resveratrol alone or in combination on the viability of MDA-MB468 cells (n = 8); *P = 0.0146. (D) Growth curve of MDA-MB468 tumors receiving the indicated treatment twice a week (n = 6); ***P < 0.0001.
Acetylated STAT3 Is Crucial for ERα Gene Methylation in Melanoma.
The ERα gene promoter is also hypermethylated in other types of cancer and antiestrogen therapy benefits some cancer patients other than those with breast cancer (16–19, 29). In the case of melanoma, ERα gene hypermethylation strongly predicts melanoma progression, with unknown underlying molecular mechanisms (16). We therefore tested whether blocking STAT3 acetylation could up-regulate ERα gene expression in the M223 melanoma cell line in which the ERα gene is methylated and silenced (16). Treating these melanoma cells with resveratrol increased ERα expression at both the mRNA and protein levels (Fig. 5A and Fig. S5A), with an accompanied reduction in methylated ERα CpG islands (Fig. 5B). This melanoma cell line was resistant to tamoxifen-induced cell death, but combinatorial treatment of the tumor cells with resveratrol and tamoxifen resulted in a fourfold stronger inhibition on cell viability (Fig. 5C). We reanalyzed tumor sections from several of the same melanoma patients used in a prior study regarding ERα gene methylation and patient survival (16) to determine acetylated STAT3 levels. Our IHC results showed that acetylated STAT3 was highly elevated in ERα-negative melanoma samples in which the ERα promoter was hypermethylated, compared with those from hypomethylated tumors positive for ERα expression (Fig. 5D and Fig. S5B). This finding suggests that acetylated STAT3 can contribute to ERα gene promoter hypermethylation and is thereby related to melanoma tumor progression.
Fig. 5.

STAT3 acetylation regulates ERα promoter methylation crucial for melanoma progression. (A) Real-time RT-PCR to quantify ERα mRNA expression in M223 human melanoma tumor cells treated with either solvent control or resveratrol (the mean and range for two experiments is shown). (B) Measurement of ERα promoter DNA methylation in human melanoma cells treated with either DMSO control (0.001%) or resveratrol; means ± SD. (C) Cell proliferation assay to show effects of the indicated treatment on melanoma cell viability; n = 8, ***P < 0.0001. (D) IHC analysis of acetylated STAT3 levels in human melanoma tissue sections prepared from 11 ERα(+) and 9 ERα(−) melanoma tumors. (Scale bars, 100 μm.)
Discussion
Protein acetylation has generally been thought to be a crucial posttranslational modification for active gene transcription through alteration of chromatin structure (30, 31). Our results provide evidence that a well-known transcriptional activator, STAT3, when acetylated, can silence gene expression and enhance DNA methylation of several important tumor-suppressor gene promoters. Many current anticancer-targeted therapeutics aim to inhibit protein tyrosine phosphorylation, which is viewed as the most important posttranslational modification regulating protein/protein and protein/DNA interactions (32, 33). Like tyrosine phosphorylation, persistent acetylation of STAT3 is generally observed in diverse human cancers. Both phosphorylation and acetylation of STAT3 are crucial for STAT3-mediated up-regulation of oncogenic genes (1, 4).
We used resveratrol to provide support that targeting STAT3 acetylation with small-molecule drugs can reverse aberrant CpG island methylation at several tumor-suppressor gene promoters in cancer cells. Although resveratrol is regarded as an inhibitor of acetylation (27), it was discovered to be a STAT3 inhibitor (34). Reveratrol was later shown to inhibit STAT3 acetylation (28), which is confirmed by our data. A potentially important therapeutic finding from our present study is that by reducing STAT3 acetylation, one can reactivate ERα gene expression in tumors where the ERα gene-promoter region is methylated. Inactivation of the ERα gene via methylation strongly correlates with poor prognosis as well as an aggressive phenotype in cancers, such as melanoma and basal subtype breast cancers (16, 20). Furthermore, up to 50% of ERα-positive primary breast cancers lose ERα expression in recurrent tumors, conferring resistance to tamoxifen therapy (15, 20). Our findings have not only identified a likely underlying mechanism for ERα gene silencing in certain cancers, but also point to the importance of developing inhibitors that selectively affect STAT3 acetylation.
It is noteworthy that HDAC inhibitors induce hyperacetylation of crucial oncogenic transcription factors, including STAT3 (1) and NF-κB (35, 36). Because acetylation of STAT3 and NF-κB can increase their oncogenic activities (36), HDAC inhibitor-induced hyperacetylation may thereby promote tumor progression. In this regard, specifically inhibiting STAT3 acetylation may boost the anticancer therapeutic efficacy of HDAC inhibitors. CpG island methylation in cancer is prevalent, and clinical use of DNA methyltransferase inhibitors also has shown great promise for cancer therapy by altering DNA methylation (30, 37–39). Although further studies are necessary to understand how acetylated STAT3 preferentially modulates CpG methylation of certain gene promoters, our studies provide mechanistic and preclinical evidence that targeting acetylation of an oncogenic transcription factor can also impact CpG island methylation to benefit cancer therapy.
Materials and Methods
Cell Lines and Reagents.
A2058 human melanoma, MCF7, MDA-MB231, and MDA-MB468 human breast cancer cell lines were obtained from ATCC. M223 is an early passage established American Joint Committee on Cancer stage IV metastatic melanoma cell line from John Wane Cancer Research Institute (JWCI). All cells were maintained in DMEM media plus 10% (vol/vol) FBS, except A2058 cells, which were maintained in RPMI media plus 10% (vol/vol) FBS. HCT human colon tumor cells expressing STAT3 K685R mutant was generated by somatic cell gene targeting, as previously described (40). TCM was prepared from U251 human glioma cell lines, as previously described (41). Conditioned medium was prepared by reconstituting fresh-cell culture medium with 20% (vol/vol) TCM. The detailed method of somatic cell gene targeting and information on reagents used in this study are described in SI Materials and Methods.
Immunofluorescent Staining, Confocal Microscopy, and IHC Staining.
Human tissue-array slides (including both normal and malignant tissues), obtained from the archive of the Pathology Laboratory at City of Hope, were used to detect acetylated STAT3. The use of the archival tissue array slides was exempt from approval by the Institutional Review Board at City of Hope Medical Center. TNBC, melanoma tumor, and normal skin tissue sections were prepared for IHC analysis as previously described (42), and were provided by the JWCI, with approval from the JWCI and Western Institutional Review Board. Immunofluorescent and IHC staining of tissue slides were performed as described previously (41).
In Vivo Xenograft Tumor Models.
Mouse care and experimental procedures were carried out under pathogen-free conditions in accordance with established institutional guidance and approved protocols from the Institutional Animal Care and Use Committee of the Beckman Research Institute at City of Hope. The in vivo tumor model was established as described in SI Materials and Methods.
Real-Time RT-PCR.
Total RNA was prepared using the RNeasy kit (Qiagen) following the manufacturer’s instructions and reverse-transcribed to cDNA using iScript cDNA Synthesis Kit (Bio-Rad), and real-time PCR reactions were performed using iQ SYBR Green supermix (Bio-Rad) on a DNA Engine thermal cycler equipped with Chromo4 detector (Bio-Rad). Gene-specific primer sets were purchased from SA Bioscience. To prepare total RNA from tumors, tissue samples were ground in liquid nitrogen using a mortar and pestle.
DNA Methylation Assay.
Genomic DNA was prepared from human cancer cell lines using the DNeasy Tissue kit following the manufacturer’s instructions (Qiagen). Tumor-suppressor gene methylation levels were analyzed by the EpiTech Methyl quantitative PCR assay (Qiagen). Unmethylated or methylated DNA is selectively digested by methylation-sensitive (cuts unmethylated and partially methylated DNA, leaving only hypomethylated DNA) and methylation-dependent restriction enzymes (cuts any methylated DNA, leaving only unmethylated DNA) according to the manufacturer’s instructions. The remaining DNA after digestion is quantified by real-time PCR. The relative concentrations of differentially methylated DNA are determined by comparing the amount of each digest with that of mock digest (no enzyme added), using the software provided by the manufacturer (Qiagen). Quantitative methylation status of the ERα promoter region of melanoma cell lines and tissues was determined as previously described (16). Methylated DNA immunoprecipitation was performed as described previously (43), with minor modifications described in SI Materials and Methods.
Western Blotting and Coimmunoprecipitation Analysis.
Cells were lysed in a modified RIPA buffer containing 50 mM Tris, PH 7.4, 1% Nonidet P-40, 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, and protease inhibitor mixture (Roche). Proteins were subjected to immunoblotting and coimmunoprecipitation, as previously described (41).
ChIP Assay.
ChIP was performed as previously described (8) and as indicated in SI Materials and Methods.
Cell Proliferation Assay.
We cultured cells at a density of 5 × 104 cells per well in flat-bottomed 96-well plates in the presence of solvent control (combination of DMSO and ethanol, 0.001%), resveratrol (10 μM), tamoxifen (2 μM), or a combination of both. We refed cells with treatment every other day for 7 d and added CellTiter 96 Aqueous One Solution Reagent (Promega) to each well according to the manufacturer’s instructions. The cell viability was determined by measuring the absorbance at 490 nm.
Statistical Analysis.
Two-way ANOVA and Bonferroni posttest were used to calculate differences in tumor growth. In some cases, one-way ANOVA or unpaired t test was used to calculate P values. Data were analyzed using Prism software (GraphPad) and shown as means ± SEM, except where indicated otherwise.
Supplementary Material
Acknowledgments
We thank the Functional Genomics Core, Bioinformatics Core, Light Microscopy Core, Pathology Core, Flow Cytometry Core, and Animal Facility Core at City of Hope Comprehensive Cancer Center for their excellent technical assistance. This work is funded by the Markel Foundation and Tim Nesviq Foundation at City of Hope Comprehensive Cancer Center; the Keck Foundation; and National Institutes of Health Grants R01 CA115815 and R01 CA115674, and P30 CA033572 to the City of Hope Comprehensive Cancer Center from the National Cancer Institute.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205132109/-/DCSupplemental.
References
- 1.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 2.Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem. 2005;280:11528–11534. doi: 10.1074/jbc.M413930200. [DOI] [PubMed] [Google Scholar]
- 3.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu H, Jove R. The STATs of cancer—New molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Q, et al. IL-2R common gamma-chain is epigenetically silenced by nucleophosphin-anaplastic lymphoma kinase (NPM-ALK) and acts as a tumor suppressor by targeting NPM-ALK. Proc Natl Acad Sci USA. 2011;108:11977–11982. doi: 10.1073/pnas.1100319108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Q, Wang HY, Liu X, Wasik MA. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat Med. 2007;13:1341–1348. doi: 10.1038/nm1659. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, et al. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci USA. 2005;102:6948–6953. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H, et al. A requirement of STAT3 DNA binding precludes Th-1 immunostimulatory gene expression by NF-κB in tumors. Cancer Res. 2011;71:3772–3780. doi: 10.1158/0008-5472.CAN-10-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niu G, et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. 2005;25:7432–7440. doi: 10.1128/MCB.25.17.7432-7440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 11.Chen ZX, Riggs AD. DNA methylation and demethylation in mammals. J Biol Chem. 2011;286:18347–18353. doi: 10.1074/jbc.R110.205286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 13.Robert MF, et al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet. 2003;33:61–65. doi: 10.1038/ng1068. [DOI] [PubMed] [Google Scholar]
- 14.Rhee I, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 15.Johnston SR, et al. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995;55:3331–3338. [PubMed] [Google Scholar]
- 16.Mori T, et al. Estrogen receptor-alpha methylation predicts melanoma progression. Cancer Res. 2006;66:6692–6698. doi: 10.1158/0008-5472.CAN-06-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Issa JP, Baylin SB, Belinsky SA. Methylation of the estrogen receptor CpG island in lung tumors is related to the specific type of carcinogen exposure. Cancer Res. 1996;56:3655–3658. [PubMed] [Google Scholar]
- 18.Issa JP, et al. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 19.Lapidus RG, et al. Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer Res. 1998;58:2515–2519. [PubMed] [Google Scholar]
- 20.Brinkman JA, El-Ashry D. ER re-expression and re-sensitization to endocrine therapies in ER-negative breast cancers. J Mammary Gland Biol Neoplasia. 2009;14:67–78. doi: 10.1007/s10911-009-9113-0. [DOI] [PubMed] [Google Scholar]
- 21.Ying J, et al. DLEC1 is a functional 3p22.3 tumour suppressor silenced by promoter CpG methylation in colon and gastric cancers. Br J Cancer. 2009;100:663–669. doi: 10.1038/sj.bjc.6604888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shively JE. CEACAM1 and hyperplastic polyps: New links in the chain of events leading to colon cancer. Oncogene. 2004;23:9303–9305. doi: 10.1038/sj.onc.1208266. [DOI] [PubMed] [Google Scholar]
- 23.Ren C, et al. mRTVP-1, a novel p53 target gene with proapoptotic activities. Mol Cell Biol. 2002;22:3345–3357. doi: 10.1128/MCB.22.10.3345-3357.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185:4912–4920. doi: 10.4049/jimmunol.1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herman JG, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 26.Xi S, et al. Decreased STAT1 expression by promoter methylation in squamous cell carcinogenesis. J Natl Cancer Inst. 2006;98:181–189. doi: 10.1093/jnci/djj020. [DOI] [PubMed] [Google Scholar]
- 27.Howitz KT, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 28.Nie Y, et al. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11:492–500. doi: 10.1038/ncb1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cocconi G, et al. Treatment of metastatic malignant melanoma with dacarbazine plus tamoxifen. N Engl J Med. 1992;327:516–523. doi: 10.1056/NEJM199208203270803. [DOI] [PubMed] [Google Scholar]
- 30.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 31.Gregory PD, Wagner K, Hörz W. Histone acetylation and chromatin remodeling. Exp Cell Res. 2001;265:195–202. doi: 10.1006/excr.2001.5187. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 33.Hunter T. Tyrosine phosphorylation: Thirty years and counting. Curr Opin Cell Biol. 2009;21:140–146. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kotha A, et al. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther. 2006;5:621–629. doi: 10.1158/1535-7163.MCT-05-0268. [DOI] [PubMed] [Google Scholar]
- 35.Chen Lf, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 36.Lee H, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–293. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 38.Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010;31:536–546. doi: 10.1016/j.tips.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoo CB, Jones PA. Epigenetic therapy of cancer: Past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, et al. Epitope tagging of endogenous proteins for genome-wide ChIP-chip studies. Nat Methods. 2008;5:163–165. doi: 10.1038/nmeth1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee H, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Maat MF, et al. Epigenetic silencing of cyclooxygenase-2 affects clinical outcome in gastric cancer. J Clin Oncol. 2007;25:4887–4894. doi: 10.1200/JCO.2006.09.8921. [DOI] [PubMed] [Google Scholar]
- 43.Mohn F, Weber M, Schübeler D, Roloff TC. Methylated DNA immunoprecipitation (MeDIP) Methods Mol Biol. 2009;507:55–64. doi: 10.1007/978-1-59745-522-0_5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.