

Abstract
Recent studies have shown that cerebellar Bergmann glia display coordinated Ca2+ transients in live mice. However, the functional significance of Bergmann glial Ca2+ signaling remains poorly understood. Using transgenic mice that allow selective stimulation of glial cells, we report here that cytosolic Ca2+ regulates uptake of K+ by Bergmann glia, thus providing a powerful mechanism for control of Purkinje cell-membrane potential. The decline in extracellular K+ evoked by agonist-induced Ca2+ in Bergmann glia transiently increased spike activity of Purkinje cells in cerebellar slices as well as in live anesthetized mice. Thus, Bergmann glia play a previously unappreciated role in controlling the membrane potential and thereby the activity of adjacent Purkinje cells.
Keywords: two-photon laser scanning microscopy, ion-sensitive microelectrode
Bergmann glial cells in cerebellum are electrically nonexcitable cells that in many ways serve the same functions as protoplasmic astrocytes in forebrain. Bergmann glia are chiefly responsible for glutamate uptake and extracellular K+ homeostasis (1). Their highly negative resting membrane potential of −80 to −85 mV combined with a large number of inwardly rectifying K+ channels helps maintain a tight control of extracellular K+ concentration (2). Bergmann glial cells also display Ca2+ transients in response to glutamate or stimulation of climbing and parallel fibers in slices and to motor activity in vivo (3). However, although glial Ca2+ signaling has been shown to regulate synaptic transmission in several brain regions, it is presently not established whether Bergmann glia can modulate the activity of Purkinje cells.
Membrane potentials of almost all neurons in mammalian brain fluctuate between a hyperpolarized state (down state) and depolarized state (up state) during sleep/anesthesia and quiet wakefulness (4, 5). These fluctuations of the membrane potentials are synchronized between neighboring cells and detected as slow (∼0.5–1 Hz), large-amplitude delta waves (4). Even though the existence of bistability of Purkinje cells in awake mice has been questioned, it is generally agreed that bistability is a common phenomenon in both anesthetized and sleeping animals (5). The bistability of the resting membrane potential in Purkinje cells was first discovered by Llinás and Sugimori in cerebellar slices (6, 7). The current model predicts that bistability of Purkinje cells, similar to thalamic neurons, relies on the balance between noninactivating inward currents (persistent Na+ currents and/or T-type Ca2+ currents) (4, 8, 9) and an outward K+ leak current (5–7, 10). However, Purkinje cell bistability is not simply an expression of intrinsic membrane properties because complex spikes generated by climbing fiber activation can toggle the membrane potential between up and down states in vivo (5). Recently, glutamate uncaging on distal dendrites of striate spiny neurons was shown to induce up states that lasted hundreds of milliseconds (11), and small hyperpolarization currents or interneuronal activity can toggle Purkinje cells between up and down states (12). Moreover, stimulating a single astrocyte can trigger up-state synchronizations of neighboring neurons in cortical slices, in a mechanism that depends on purinergic and glutamatergic signaling (13).
Our earlier studies have shown that astrocytes in hippocampus and cortex increase K+ uptake in response to agonist-induced Ca2+ signaling (14). Because the extracellular concentration of K+ is a critical determinant of Purkinje cell-membrane potential, we here asked whether Bergmann glia can modulate Purkinje cell bistability. We found that selectively induced Ca2+ increases in Bergmann glia also evoked a transient reduction of extracellular K+ in cerebellar slices and cerebellum in live adult mice. Moreover, Bergmann glial Ca2+ signaling consistently triggered a switch to up states of longer duration. All of the effects of Bergmann glial Ca2+ signaling on Purkinje cell bistability could be reproduced by simply reducing K+ in the bath solution or depolarizing a single Bergmann glial cell. Thus, our observations indicate that Bergmann glia can regulate cerebellar network activity by a pathway involving Ca2+-mediated K+ uptake. Because Purkinje cells fire continuously in up state, Ca2+ signaling in Bergmann glia is associated with a sharp increase in output from the cerebellar cortex.
Results
Bergmann Glial Ca2+ Transients Changed Purkinje Cell Bistability.
To address a possible role of Bergmann glia in regulation of Purkinje cell activity, we combined whole-cell recordings of Purkinje cells with imaging of intracellular Ca2+ signaling in Bergmann glia loaded with the Ca2+ indicator rhod2/am in cerebellar slices. Whole-cell recordings showed that the membrane potentials of almost all recorded Purkinje cells spontaneously fluctuated with a frequency of 0.5 ± 0.1 Hz between two stable states of 63.4 ± 2.16 mV (down state) and 47.2 ± 3.04 mV (up state), giving rise to a bimodal time distribution (n = 83; Fig. S1). Similar oscillations were recorded by extracellular electrodes, demonstrating that the oscillations are not a result of dialysis of intracellular fluid with the patch electrode (Fig. S1). To selectively stimulate Bergmann glial Ca2+ transients, cerebellar slices were prepared from mice selectively expressing the MrgA1 receptor under the human GFAP promoter (15). The MrgA1 receptor is a member of the Gq-coupled receptor family with expression restricted to a subset of nociceptive sensory neurons outside the CNS. Thus, the peptide ligand Phe-Met-Arg-Phe amide (FMRF) does not activate endogenous brain receptors (15). As previously reported, bath application of FMRF peptide induced robust Ca2+ increases in EGFP+ Bergmann glia (Fig. 1A) concomitant with a sharp increase in the duration of Purkinje cell up state (Fig. 1 B and C). The FMRF-induced changes in Purkinje cell-membrane potential were transient, lasting 20–120 s. The regular pattern of bistability returned with a delay of ∼10–60 s after normalization of Ca2+ in surrounding Bergmann glial cells. Of note, FMRF did not evoke Ca2+ increases in Bergmann glia and did not reduce extracellular K+ in slices prepared from WT mice (Fig. S2). We next assessed the effect of ATP because it has previously been shown that ATP mediates Ca2+ increases in Bergmann glia after parallel fiber stimulation. Bath application of ATP also triggered elevations in Bergmann glial Ca2+ accompanied by a transient increase in Purkinje cell up state in both MrgA1+ mice and WT mice (Fig. 1 D and E and Fig. S2). Of note, Purkinje cells only fired action potentials during up state. As a consequence, Ca2+ signaling in Bergmann glia led to a significant increase in Purkinje cell output detected as an increased frequency of spiking (Fig. 1 F and G). Interspike interval (ISI) and coefficient of variance (CV) of action potential firing during up state did not change significantly after application of either FMRF or ATP (Fig. 1 H and I and Fig. S2), suggesting that neither FMRF nor ATP directly modified the properties of the voltage-gated channels generating Purkinje cells action potentials.
Fig. 1.

Agonist-induced Ca2+ signaling in Bergmann glia modifies Purkinje cell bistability and increases their spiking activity. (A) Two-photon imaging of freshly prepared vibratome sections from a MrgA1+ mouse expressing EGFP under the GFAP promoter (green; Left) loaded with the Ca2+ indicator rhod2/am (red). EGFP and rhod2 signal overlap, indicating that rhod2 primarily is taken up by Bergmann glia. Right three images display rhod2 emission changes in response to exposure to the MrgA1 receptor agonist FMRF (15 μM). (B) Whole-cell recording of a Purkinje cell (PC) combined with imaging rhod2 in Bergmann glia in a slice from a MrgA1+ mouse. The durations of up states increase concurrently with a Ca2+ increase in Bergmann glia evoked by FMRF. (C) Histogram depicting the bimodal distribution of Purkinje cell-membrane potential before (gray) and after (red) FMRF exposure (10 s were analyzed, bin = 1 mV). (D) ATP (100 μM) increased Purkinje cell spiking and rhod2 signal in neighboring Bergmann glial cells. (E) Histogram depicting the bimodal distribution of Purkinje cell-membrane potential before (gray) and after (pink) ATP exposure (10 s were analyzed, bin = 1 mV). (F) Percentage of time Purkinje cells spent in up state before, during, and after exposure to FMRF or ATP (**P < 0.01, n = 6–7). (G) Action potential frequency before, during, and after exposure to FMRF and ATP (**P < 0.01, n = 6–7). (H) Comparison of ISIs of action potentials before, during, and after exposure to FMRF or ATP. ISI was analyzed during up state only (P = 0.13 for FMRF, P = 0.9 for ATP, n = 6–7). (I) CVs for action potentials before, during, and after application of FMRF and ATP (P = 0.6 for FMRF, P = 0.3 for ATP, n = 6–7).
Because it cannot be excluded that ATP activates Purkinje cell P2Y1 receptors, the P2Y2 and P2Y4 receptor-specific agonist UTP was also evaluated. UTP-induced Ca2+ increases in Bergmann glia were directly comparable to those of ATP and FMRF and were also associated with a transient increase in the duration of Purkinje cell up states (Fig. 2 A and B). To address whether agonist-induced increases in cytosolic Ca2+ of Bergmann glia was required for receptor-mediated changes in Purkinje cell output, UTP was next applied to slices prepared from inositol 1,4,5-trisphosphate (IP3) receptor subtype 2 knockout mice (IP3R2−/−). In contrast, Bergmann glia did not exhibit Ca2+ transients in response to UTP in slices prepared from IP3R2−/− mice (Fig. 2C), consistent with the report that Bergmann glia only expresses IP3R2 and not subtypes 1 or 3 (16). UTP also failed to trigger significant changes in Purkinje cell bistability in slices prepared from IP3R2−/− mice (P = 0.8, n = 6; Fig. 2 C–E). Similarly, whereas the spiking frequency increased in IP3R2+/+ mice (P < 0.01), no significant change was observed in IP3R2−/− mice (P = 0.437; Fig. 2F). Purkinje cell up state ISI and CV were unaffected by the UTP exposure in both genotypes, suggesting that UTP did not alter the membrane properties of Purkinje cells (Fig. 2 G and H). Similarly, ATP increased bistability in Purkinje cells from WT mice but failed to induced changes in IP3R2−/− mice too (Fig. S2).
Fig. 2.

Ca2+ dependency of Bergmann glial modulation of Purkinje cell bistability. (A) The P2Y2 and P2Y4 receptor agonist UTP (100 μM) induced increases in cytosolic Ca2+ in Bergmann glia and an increase in up state of Purkinje cells (PC) in a IP3R2+/+ (WT) mouse. (B) Histogram plots the membrane potential distributions before (gray) and after (pink) UTP exposure (10 s, bin = 1 mV). (C) UTP failed to induce increases in Ca2+ in an IP3R2−/− mouse and failed to change the regular pattern of bistability in Purkinje cells. (D) Histogram shows the membrane potential distributions before (gray) and after (green) UTP exposure in the IP3R2−/− recording in C (10 s, bin = 1 mV). (E) Time spent in up state before, during, and after exposure to UTP in IP3R2+/+ and IP3R2−/− littermates (**P < 0.01, n = 6–7). (F) The spiking frequency before, during, and after exposure to UTP in IP3R2+/+ and IP3R2−/− littermates (**P < 0.01, n = 6–7). (G) Comparison of ISIs of action potentials during up states before, during, and after exposure to UTP in IP3R2+/+ and IP3R2−/− littermates (P = 0.14 for IP3R2+/+, P = 0.29 for IP3R2−/−, n = 6–7). (H) CVs before, during, and after exposure to UTP in IP3R2+/+ and IP3R2−/− littermates (P = 0.65 for IP3R2+/+, P = 0.81 for IP3R2−/−, n = 6–7).
Overall, the analysis showed that agonist-induced Ca2+ increases in Bergmann glia were linked to a sharp increase in the duration of Purkinje cell up state and thereby in spiking activity. The increase in up state was transient because normalization of Ca2+ in Bergmann glia after a delay of 10–60 s was followed by a return to normal periodic oscillations of the membrane potential in Purkinje cells.
Ca2+ Signaling in Bergmann Glia Reduced Extracellular K+ Concentration.
Because buffering extracellular K+ is a well-known function of astrocytes, and Ca2+ signaling in hippocampal astrocytes has been shown to increase their uptake of K+ (14, 17, 18), we next tested whether agonist-induced Ca2+ signaling in Bergmann glia triggered a transient decrease in extracellular K+ in cerebellar slices. We combined two-photon imaging of Ca2+ and whole-cell recording of Bergmann glia with measurements of extracellular K+ by using ion-sensitive microelectrodes.
Bath application of FMRF in MrgA1+ mouse cerebellar slices triggered a robust increase in Bergmann glial Ca2+ accompanied by a transient reduction of extracellular K+ and a minor hyperpolarization of Bergmann glial membrane potential (Fig. 3A). Similarly, application of ATP consistently triggered Ca2+ increases and hyperpolarization of Bergmann glia concomitant with a significant drop in extracellular K+ (Fig. 3B), providing strong support for a role of Bergmann glia in active K+ uptake. To address whether Bergmann glial Ca2+ was required for receptor-mediated K+ uptake, UTP was applied to slices prepared from IP3R2−/− mice and WT littermates (IP3R2+/+). UTP-induced Ca2+ increases in Bergmann glia were also linked to a transient decrease in extracellular K+ and hyperpolarization of Bergmann glia in IP3R2+/+ mice (Fig. 3C). Consistent with the notion that agonist-induced K+ uptake in Bergmann glia is Ca2+-dependent, slices prepared from IP3R2−/− mice failed to exhibit UTP-mediated increases in Ca2+ and reduction in extracellular K+ concentration (Fig. 3D). A comparison of FMRF, ATP, and UTP showed comparable increases in Ca2+, extracellular K+, and hyperpolarization of Bergmann glia (Fig. 3 E–G). In addition, UTP had no significant effect on extracellular K+ or Bergmann glial membrane potential in slices prepared from IP3R2−/− mice (Fig. 3 F and G). UTP also failed to alter bistability and thereby the output of Purkinje cells in IP3R2−/− mice (Fig. 2). Importantly, the Ca2+-mediated change in extracellular K+ was positively linked to hyperpolarization of Bergmann glia (R2 = 0.486, n = 24; Fig. 3H). Thus, these experiments showed that agonist-induced Ca2+ signaling in Bergmann glia is linked to a transient reduction of extracellular K+. Of note, the relative large tip of the K+-selective microelectrode (∼3 μm) creates an artificially large extracellular space, and the actual K+ decreases might be of a larger amplitude than those recorded, as suggested in refs. 19 and 20.
Fig. 3.
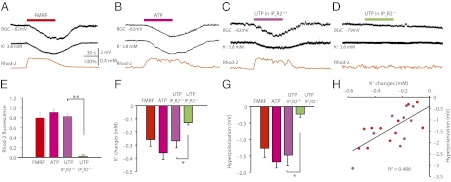
Ca2+ signaling in Bergmann glia is linked to membrane hyperpolarization and a reduction of extracellular K+. (A) Combined whole-cell recordings of Bergmann glia with measurement of extracellular K+ and cytosolic Ca2+ in slices from a MrgA1+ mouse. The MrgA1 agonist FMRF (15 μM) triggered hyperpolarization of the Bergmann glia concomitantly with a decrease in extracellular K+ and an increase in rhod2 signal. (B) Exposure to ATP (100 μM) also triggered Ca2+ signaling and hyperpolarization of Bergmann glia concomitant with a decrease in extracellular K+. (C) UTP evoked increases in cytosolic Ca2+ and a hyperpolarization of Bergmann glia as well as a decrease in extracellular K+ in slices prepared from IP3R2+/+ mice. (D) UTP had little effect in slices prepared from IP3R2−/− littermates. (E) Histogram comparing increases in rhod2 signal in slices from MrgA1+ mice exposed to FMRF (15 μM, n = 19), slices from WT mice exposed to ATP (100 μM, n = 16), and slices from either IP3R2+/+ (n = 13) or IP3R2−/− (n = 13) mice exposed to UTP (100 μM) (**P < 0.01, n = 12). (F) Changes in extracellular K+ in slices from MrgA1+ mice exposed to FMRF (n = 9), slices from WT mice exposed to ATP (n = 9), and slices from either IP3R2+/+ (n = 9) or IP3R2−/− (n = 6) mice exposed to UTP (*P < 0.05, n = 6–9). (G) Changes in Bergmann glia membrane potential in slices from MrgA1+ mice exposed to FMRF, slices from WT mice exposed to ATP, and slices from either IP3R2+/+ or IP3R2−/− mice exposed to UTP (*P < 0.05, n = 6–9). (H) Scatter histogram displaying Bergmann glia membrane potential as a function of changes in extracellular K+. Data are combined from the agonists mentioned before: ATP, FMRF, and UTP (R2 = 0.486, n = 24).
Hyper- and Depolarization of Bergmann Glia Modulate Purkinje Neuron Bistability.
The passive membrane permeability of Bergmann glial cells is predominantly attributable to K+. Hyper- and depolarization currents are almost exclusively carried by K+ influx or efflux and are accompanied by changes of extracellular K+ in a symmetrical manner. Previous analysis shows that clamping the resting membrane potential of Bergmann glial cells offers an alternative approach to assessing the role of glial K+ buffering on Purkinje cell bistability (2). We repeated the original observations by Hounsgaard and Nicholson (2) and confirmed that injecting a hyperpolarizing current (−100 pA) in Bergmann glial cells caused a drop in the resting membrane potential, averaging −7.04 ± 0.69 mV (n = 6), which was paralleled by a decrease in extracellular K+ of 0.288 ± 0.017 mM (n = 6; Fig. S3A). Conversely, injecting a depolarizing current (+100 pA) triggered a symmetric increase in membrane potential, −7.00 ± 0.68 mV (n = 6), and an increase in extracellular K+, averaging 0.290 ± 0.021 mM (n = 6; Fig. S3A).
To assess the effect of Bergmann glial cell-membrane potential on Purkinje cell up and down states, we next used dual whole-cell recordings of neighboring Purkinje cells and Bergmann glial cells. As shown in Fig. S3 B and C, the relative time Purkinje cells spent in up state increased from 39.1% ± 8.5% to 68.2% ± 11.4% (n = 6) when an adjacent Bergmann glial cell was hyperpolarized by injecting a negative current (−100 pA). Depolarization of Bergmann glial cells (+100-pA current) had the opposite effect (Fig. S3 D–F). Concomitantly, the gap in membrane potential between the two states narrowed when Bergmann glial cells were hyperpolarized and widened when Bergmann glial cells were depolarized (Fig. S3G). The ISI and CV of action potentials did not change significantly in response to either hyperpolarization or depolarization of Bergmann glia cells (Fig. S3H).
Extracellular K+ Directly Modulated Purkinje Cell Bistability.
The analysis above indicates that Ca2+ signaling in Bergmann glia induced both a decrease in extracellular K+ and an increase in the duration of Purkinje cell up states. To critically evaluate whether a reduction in extracellular K+, in the absence of Bergmann glial Ca2+ signaling, is sufficient to trigger an increase in the duration of up states, we next systematically changed extracellular K+ while continuously recording from Purkinje cells. Whole-cell recording from Purkinje cells showed that, by decreasing bath K+ from 2.5 mM to 1 mM, the duration of up state increased (P < 0.01, n = 6; Fig. 4 A and B). The increase in duration of up state was linked to a twofold increase in the frequency of simple spikes (Fig. 4 C and D) without affecting ISI or CV of action potentials during up state (Fig. 4 E and F). The normal pattern of bistability returned with a delay of 10–30 s after bath K+ was returned to 2.5 mM (Fig. 4A). Testing a range of physiological K+ concentrations (1–5 mM) revealed that up state was prevalent when K+ concentration was reduced in the bath solution (Fig. 4G). The prevalence of up state with lower K+ was associated with a narrowed gap between up and down states of Purkinje cell-membrane potentials (Fig. 4H). Of note, K+-sensitive microelectrodes inserted ∼80–100 μm below the surface of the slice showed that changes in extracellular K+ within cerebellar slices were of much smaller amplitude than the changes in bath K+ (Fig. 4H, compare bottom axes) and the depth of the recordings affected how much changes in bath K+ changed extracellular K+ (Fig. S4). The most significant effect on Purkinje cell bistability occurred at 3 mM extracellular K+, which is within the physiological concentration range (2).
Fig. 4.
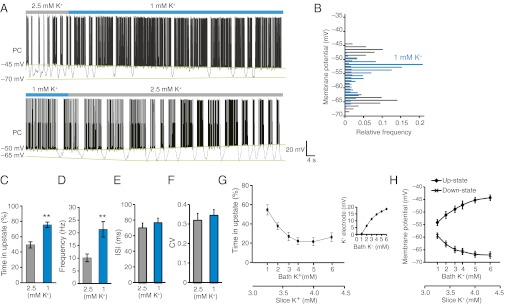
Purkinje cell bistability is modulated by extracellular K+ concentration. (A) Whole-cell recording of a Purkinje cell (PC) superfused with 2.5 mM or 1 mM K+. (B) Distribution of membrane potential when superfused with 2.5 mM or 1 mM K. (C and D) Time spent in up state (C) and the Purkinje cell spiking frequency (D) in 2.5 and 1 mM K+ (**P < 0.01, n = 5). (E) ISI of action potential firing analyzed during up state. (F) CV of action potential frequency. (G) Time in up state plotted as a function of increasing concentration of extracellular K+. The two x axes show K+ in the bath solution and the actual changes in extracellular K+ detected by a K+-sensitive microelectrode at 100 μm below the slice surface (n = 9). (Inset) Calibration of a double-barreled K+-sensitive microelectrode. (H) Membrane potential of up and down states in Purkinje cells plotted as a function of bath (upper trace) and extracellular K+ (lower trace) (n = 9). The Purkinje cell recordings were similar to all other recordings in this study obtained in neurons located 80–100 μm below the surface of the slice.
To address the alternative hypothesis that Bergmann glia modulate Purkinje cell bistability via Ca2+-mediated release of gliotransmitters, we analyzed the effect of glutamatergic, GABAergic, and purinergic receptor antagonists on Purkinje cell bistability. Mixtures of glutamate, GABA, and purinergic receptors had no detectable effects on Purkinje cell bistability in rat cerebellar slices (P > 0.5; Fig. S5). Moreover, the same combination of receptor antagonists did not significantly reduce FMRF-induced changes in Purkinje cell bistability in slices prepared from MrgA1+ mice (P > 0.5; Fig. S5), suggesting that FMRF-induced Ca2+ signaling is not associated with gliotransmitter release. This finding is consistent with a study that found that FMRF-induced Ca2+ signaling in hippocampal astrocytes did not result in gliotransmitter release (15).
Agonist-Induced Bergmann Glial Ca2+ Signaling in Vivo Triggered a Reduction in Extracellular K+ and Increased Neural Activity.
Bergmann glia exhibit spontaneous Ca2+ events in vivo that are expressed as highly localized Ca2+ increases within individual processes or radially expanding waves encompassing up to ∼40 cells (3). The functional role of Ca2+ signaling in Bergmann glia has not yet been established, although locomotion is linked to an increased frequency of Ca2+ events (3). We here investigated the physiological significance of agonist-induced Ca2+ increases in Bergmann glia in vivo, as far as Purkinje cell bistability/slow oscillations are concerned. Acute cranial windows were prepared over the vermis, and the cerebellar cortex bolus was loaded with the Ca2+ indicator rhod2/am. Ca2+ imaging was combined with recordings of cerebellar electrocortical activity (ECoG). To identify processes of Bergmann glia, MrgA1 receptor mice expressing EGFP under the human GFAP promoter (15) or reporter mice expressing EGFP under the glutamate transporter 1 (GLT1) promoter (GLT1-EGFP) (21) were used. The EGFP+ palisading Bergmann glial processes labeled more intensively with rhod2 than the surrounding neuropil (Fig. 5A). Time-lapse imaging documented that FMRF (15 μM) in MrgA1+ mice or ATP (100 μM) in GLT1-EGFP mice triggered robust increases in Ca2+. The Ca2+ increases engaged all EGFP+ glial processes (Fig. 5 A–C). Concurrent with the increase in Ca2+, extracellular K+ decreased from 3.98 ± 0.05 mM to 3.56 ± 0.07 mM (n = 5) in response to FMRF and from 3.91 ± 0.04 mM to 3.45 ± 0.05 mM for ATP (n = 7; Fig. 5 B and D). Thus, agonist-induced K+ in the live intact cerebellum was directly comparable to observations in cerebellar slices (Fig. 3). Recording of cerebellar ECoG revealed that agonist exposure triggered a transient decrease in the power of low-frequency activity (≤4-Hz delta waves), whereas higher frequencies (>4–32 Hz) increased (Fig. 5E, Right) (22). Agonist-induced changes in ECoG were transients as the relative power of slow and high-frequency activity before and after the Ca2+ increases did not differ (Fig. 5 B and C; P > 0.01, paired t test, n = 7 for ATP or FMRF). Thus, in vivo observations in adult mice (Fig. 5) replicated all of the major findings in cerebellar slices (Figs. 1–4).
Fig. 5.

Agonist-induced Ca2+ signaling in Bergmann glia reduces extracellular K+ and alters cerebellar ECoG in vivo. (A) Two-photon imaging of EGFP and rhod2 in vermis of an adult MrgA1+ mouse. FMRF exposure transiently increases rhod2 signal in EGFP+ Bergmann glia processes. Single optical sections at 100 μm below the pial surface are shown. (Scale bar: 30 μm.) (B) Cerebellar ECoG combined with imaging of rhod2 signal and recordings of K+ with K+-sensitive microelectrodes at a depth of 100 μm. (C) rhod2 fluorescence emission before, during, and after exposure to FMRF (in MrgA1+ mice) or to ATP. (D) Changes in extracellular K+ before, during, and after exposure to FMRF or ATP (P < 0.01, n = 5–7). (E) Power spectrum analysis of ECoG. The relative power of slow activity (≤4 Hz) decreased, whereas the relative power of higher frequencies (<4–32 Hz) increased during agonist-induced Ca2+ signaling (*P < 0.05, **P < 0.01, n = 5–7).
Discussion
It is generally acknowledged that Ca2+ signaling in many brain regions can modulate synaptic activity. We here describe that in cerebellum, Ca2+ signaling in Bergmann glia transiently increased their K+ uptake, reduced the extracellular concentration of K+, and prolonged the duration of Purkinje cell up states that resulted in an increase in spike activity. By combining measurements of extracellular potassium with two-photon imaging in genetically altered mice, we show that selectively stimulating Ca2+ transients in Bergmann glia expressing MrgA1 receptors lowers extracellular K+ with concomitant increases in Purkinje cell up state (Fig. 1). The process is Ca2+-dependent as the agonist-induced increase in Purkinje cell up state (Fig. 2) or reduction in extracellular K+ (Fig. 3) was minimal or absent when Bergmann glia cells were unable to mobilize intracellular Ca2+ stores (IP3R2 deletion). Simply lowering extracellular K+ (Fig. 4) or depolarizing a single Bergmann glial cell (Fig. S3) replicated all of the effects of agonist-induced Ca2+ signaling, supporting the notion that Bergmann glial K+ uptake provides a simple, yet powerful, mechanism for modulation of Purkinje cell output. In vivo experiments confirmed that this glia–neuronal signaling pathway functions in the intact adult cerebellum because agonist-induced Ca2+ signaling in Bergmann glia triggered a transient drop in extracellular K+ concentration concomitant with a decrease in low-frequency power (1–4 Hz) of ECoG (Fig. 5, Left). The ability of Bergmann glia to dynamically regulate extracellular K+ in a Ca2+-dependent manner may be central to our understanding of how these electrically nonexcitable cells participate in cerebellar network activity. A recent publication observed, similar to findings in this study, that stimulation of astrocytic Ca2+ signaling in cortical slices triggered an increase in up states (13). However, cortical astrocytes modulated bistability by release of gliotransmitters, as P2 receptors antagonists increased, whereas P1 (adenosine A1) receptor antagonists decreased up states. It is not surprising that the observations in cortex and cerebellum differ (13). Although bistability in cerebellar Purkinje cells is generated by intrinsic membrane properties and depends on persistent currents (6, 7), neocortex is extensively interconnected by recurrent excitatory collaterals, and bistability operates through a balance between excitation and inhibition (8). Moreover, cerebellar Purkinje cells are inhibitory neurons that do not express adenosine A1 receptors (1, 23). It is, however, of interest that both studies concluded that glia Ca2+ signaling triggers an increase in up states and may reflect that glia use different strategies to modulate neural circuits depending on their location in CNS. Of note, our study excludes that gliotransmitter played a role in the increase in up states during Ca2+ signaling in Bergmann glia cells, because a broad range of receptor antagonists applied either individually or as a mixture failed to affect Bergmann glia modulation of bistability (Fig. S5).
Extracellular K+ Modulates Purkinje Cell Bistability.
During sleep and anesthesia, the membrane potentials of Purkinje cells oscillate between up and down states (5), whereas Purkinje cells in awake animals operate primarily in the up state (24). The bistability persists without synaptic input and is also present in cerebellar slices, suggesting that it is driven by intrinsic membrane properties (6, 7). Similar to other neurons, it is thought that Purkinje cell bistability is generated by the balance between inward currents, consisting of T-type Ca2+ currents and/or persistent Na+ currents in combination with K+ leak currents, and hyperpolarization-activated cation channels (6, 7, 12).
The Nernst equation predicts that reducing extracellular K+ will result in membrane hyperpolarization by increasing the driving force for K+ leaking out of the cell. Why then, does a reduction in extracellular K+ lead to an increase in up states in Purkinje cells in both cerebellar slices and in vivo preparations? A recent study showed that hyperpolarization of Purkinje cells paradoxically increased the duration of up states by activating Ih channels (12). However, it is unlikely that this mechanism is responsible for the observations reported here because Ca2+ signaling in Bergmann glia was linked to hyperpolarization of up states and depolarization in down states (Figs. 1 and 2). A more likely explanation is that the hyperpolarization of up states (Fig. 4H) decreased the activity of the Ca2+-dependent K+ channels, which are necessary for driving the membrane potential back to the down state. Conversely, the depolarization of down states increased T-type Ca2+ and Na+ channel activation and thereby destabilized, and shortened, the duration of down state (10). In all, although a reduction in extracellular K+ results in membrane hyperpolarization at up states and depolarization at down states, the net effect is a stabilization and prolongation of up state combined with depolarization and destabilization of down state.
Implication of Ca2+-Dependent Glial K+ Buffering.
Our results show that Bergmann glia cells have the ability to transiently boost their K+ uptake capacity via receptor-mediated increases in cytosolic Ca2+. We have shown in a previous study that the Na+,K+ATPase activity is increased during Ca2+ signaling in hippocampal glia cells, resulting in a reduction in extracellular K+ (14). We speculate that a similar mechanism exists in cerebellum and that the transient reduction of extracellular K+ hyperpolarized Purkinje cells, which paradoxically prolonged up states. An in vivo analysis in anesthetized mice confirmed that agonist-induced Ca2+ signaling in Bergmann glia was linked to a transient reduction in extracellular K+ and an increased duration of time spent in up state (Fig. 5). We speculate that the increased spontaneous Ca2+ signaling in nonanesthetized mice may cause a persistent reduction in extracellular K+ that contributes to the lack of bistability in awake animals (24). If these observations can be generalized to other brain regions, our report provides a framework for dissecting the role of Ca2+-dependent K+ uptake on synaptic transmission and neural network activity. Neuromodulators are released diffusely across most of the forebrain during arousal, and neuromodulators directly induce Ca2+ signaling in cortical astrocytes (25). Ca2+-dependent K+ uptake in glia may therefore support the tonic up state characteristic of the awake state and affect functional networks on a global scale.
Materials and Methods
Full details of all methods are available in SI Materials and Methods.
Animals.
All slice experiments used cerebellum from 14- to 23-d-old Sprague-Dawley rat pups or C57BL/6 mouse pups (Charles River).
Two-Photon Laser Scanning Microscopy.
A custom-built microscope attached to a Tsunami/Millenium laser (10 W; Spectra-Physics) and scan box (FV300; Olympus) using FluoView software was used for two-photon laser scanning microscopy.
K+-Sensitive Microelectrodes.
Ion-sensitive microelectrodes were fabricated from double-barreled pipette glass (PB150F-6; WPI).
Supplementary Material
Acknowledgments
We thank Charles Nicholson and Sabina Hrabetova for technical assistance with fabrication of K+-sensitive microelectrodes; J. Chen, K. McCarthy, and J. Rothstein for generously sharing genetically modified mice; and Lane Bekar for valuable comments on the manuscript. This study was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke Grants NS075177 and NS078304; the W. M. Keck Foundation; and the Dana Foundation.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1120380109/-/DCSupplemental.
References
- 1.Bellamy TC. Interactions between Purkinje neurones and Bergmann glia. Cerebellum. 2006;5:116–126. doi: 10.1080/14734220600724569. [DOI] [PubMed] [Google Scholar]
- 2.Hounsgaard J, Nicholson C. Potassium accumulation around individual Purkinje cells in cerebellar slices from the guinea-pig. J Physiol. 1983;340:359–388. doi: 10.1113/jphysiol.1983.sp014767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoogland TM, et al. Radially expanding transglial calcium waves in the intact cerebellum. Proc Natl Acad Sci USA. 2009;106:3496–3501. doi: 10.1073/pnas.0809269106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steriade M, Contreras D, Curró Dossi R, Nuñez A. The slow (< 1 Hz) oscillation in reticular thalamic and thalamocortical neurons: Scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. 1993;13:3284–3299. doi: 10.1523/JNEUROSCI.13-08-03284.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loewenstein Y, et al. Bistability of cerebellar Purkinje cells modulated by sensory stimulation. Nat Neurosci. 2005;8:202–211. doi: 10.1038/nn1393. [DOI] [PubMed] [Google Scholar]
- 6.Llinás R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol. 1980;305(2):197–213. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Llinás R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol. 1980;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCormick DA. Neuronal networks: Flip-flops in the brain. Curr Biol. 2005;15:R294–R296. doi: 10.1016/j.cub.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Kay AR, Sugimori M, Llinás R. Kinetic and stochastic properties of a persistent sodium current in mature guinea pig cerebellar Purkinje cells. J Neurophysiol. 1998;80:1167–1179. doi: 10.1152/jn.1998.80.3.1167. [DOI] [PubMed] [Google Scholar]
- 10.Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plotkin JL, Day M, Surmeier DJ. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat Neurosci. 2011;14:881–888. doi: 10.1038/nn.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldfield CS, Marty A, Stell BM. Interneurons of the cerebellar cortex toggle Purkinje cells between up and down states. Proc Natl Acad Sci USA. 2010;107:13153–13158. doi: 10.1073/pnas.1002082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poskanzer KE, Yuste R. Astrocytic regulation of cortical UP states. Proc Natl Acad Sci USA. 2011;108:18453–18458. doi: 10.1073/pnas.1112378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F, et al. Astrocytes modulate neural network activity by Ca2+ dependent uptake of extracellular K+ Science Signalling. 2012. 5(218):ra26. Available at http://stke.sciencemag.org/cgi/content/summary/sigtrans;5/218/ra26. [DOI] [PMC free article] [PubMed]
- 15.Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327:1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- 16.Holtzclaw LA, Pandhit S, Bare DJ, Mignery GA, Russell JT. Astrocytes in adult rat brain express type 2 inositol 1,4,5-trisphosphate receptors. Glia. 2002;39(1):69–84. doi: 10.1002/glia.10085. [DOI] [PubMed] [Google Scholar]
- 17.Kuffler SW, Nicholls JG, Orkand RK. Physiological properties of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:768–787. doi: 10.1152/jn.1966.29.4.768. [DOI] [PubMed] [Google Scholar]
- 18.Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36(4–5):291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- 19.Wen R, Oakley B., 2nd K+-evoked Müller cell depolarization generates b-wave of electroretinogram in toad retina. Proc Natl Acad Sci USA. 1990;87(6):2117–2121. doi: 10.1073/pnas.87.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman EA. Electrophysiology of retinal glial cells. Prog Retinal Res. 1988;8(4–5):153–171. [Google Scholar]
- 21.Regan MR, et al. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci. 2007;27:6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thakor NV, Tong S. Advances in quantitative electroencephalogram analysis methods. Annu Rev Biomed Eng. 2004;6:453–495. doi: 10.1146/annurev.bioeng.5.040202.121601. [DOI] [PubMed] [Google Scholar]
- 23.Yoon KW, Rothman SM. Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J Neurosci. 1991;11:1375–1380. doi: 10.1523/JNEUROSCI.11-05-01375.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schonewille M, et al. Purkinje cells in awake behaving animals operate at the upstate membrane potential. Nat Neurosci. 2006;9:459–461, author reply 461. doi: 10.1038/nn0406-459. [DOI] [PubMed] [Google Scholar]
- 25.Bekar LK, He W, Nedergaard M. Locus coeruleus α-adrenergic-mediated activation of cortical astrocytes in vivo. Cereb Cortex. 2008;18:2789–2795. doi: 10.1093/cercor/bhn040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.