

Abstract
OBJECTIVE
The human aldehyde dehydrogenase (ALDH) gene superfamily consists of 19 genes encoding enzymes critical for NAD(P)+-dependent oxidation of endogenous and exogenous aldehydes, including drugs and environmental toxicants. Mutations in ALDH genes are the molecular basis of several disease states (e.g. Sjögren-Larsson syndrome, pyridoxine-dependent seizures, and type II hyperprolinemia) and may contribute to the etiology of complex diseases such as cancer and Alzheimer’s disease. The aim of this nomenclature update was to identify splice transcriptional variants principally for the human ALDH genes.
METHODS
Data-mining methods were used to retrieve all human ALDH sequences. Alternatively-spliced transcriptional variants were determined based upon: a) criteria for sequence integrity and genomic alignment; b) evidence of multiple independent cDNA sequences corresponding to a variant sequence; and c) if available, empirical evidence of variants from the literature.
RESULTS AND CONCLUSION
Alternatively-spliced transcriptional variants and their encoded proteins exist for most of the human ALDH genes; however, their function and significance remain to be established. When compared with the human genome, rat and mouse include an additional gene, Aldh1a7, in the ALDH1A subfamily. In order to avoid confusion when identifying splice variants in various genomes, nomenclature guidelines for the naming of such alternative transcriptional variants and proteins are recommended herein. In addition, a web database (www.aldh.org) has been developed to provide up-to-date information and nomenclature guidelines for the ALDH superfamily.
Keywords: Aldehyde Dehydrogenase, ALDH, Alternatively-Spliced Variants, Nomenclature, Human
Introduction
Aldehydes are highly reactive compounds capable of exerting a variety of toxic cellular events including adduct formation with DNA and proteins. Endogenous aldehydes are formed during the metabolism of numerous compounds including alcohols, amino acids, biogenic amines, vitamins, steroids and lipids. Exogenous aldehydes are often generated from the biotransformation of drugs and environmental agents [1, 2]. The mammalian ALDH gene superfamily encodes a group of evolutionarily-related sequences whose protein products all have pyridine nucleotide-dependent oxidation activity catalyzing the irreversible oxidation of aldehydic substrates to their corresponding carboxylic acids [3-5].
Although many ALDH enzymes display broad substrate specificity and oxidize a variety of aliphatic and aromatic aldehydes, others retain unique substrate preferences. In addition to their primary role in aldehyde oxidation, many ALDH enzymes possess multiple catalytic and non-catalytic functions. For example, ALDH1A1, ALDH2, ALDH3A1 and ALDH4A1 catalyze ester hydrolysis; in the case of ALDH2, this hydrolytic activity has been implicated in the bioactivation of nitroglycerin to nitric oxide [6, 7]. ALDH1A1 is capable of binding androgens, cholesterol, thyroid hormone and flavopyridol whereas ALDH2 has been identified as an acetaminophen-binding protein [4, 8]. ALDH proteins have been hypothesized to play a critical role in cellular homeostasis by maintaining redox balance [9]. For example, ALDH enzymes contribute to the antioxidant capacity of a cell by generating NAD(P)H, which can be used for the regeneration of reduced glutathione (GSH). Furthermore, it has been proposed that ALDH3A1 may scavenge hydroxyl radicals via reduction of its cysteine and methionine thiol groups [10, 11]. The ALDH proteins not only differ with regard to their catalytic/non-catalytic properties and tissue distribution but also in relation to their sensitivity to inhibitors, suppressors and inducers.
The clinical importance of ALDH enzymes is evident from the observation that mutations and polymorphisms in ALDH genes (leading to loss of function) are associated with distinct phenotypes in humans [8, 12]—including Sjögren-Larsson syndrome [13], type II hyperprolinemia [14], γ-hydroxybutyric aciduria [15], pyridoxine-dependent seizures [16], hyperammonemia [17], alcohol-related diseases [18], cancer [19] and late-onset Alzheimer’s disease [20]. Aside from the clinical phenotypes associated with mutations in ALDH genes, knockout mouse models have suggested a crucial role of ALDH enzymes in physiological functions and processes, such as embryogenesis and development [21, 22] as well as protection against oxidative stress [23].
A growing body of evidence supports the expression of alternatively-spliced transcriptional variants for many of the ALDH genes. However, the spatiotemporal factors affecting this expression (as well as their physiologic roles) remain unclear. In the present paper, we describe and classify alternatively-spliced transcript products within the human ALDH gene superfamily. These alternatively-spliced variants were identified within the molecular sequence libraries from the National Center for Biotechnology Information (NCBI) and the European Bioinformatics Institute (EBI) and classified in accordance with recommended nomenclature guidelines for the naming of such alternative transcriptional variants and their proteins.
To assist readers and to provide a detailed resource for the ALDH gene superfamily, an ALDH database is located on the web at www.aldh.org. Extensive information for each ALDH gene found in human, other animals, archaebacteria, eubacteria, fungi, plant, and yeast genomes is available—including information on the current practices of the ALDH nomenclature system. There are also links to other informational databases and programs for analyzing protein and DNA sequences, such as those maintained by NCBI. Furthermore, graphical and tabular representation of all transcriptional variants and corresponding proteins described in this present report are available at www.aldh.org for visual reference.
Methods
Data mining was employed to identify new (and existing), putatively-functional ALDH protein-coding sequences and relevant information for the genes, transcripts, and corresponding proteins of mammalian genomes from the human, mouse, rat, rhesus monkey, chimpanzee, cow, dog, rabbit and opossum. Transcript and peptide sequence orthologs were identified utilizing the Basic Local Alignment Search Tool (BLAST) program [24]. Multiple sequence alignments using Clustal W [25] and T-Coffee [26] were used to compare and catalog ALDH genes across species. We also created an evolutionary dendrogram of known human, mouse and rat ALDH sequences (Figure 1).
Figure 1.
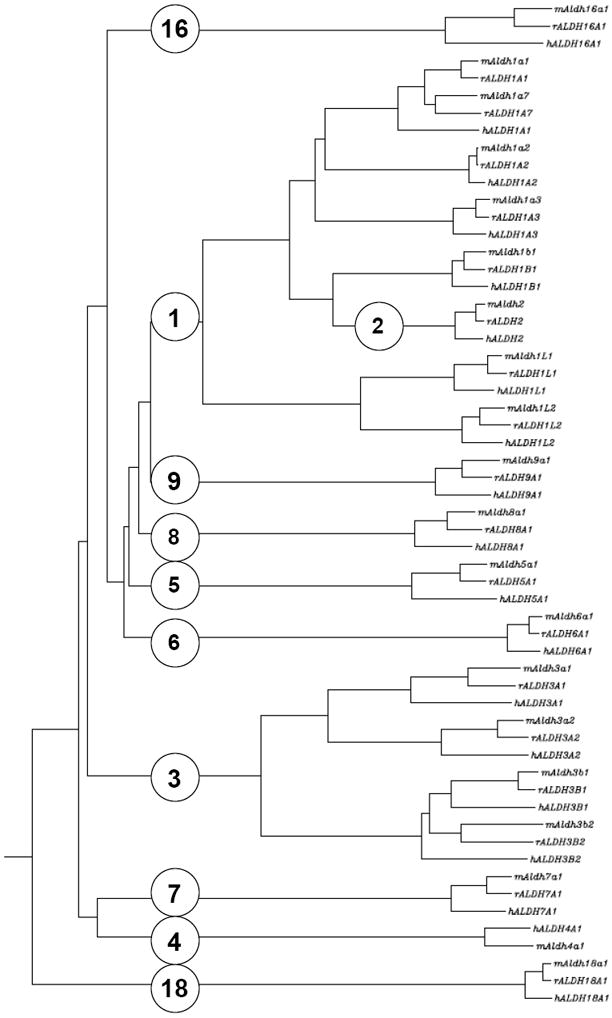
Dendrogram illustrating the evolutionary relationship of ALDH protein sequences from human, mouse, and rat. Accession numbers for ALDH sequences are provided at www.aldh.org.
Sequences for all transcript and peptide translations of accession identification numbers referenced within are available from the NCBI and European Molecular Biology Laboratory (EMBL)-EBI databases. These entities were analyzed for sequence integrity and genomic alignment based upon the most recent build assemblies available from these institutes at the time of this writing. Transcript sequences were aligned with their corresponding genomic assembly using our proprietary SAST alignment software (2009, W. Black and V. Vasiliou, manuscript in preparation) and confirmed with NCBI’s Splign utility [27].
The structural integrity of all transcript sequences was determined to have a coding sequence beginning with a 5’methionine initiation codon (ATG) and a 3’ termination codon (TGA, TAG or TAA). Translation of this coding sequence was then analyzed to confirm that the corresponding reading frame retained an ALDH peptide domain according to the Hidden Markov Model (HMM) for this domain, termed “aldedh”, available from Pfam [28]. Alternatively-spliced transcriptional variants (described herein) were determined based upon: a) criteria for sequence integrity and genomic alignment; b) evidence of multiple independent cDNA sequences corresponding to a variant sequence; and c) if available, empirical evidence of variants from the literature. Multiple independent cDNA sequences that were associated with a particular variant were considered indicative of a potential alternatively-spliced transcriptional variant; unique sequences were not described but were shelved for further analysis and data support.
The identification of splice transcripts and the resulting proteins raises the issue of nomenclature for these entities within existing and future literature, as they are identified in various genomes. In keeping with the Human Gene Nomenclature Guidelines, alternatively-spliced transcriptional variants and corresponding proteins are denoted by a “_v” symbol followed by a number indicating the variant (e.g. ALDH3A1_v2). Manuscripts describing an ALDH entity subject to alternative splicing should clearly state the variant being studied. In this regard, different alternative transcriptional variants and corresponding proteins may prove to have vastly different properties and functionalities. In the human genome, evidence for alternative transcripts exists for most of the 19 ALDH genes—with the exception of ALDH1B1, ALDH2, ALDH7A1 and ALDH9A1.
The ALDH-like Clan and the Mammalian ALDH Gene Superfamily
The ALDH gene superfamily is included in the ALDH-like clan (Pfam CL0099) which consists of four members; the ALDH gene superfamily (Pfam “Aldedh”), a family of uncharacterized proteins from Drosophila melanogaster (Pfam DUF1487; PF07368), a histidinol dehydrogenase family (Pfam “Histidinol_dh”; PF00815), and an acyl-CoA reductase family (Pfam “LuxC”; PF05893). Members of the ALDH gene superfamily are widely expressed among eukaryotes and prokaryotes. Analysis of mammalian genomes has revealed the presence of 19 or 20 ALDH gene orthologs per species. A clustering dendrogram of the human, mouse and rat ALDHs is shown in Figure 1. To date, 19 putatively functional ALDH genes exist in the human genome and a brief description of the function of these gene products is provided in Table 1.
Table 1.
Human ALDH genes and gene products
| Gene | Protein Description |
|---|---|
| ALDH1A1 | ALDH1A1 is a cytosolic enzyme that oxidizes retinal, acetaldehydes and 3-deoxyglucosone (a product of protein deglycation and a potent glycating agent). |
| ALDH1A2 | ALDH1A2 is a cytosolic enzyme that is integrally involved in the oxidation of retinal to retinoic acid during embryonic development. Aldh1a2(-/-) mice are embryolethal. |
| ALDH1A3 | ALDH1A3 is a cytosolic retinaldehyde-metabolizing enzyme. |
| ALDH1B1 | ALDH1B1 is a mitochondrial enzyme that metabolizes acetaldehyde. |
| ALDH1L1 | ALDH1L1 is a fusion protein comprising three domains: a formyl transferase domain at the amino terminal, a centrally-located formyltransferase carboxyl terminal domain and an aldehyde dehydrogenase domain at its carboxyl terminal (Figure 2). |
| ALDH1L2 | ALDH1L2 shares ≈73% identity with ALDH1L1; no functional data have been reported for this protein. |
| ALDH2 | ALDH2 is a mitochondrial enzyme involved in the oxidation of acetaldehyde and the metabolites of dopamine and norepinephrine, DOPAL and DOPEGAL, respectively. |
| ALDH3A1 | ALDH3A1 is a multifunctional enzyme that plays a significant role in the cellular response to oxidative stress. |
| ALDH3A2 | ALDH3A2 is a microsomal enzyme that oxidizes medium to long-chain fatty aldehydes. |
| ALDH3B1 | ALDH3B1 is a cytosolic protein that oxidizes medium- and long-chain saturated and unsaturated aliphatic aldehydes. |
| ALDH3B2 | ALDH3B2 is a putative ALDH with no functional data available. |
| ALDH4A1 | ALDH4A1 catalyzes the irreversible conversion of Δ1-pyrroline-5-carboxylate (derived from either proline or ornithine) to glutamate, necessary to connect the urea cycle with the tricarboxylic acid cycle. |
| ALDH5A1 | ALDH5A1 is the succinate semialdehyde dehydrogenase involved in the last step of GABA catabolism, converting GABA to succinate semialdehyde. |
| ALDH6A1 | ALDH6A1 is the methylmalonate semialdehyde dehydrogenase that catalyzes the irreversible oxidative decarboxylation of malonate and methylmalonate semialdehydes to acetyl- and propionyl-CoA, respectively. |
| ALDH7A1 | ALDH7A1 metabolizes α-aminoadipic semialdehyde, generated during lysine catabolism. |
| ALDH8A1 | ALDH8A1 appears to be involved in 9-cis-retinoic acid biosynthesis. |
| ALDH9A1 | ALDH9A1 catalyzes the oxidation of γ-aminobutyraldehyde and betaine aldehyde, a γ-trimethylaminobutyraldehyde. |
| ALDH16A1 | No functional information exists in the literature for this enzyme. |
| ALDH18A1 | ALDH18A1 is a bi-functional ATP- and NAD(P)H-dependent mitochondrial inner-membrane protein having both γ-glutamyl kinase and γ-glutamyl phosphate reductase activities |
ALDH1 Family
The ALDH1 family consists of six human ALDH genes: ALDH1A1, ALDH1A2, ALDH1A3, ALDH1B1, ALDH1L1 and ALDH1L2. The genomes of Rattus norvegicus (rat) and Mus musculus (mouse) contain an additional gene, Aldh1a7 that is 92% identical to mouse Aldh1a1. Therefore, the rodent Aldh1a7 very likely arose as a gene duplication event after the mammalian radiation ~70 million years ago (MYA) and then became fixed in the genome before the rat-mouse divergence ~17 MYA.
ALDH1A1
Two transcriptional variants identified for the human ALDH1A1 gene consist of 13 and 8 exons for the consensus ALDH1A1_v1 and ALDH1A1_v2, respectively (Table 2). Relative to the native ALDH1A1_v1, the ALDH1A1_v2 variant lacks the 3’ end of exon 7, a portion of the 5’ and 3’ ends of exon 9, and is missing exons 8, 10, 11, 12 and 13. This translates to a protein splice-variant missing 271 amino acids from the COOH-terminus, relative to the native form. Pfam analysis revealed this protein splice-variant retains an ALDH peptide domain—although truncated. The predicted active-site cysteine and glutamate residues of the primary variant ALDH1A1_v1 at positions 303 and 269, respectively, are not apparent within the ALDH1A1_v2 variant, strongly suggesting that this protein likely has no ALDH activity.
Table 2.
Human ALDH Alternative Transcripts
| Gene | Transcript | Exons | Clones* | Transcript Accession ‡ | Peptide | Peptide Accession ‡ | Length (amino acids) | M.W. (kDa) |
|---|---|---|---|---|---|---|---|---|
| ALDH1A1 | ||||||||
| ALDH1A1 | 13 | 236 | NM_000689 | ALDH1A1 | NP_000680 | 501 | 54.7 | |
| ALDH1A1_v2 | 8 | 16 | ENST00000376939 | ALDH1A1_v2 | ENSP00000366138 | 230 | 25.3 | |
| ALDH1A2 | ||||||||
| ALDH1A2 | 13 | 135 | NM_003888 | ALDH1A2 | NP_003879 | 518 | 56.5 | |
| ALDH1A2_v2 | 12 | 5 | NM_170696 | ALDH1A2_v2 | NP_733797 | 480 | 52.9 | |
| ALDH1A2_v3 | 11 | 5 | NM_170697 | ALDH1A2_v3 | NP_733798 | 422 | 46.0 | |
| ALDH1A2_v4 | 12 | 5 | ALDH1A2.cApr07 | ALDH1A2_v4 | ALDH1A2.cApr07 | 384 | 42.4 | |
| ALDH1A3 | ||||||||
| ALDH1A3 | 13 | 153 | NM_000693 | ALDH1A3 | NP_000684 | 512 | 55.9 | |
| ALDH1A3_v2 | 10 | 158 | ENST00000346623 | ALDH1A3_v2 | ENSP00000343294 | 416 | 45.4 | |
| ALDH1B1 | ||||||||
| ALDH1B1 | 2 | 213 | NM_000692 | ALDH1B1 | NP_000683 | 517 | 57.2 | |
| ALDH1L1 | ||||||||
| ALDH1L1 | 23 | 190 | NM_012190 | ALDH1L1 | NP_036322 | 902 | 98.6 | |
| ALDH1L1_v2 | 23 | 1 | ENST00000273450 | ALDH1L1_v2 | ENSP00000273450 | 912 | 99.7 | |
| ALDH1L1_v3 | 22 | N.A. | ENST00000393431 | ALDH1L1_v3 | ENSP00000377081 | 505 | 55.3 | |
| ALDH1L1_v4 | 7 | 7 | ALDH1L1.hApr07 | ALDH1L1_v4 | ALDH1L1.hApr07 | 333 | 36.4 | |
| ALDH1L1_v5 | 6 | 6 | ALDH1L1.jApr07 | ALDH1L1_v5 | ALDH1L1.jApr07 | 259 | 28.5 | |
| ALDH1L2 | ||||||||
| ALDH1L2 | 23 | 10 | NM_001034173 | ALDH1L2 | NP_001029345 | 923 | 101.6 | |
| ALDH1L2_v2 | 11 | 37 | ALDH1L2.cApr07 | ALDH1L2_v2 | ALDH1L2.cApr07 | 378 | 41.4 | |
| ALDH1L2_v3 | 22 | 34 | ALDH1L2.aApr07 | ALDH1L2_v3 | ALDH1L2.aApr07 | 810 | 89.1 | |
| ALDH2 | ||||||||
| ALDH2 | 13 | 222 | NM_000690 | ALDH2 | NP_000681 | 517 | 56.3 | |
| ALDH3A1 | ||||||||
| ALDH3A1 | 11 | 325 | NM_000691 | ALDH3A1 | NP_000682 | 453 | 50.4 | |
| ALDH3A1_v2 | 9 | 63 | ALDH3A1.aApr07 | ALDH3A1_v2 | ALDH3A1.aApr07 | 570 | 61.6 | |
| ALDH3A1_v3 | 11 | 44 | ALDH3A1.dApr07 | ALDH3A1_v3 | ALDH3A1.dApr07 | 452 | 50.3 | |
| ALDH3A1_v4 | 9 | 31 | ALDH3A1.hApr07 | ALDH3A1_v4 | ALDH3A1.hApr07 | 323 | 35.7 | |
| ALDH3A1_v5 | 8 | N.A. | ENST00000333946 | ALDH3A1_v5 | ENSP00000334590 | 570 | 61.5 | |
| ALDH3A1_v6 | 10 | 1 | ENST00000395555 | ALDH3A1_v6 | ENSP00000378923 | 389 | 43.3 | |
| ALDH3A1_v7 | 10 | N.A. | ALDH3A1.eApr07 | ALDH3A1_v7 | ALDH3A1.eApr07 | 380 | 41.9 | |
| ALDH3A2 | ||||||||
| ALDH3A2 | 10 | 191 | NM_000382 | ALDH3A2 | NP_000373 | 485 | 54.9 | |
| ALDH3A2_v2 | 11 | 18 | NM_001031806 | ALDH3A2_v2 | NP_001026976 | 508 | 57.5 | |
| ALDH3A2_v3 | 11 | 11 | ENST00000395575 | ALDH3A2_v3 | ENSP00000378942 | 485 | 54.8 | |
| ALDH3A2_v4 | 10 | N.A. | ENST00000404114 | ALDH3A2_v4 | ENSP00000385699 | 508 | 57.6 | |
| ALDH3A2_v5 | 7 | 38 | ALDH3A2.eApr07 | ALDH3A2_v5 | ALDH3A2.eApr07 | 292 | 33.0 | |
| ALDH3A2_v6 | 3 | 5 | ALDH3A2.lApr07 | ALDH3A2_v6 | ALDH3A2.lApr07 | 97 | 10.9 | |
| ALDH3B1 | ||||||||
| ALDH3B1 | 10 | 45 | NM_000694 | ALDH3B1 | NP_000685 | 468 | 51.7 | |
| ALDH3B1_v2 | 9 | 18 | NM_001030010 | ALDH3B1_v2 | NP_001025181 | 431 | 47.5 | |
| ALDH3B1_v3 | 9 | 98 | ALDH3B1.dApr07 | ALDH3B1_v3 | ALDH3B1.dApr07 | 248 | 27.6 | |
| ALDH3B1_v4 | 7 | 3 | ALDH3B1.eApr07 | ALDH3B1_v4 | ALDH3B1.eApr07 | 223 | 24.7 | |
| ALDH3B1_v5 | 9 | 4 | ALDH3B1.kApr07 | ALDH3B1_v5 | ALDH3B1.kApr07 | 88 | 9.6 | |
| ALDH3B2 | ||||||||
| ALDH3B2 | 10 | 89 | NM_000695 | ALDH3B2 | NP_000686 | 385 | 42.4 | |
| ALDH3B2_v2 | 10 | 101 | NM_001031615 | ALDH3B2_v2 | NP_001026786 | 385 | 42.4 | |
| ALDH3B2_v3 | 9 | 2 | ALDH3B2.cApr07 | ALDH3B2_v3 | ALDH3B2.cApr07 | 357 | 39.3 | |
| ALDH4A1 | ||||||||
| ALDH4A1 | 15 | 203 | NM_003748 | ALDH4A1 | NP_003739 | 563 | 61.7 | |
| ALDH4A1_v2 | 16 | 2 | NM_170726 | ALDH4A1_v2 | NP_733844 | 563 | 61.7 | |
| ALDH4A1_v3 | 14 | N.A. | ENST00000375335 | ALDH4A1_v4 | ENSP00000364484 | 547 | 59.8 | |
| ALDH4A1_v4 | 8 | N.A. | ENST00000375334 | ALDH4A1_v3 | ENSP00000364483 | 195 | 21.2 | |
| ALDH4A1_v5 | 9 | 2 | ALDH4A1.eApr07 | ALDH4A1_v5 | ALDH4A1.eApr07 | 195 | 21.2 | |
| ALDH5A1 | ||||||||
| ALDH5A1 | 10 | 216 | NM_001080 | ALDH5A1 | NP_001071 | 535 | 57.2 | |
| ALDH5A1_v2 | 11 | 10 | NM_170740 | ALDH5A1_v2 | NP_733936 | 548 | 58.6 | |
| ALDH5A1_v3 | 4 | 5 | ALDH5A1.cApr07 | ALDH5A1_v3 | ALDH5A1.cApr07 | 172 | 18.5 | |
| ALDH6A1 | ||||||||
| ALDH6A1 | 12 | 427 | NM_005589 | ALDH6A1 | NP_005580 | 535 | 57.8 | |
| ALDH6A1_v2 | 7 | 8 | ALDH6A1.bApr07 | ALDH6A1_v2 | ALDH6A1.bApr07 | 293 | 31.6 | |
| ALDH6A1_v3 | 5 | 3 | ALDH6A1.cApr07 | ALDH6A1_v3 | ALDH6A1.cApr07 | 179 | 19.6 | |
| ALDH6A1_v4 | 4 | 5 | ALDH6A1.jApr07 | ALDH6A1_v4 | ALDH6A1.jApr07 | 117 | 12.7 | |
| ALDH7A1 | ||||||||
| ALDH7A1 | 18 | 187 | NM_001182 | ALDH7A1 | NP_001173 | 511 | 55.2 | |
| ALDH8A1 | ||||||||
| ALDH8A1 | 7 | 68 | NM_022568 | ALDH8A1 | NP_072090 | 487 | 53.2 | |
| ALDH8A1_v2 | 6 | 3 | NM_170771 | ALDH8A1_v2 | NP_739577 | 433 | 47.1 | |
| ALDH9A1 | ||||||||
| ALDH9A1 | 11 | 246 | NM_000696 | ALDH9A1 | NP_000687 | 518 | 56.1 | |
| ALDH16A1 | ||||||||
| ALDH16A1 | 17 | 153 | NM_153329 | ALDH16A1 | NP_699160 | 802 | 84.9 | |
| ALDH16A1_v2 | 15 | 1 | ALDH16A1andFLT3LG.cApr07 | ALDH16A1_v2 | ALDH16A1andFLT3LG.cApr07 | 292 | 31.6 | |
| ALDH18A1 | ||||||||
| ALDH18A1 | 18 | 434 | NM_002860 | ALDH18A1 | NP_002851 | 795 | 87.1 | |
| ALDH18A1_v2 | 18 | 11 | NM_001017423 | ALDH18A1_v2 | NP_001017423 | 793 | 86.9 | |
Number of clones, as provided by the NCBI-AceView database.
Accession identification numbers from NCBI – GenBank have the format “NM_…”, “NP_…”, “XM_…”, or “XP_…”; from EBI – Ensembl have the format “ENS…”; and from NCBI – AceView have the format “ALDH#X#.xApr07”.
ALDH1A2
Four distinct human ALDH1A2 transcriptional variants have been identified (Table 2). The consensus ALDH1A2 variant, ALDH1A2_v1, represents the longest and most prevalent transcript and protein. Interestingly, intron 1 of both ALDH1A2_v1 and ALDH1A2_v2 is quite large (51.4 kb). ALDH1A2_v2 lacks the exon 7 segment present in the primary variant ALDH1A2_v1. Exon 7 is within the coding region of the transcript; the lack of this segment translates to a shorter protein. Variant ALDH1A2_v3, a derivative of ALDH1A2_v1, lacks exons 1 and 2 of ALDH1A2_v1. Relative to ALDH1A2_v1, the first exon of ALDH1A2_v3 contains a distinct 5’-untranslated region (UTR) comprising an additional 15-bp segment upstream of exon 3. The resulting protein variant has a shorter NH2-terminus in comparison to the major variant ALDH1A2_v1. A fourth variant identified within the sequence databases, ALDH1A2_v4, is a derivative of the ALDH1A2_v2 variant and lacks the 114-bp exon 7 of ALDH1A2_v1. This variant, however, utilizes an alternate exon 1 leading to a modified 5’ coding region.
ALDH1A3
The human ALDH1A3 gene includes two variant transcripts (Table 2). Although only a single transcript is reported by RefSeq in the NCBI Entrez Gene database (GeneID 220), a second variant, ALDH1A3_v2 is readily apparent according to cDNA evidence (Table 2) and as described by EMBL-EBI’s Ensembl (ENST00000346623). The ALDH1A3_v2 variant transcript lacks exons 4, 5, and 6—compared with ALDH1A3_v1—and encodes a splice-variant that is missing an internal segment within the ALDH peptide domain 5’ to the predicted cysteine and glutamate residues in the active-site.
Aldh1a7
Mouse Aldh1a7 most closely resembles an ancestral Aldh1a1 homolog when examined using evolutionary divergence (Figure 1). Comparing Aldh1a7 exon segments to other mammalian genomes using BLAST analysis does not produce significant correlations, suggesting speciation is limited. Details of alternatively-spliced transcriptional variants for the mouse and rat are beyond the scope of this manuscript. However, preliminary evidence suggests there are two transcriptional variants within NCBI’s AceView database accession identification numbers Aldh1a7.aSep07 and Aldh1a7.bSep07.
ALDH1B1
To date, no human transcriptional variants have been identified for this gene.
ALDH1L1
Five transcriptional variants have been identified for the ALDH1L1 gene (Figure 2, Table 2). The major transcript ALDH1L1_v1 encodes a 902-residue protein, and ALDH1L1_v2 encodes a 912-residue variant. ALDH1L1_v1 and ALDH1L1_v2 differ by an alternative exon 1—resulting in varied translation initiation points on exons 2 and 1 for ALDH1L1_v1 and ALDH1L1_v2, respectively. The ten additional amino acids at the NH2-terminus of ALDH1L1_v2 are not within any of the three peptide domains previously described for this protein; as such, functional relevance, if any, is unclear. The ALDH1L1_v3 transcript lacks the 151-bp exon 13 present in the other two variants. This represents a significant alteration in the reading frame that introduces an early termination signal and subsequent truncation in peptide translation. This truncation ablates most of the ALDH peptide domain, including its active-site cysteine and glutamate residues; accordingly, ALDH activity for this variant would presumably be null. ALDH1L1_v4 and ALDH1L1_v5 are truncated transcripts with no ALDH peptide domain in either of their resultant translated products.
Figure 2.
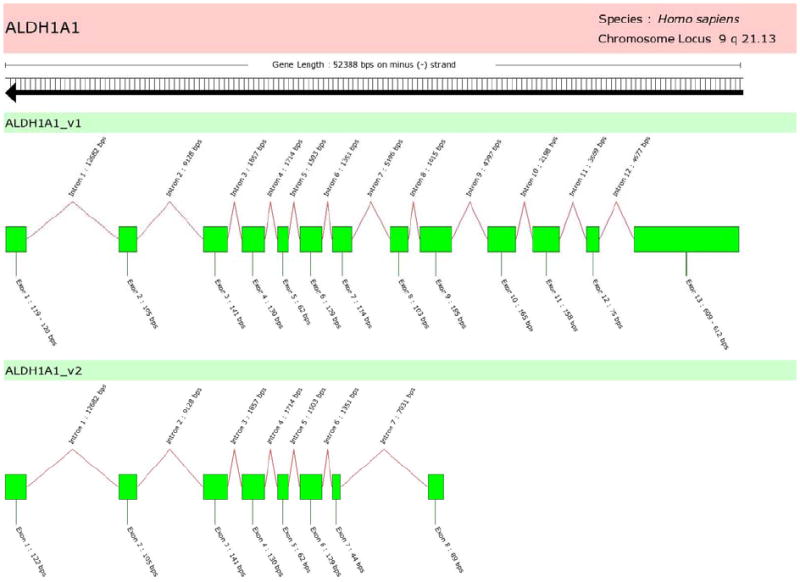
Human ALDH1A1 alternatively-spliced variant exon structures. The consensus variant ALDH1A1_v1 is a 13-exon transcript, whereas ALDH1A1_v2 has a shorter sequence due to truncation at its 3’ end. Specifically, ALDH1A1_v2 has a truncated exon 7 and a longer intron 8; its last exon (exon 8) is a 5’ and 3’ truncated subset of the ALDH1A1_v1 exon 9. The translation of ALDH1A1_v2 retains an ALDH peptide domain; however, no active-site residues are readily apparent.
ALDH1L2
The ALDH1L2 gene has three transcriptional variants (Table 2). The major transcript ALDH1L2_v1 encodes a 923-amino-acid protein. ALDH1L2_v2 utilizes an alternate exon 1, a 5’extended derivative of ALDH1L2_v1 exon 13, and lacks exons 1 to 12 of the ALDH1L2_v1 variant. The translation of this variant retains a central portion of ALDH peptide domain but the NH2-terminal and COOH-terminal formyl transferase peptide domains are ablated. The variant ALDH1L2_v3 lacks the 70-bp exon 1 of the ALDH1L2_v1 variant and encodes an 810-residue protein.
ALDH2 Family
To date, no human transcriptional variants have been identified for this gene.
ALDH3 Family
ALDH3A1
Several alternative splice variants exist within the molecular sequence databases for human ALDH3A1. The consensus gene product is an 11-exon transcript encoding a 50.4-kDa, 453-residue protein. Analysis of cDNA sequences for ALDH3A1 demonstrates a prevalence of three additional variants: ALDH3A1_v2, _v3 and _v4 relative to the ALDH3A1_v1 Reference Sequence (Table 2).
ALDH3A1_v2 comprises only nine exons, but encodes a larger 570-amino-acid variant due to its second exon being a fusion of exon 3, intron 3 and exon 4 (relative to the wild-type ALDH3A1_v1).. ALDH3A1_v3 is also an 11-exon transcript but it differs slightly from the ALDH3A1_v1 transcript by having a 5’ truncation of “GAG” from exon 7 within the coding region.. ALDH3A1_v4 is a 9-exon variant lacking the ALDH3A1_v1 exons 2 and 9. ALDH3A1_v5 is an 8-exon variant resembling ALDH3A1_v2, with regard to the “fusion” exon. However, this variant lacks exon 1 and the “fusion” exon has a 5’ truncation of the 88-bp exon 3 of ALDH3A1_v1. ALDH3A1_v6 is a 10-exon variant lacking the ALDH3A1_v1 exon 7 and truncation of 50 bp from the 5’ portion of exon 8. Lastly, ALDH3A1_v7 is a 10-exon variant lacking the ALDH3A1_v1 exon 2 encoding a functional ALDH peptide domain.
ALDH3A2
Similar to human ALDH3A1, ALDH3A2 has a number of transcriptional variants (Table 2). The primary variant ALDH3A2_v1 is a 10-exon transcript encoding a 485-residue protein expressed in microsomes. ALDH3A2_v2 includes an additional 125-bp exon between exons 9 and 10 (relative to the ALDH3A2_v1 variant), thus encoding a longer protein of 508 amino acids that is expressed in the peroxisomes [29]. The ALDH3A2_v3 and ALDH3A2_v4 variants have coding regions identical to that of the ALDH3A2_v1 and ALDH3A2_v2 variants, respectively, and differ only in exon structure. A number of independent cDNAs within the molecular sequence databases suggest the existence of ALDH3A2_v5—which uses an alternative exon 1 beginning upstream to and including exon 4 of the ALDH3A2_v1 variant.
ALDH3B1
Human ALDH3B1 may have as many as five transcriptional variants, according to the molecular sequence databases for the human ALDH3B1 gene (Table 2). The consensus product is a 10-exon transcript encoding a 468-residue protein. The ALDH3B1_v2 variant lacks exon 3 relative to ALDH3B1_v1; although exon 3 is within the coding region of the peptide, its translation is not associated with the ALDH peptide domain. Therefore, this variant encodes a shorter protein with a complete ALDH peptide domain. The ALDH3B1_v3 transcript has a 3340-bp exon 2—which is a fusion of exon 2, intron 2 and exon 3 of the ALDH3B1_v1 variant. This fusion results in a 3’ shift in the transcript coding sequence and subsequent NH2-terminal truncation of the peptide. ALDH3B1_v4 lacks exons 1 and 2, plus a 54-base segment from the 5’ end of exon 3 (relative to ALDH3B1_v1) resulting in an NH2-terminal truncation of the ALDH peptide domain for this protein. ALDH3B1_v5 utilizes a distinct exon 1 and lacks the ALDH3B1_v1 exon 6. There is evidence suggesting a sixth variant, ALDH3B1_v6; the first exon of ALDH3B1_v6 is a 2516-bp fusion of intron 2 and exon 3 of the ALDH3B1_v1 variant and results in an NH2-terminal truncated protein.
ALDH3B2
Three transcriptional variants have been identified in the sequence databases. ALDH3B2_v1 and ALDH3B2_v2 differ by an alternative exon 1. ALDH3B2_v3 lacks the 100-bp exon 9 present in ALDH3B2_v1, resulting in a shorter protein truncated at the COOH-terminus portion of the ALDH peptide domain.
ALDH4 Family
ALDH4A1_v1 is a 15-exon transcript encoding a 563-amino-acid variant. ALDH4A1_v1 and ALDH4A1_v2 have identical coding regions and subsequently yield identical proteins. The variation between these two transcripts occurs in the last exon (relative to ALDH4A1_v1), because it is transcribed as two separate exons in ALDH4A1_v2: a 154-bp exon 15 and a 359-bp exon 16—both separated by a 1013-bp intron 15, thus yielding a variably sized 3’-UTR. A third variant (described by EMBL-EBI’s Ensembl) lacks the ALDH4A1_v1 exon 4, resulting in a 5’ truncation of the protein’s ALDH peptide domain. ALDH4A1_v4 and ALDH4A1_v5 represent shorter transcripts, yielding peptides truncated at the COOH-terminus with partial ALDH domains and no apparent active site residues (according to Pfam analysis). Another variant, ALDH4A1_v6, has been identified in our laboratory and is being further characterized (W. Black, D. Stagnos, and V. Vasiliou; manuscript in preparation); this transcript lacks exon 12 (relative to ALDH4A1_v1), yet is translated as a splice variant that is missing an internal 51-amino-acid segment.
ALDH5 Family
ALDH5A1_v1 is a 10-exon transcript encoding a 535-amino-acid peptide. ALDH5A1_v2 variant has an additional 39-bp exon transcribed from within intron 4. This exon accounts for 13 additional amino acids within the ALDH peptide domain region of ALDH5A1_v2 (relative to the ALDH5A1_v1 protein). Evidence exists for a third and shorter variant, ALDH5A1_v3, which lacks both 5’ and 3’ exon segments (relative to ALDH5A1_v1). This translates into an NH2- and COOH-terminal truncated protein that retains a partial ALDH peptide domain, although with no apparent active-site residues.
ALDH6 Family
ALDH6A1_v1 is a 12-exon transcript encoding a 535-amino-acid protein. ALDH6A1_v2 lacks exons 1 through 6 and begins 6-bp upstream from exon 7 (relative to ALDH6A1_v1). The last exon of ALDH6A1_v1 is transcribed as two separate exons in ALDH6A1_v2: a 442-bp exon 6 and a 404-bp exon 7, both separated by a 2237-bp intron. The coding sequence for this transcript ends within exon 6 at the same stop codon as the primary variant, thereby rendering exon 7 irrelevant to the protein’s amino-acid sequence. ALDH6A1_v3 and ALDH6A1_v4 are truncated transcripts at their 3’ ends and comprise exons 1 to 5 and exons 1 to 4 of ALDH6A1_v1, respectively. Both of these variants encode truncated proteins at their COOH-termini; however, they retain a 5’ portion of the ALDH peptide domain.
ALDH7 Family
To date, no human transcriptional variants have been identified for this gene.
ALDH8 Family
Human ALDH8A1 has two transcriptional variants so far identified (Table 2). ALDH8A1_v1 represents the longer transcript encoding a 487-residue protein. ALDH8A1_v2 lacks an in-frame segment within the coding region (exon 6 of ALDH8A1_v1); this translates into a 433-amino-acid splice variant, which has no apparent active-site residues within the ALDH peptide domain.
ALDH9 Family
To date, no human transcriptional variants have been identified for this gene.
ALDH16 Family
Perhaps two transcriptional variants exist for human ALDH16A1 (Table 2). ALDH16A1_v1 is a 17-exon transcript encoding an 802-amino-acid protein. A second variant may be present, although evidence is limited. ALDH16A1_v2 comprises 15 exons. Its exon 6 is a fusion of exon 6, intron 6 and exon 7; its exon 15 is a fusion of exon 16, intron 16 and exon 17 (relative to ALDH16A1_v1). This fusion alters the reading frame of the coding sequence and introduces an early termination codon with subsequent truncation in translation of the peptide.
ALDH18 Family
Alternative splicing of human ALDH18A1 and mouse Aldh18a1 generates two proteins that differ by a 2-amino-acid insertion at the NH2-terminus of the γ-glutamyl kinase active-site [30]. Exon 6 is 159- and 153-bp in length for ALDH18A1_v1 and ALDH18A1_v2, respectively, yielding the two additional amino acid residues. The shorter variant, ALDH18A1_v2, has high activity in the gut and catalyzes an essential step in arginine biosynthesis. It is inhibited by ornithine, a mechanism by which arginine synthesis can be regulated. The widely expressed longer enzyme ALDH18A1_v1 is necessary for synthesis of proline from glutamate and is insensitive to ornithine inhibition. Impaired function of both the long and short forms, by way of mutations in the human ALDH18A1 gene, may be associated with neurodegeneration, cataracts, and connective tissue diseases [17]. Further studies of these and other ALDH alternative transcripts and protein products will be needed to elucidate their physiological function and significance.
Concluding Remarks
The mammalian ALDH genes identified to date appear to be comprehensive for human, mouse and rat because these genomes are virtually complete. As a result, additional ALDH genes are unlikely to be found in these species, although orthologs and paralogs will continue to be identified in other species as the completion of additional genomes occurs. The human ALDH gene superfamily comprises 19 genes in eleven families and four subfamilies. When compared with the human genome, rat and mouse include an additional gene in the ALDH1A subfamily, namely Aldh1a7. In addition, whereas the human and mouse genomes contain the human ALDH4A1 and mouse Aldh4a1 gene, a rat ortholog has yet to be identified or documented. However, strong evidence for the presence of rat Aldh4a1 exists, located at rat chromosome 5q36. Whereas many mammalian ALDH genes have been identified, several of the protein products encoded by these genes are not yet fully characterized.
Genomic alignment of existing transcript sequences from the molecular sequence databases reveals a number of potential alternatively-spliced transcriptional variants of human, mouse and rat ALDH genes. Yet, little empirical evidence has been reported for these variants in the literature. Further studies will be needed to assess the cell-specific existence of these variants and, ultimately, the functional relevance of such spliced gene products.
Figure 3.
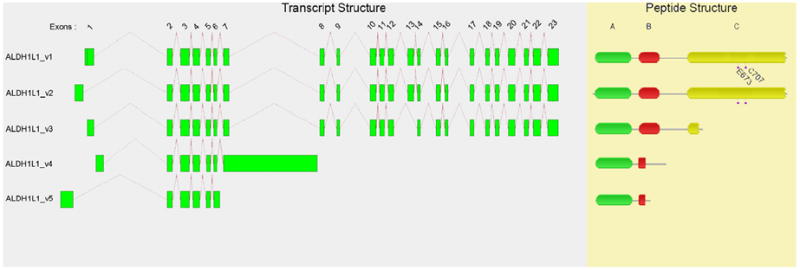
Human ALDH1L1 exon and protein structures for alternatively-spliced transcriptional variants. Most, or all, of the ALDH peptide domain in variants _v3, _v4 and _v5 are ablated and thus ALDH activity is presumed to be nil.
Acknowledgments
We thank our colleagues, especially Dr. David Thompson, for valuable discussions and critical reading of this manuscript. This work was supported by NIH/NEI grants EY11490 and EY17963 (V.V.) and P30 ES06096 (D.W.N.). S.A.M. was supported by an NIH/NIAAA Pre-doctoral Fellowship AA016875.
Reference List
- 1.Lindahl R. Aldehyde dehydrogenases and their role in carcinogenesis. Crit Rev Biochem Mol Biol. 1992;27:283–335. doi: 10.3109/10409239209082565. [DOI] [PubMed] [Google Scholar]
- 2.Sladek NE. Human aldehyde dehydrogenases: potential pathological, pharmacological, and toxicological impact. J Biochem Mol Toxicol. 2003;17:7–23. doi: 10.1002/jbt.10057. [DOI] [PubMed] [Google Scholar]
- 3.Vasiliou V, Bairoch A, Tipton KF, Nebert DW. Eukaryotic aldehyde dehydrogenase (ALDH) genes: human polymorphisms, and recommended nomenclature based on divergent evolution and chromosomal mapping. Pharmacogenetics. 1999;9:421–434. [PubMed] [Google Scholar]
- 4.Vasiliou V, Pappa A, Petersen DR. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem Biol Interact. 2000;129:1–19. doi: 10.1016/s0009-2797(00)00211-8. [DOI] [PubMed] [Google Scholar]
- 5.Perozich J, Nicholas H, Wang BC, Lindahl R, Hempel J. Relationships within the aldehyde dehydrogenase extended family. Protein Sci. 1999;8:137–146. doi: 10.1110/ps.8.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Z, Stamler JS. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc Med. 2006;16:259–265. doi: 10.1016/j.tcm.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Vasiliou V, Pappa A, Estey T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug Metab Rev. 2004;36:279–299. doi: 10.1081/dmr-120034001. [DOI] [PubMed] [Google Scholar]
- 9.Lassen N, Black WJ, Estey T, Vasiliou V. The role of corneal crystallins in the cellular defense mechanisms against oxidative stress. Semin Cell Dev Biol. 2008;19:100–112. doi: 10.1016/j.semcdb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Lassen N, Pappa A, Black WJ, Jester JV, Day BJ, Min E, et al. Antioxidant function of corneal ALDH3A1 in cultured stromal fibroblasts. Free Radic Biol Med. 2006;41:1459–1469. doi: 10.1016/j.freeradbiomed.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Uma L, Hariharan J, Sharma Y, Balasubramanian D. Corneal aldehyde dehydrogenase displays antioxidant properties. Exp Eye Res. 1996;63:117–120. doi: 10.1006/exer.1996.0098. [DOI] [PubMed] [Google Scholar]
- 12.Vasiliou V, Pappa A. Polymorphisms of human aldehyde dehydrogenases. Consequences for drug metabolism and disease. Pharmacology. 2000;61:192–198. doi: 10.1159/000028400. [DOI] [PubMed] [Google Scholar]
- 13.Rizzo WB, Carney G. Sjogren-Larsson syndrome: diversity of mutations and polymorphisms in the fatty aldehyde dehydrogenase gene (ALDH3A2) Hum Mutat. 2005;26:1–10. doi: 10.1002/humu.20181. [DOI] [PubMed] [Google Scholar]
- 14.Onenli-Mungan N, Yuksel B, Elkay M, Topaloglu AK, Baykal T, Ozer G. Type II hyperprolinemia: a case report. Turk J Pediatr. 2004;46:167–169. [PubMed] [Google Scholar]
- 15.Akaboshi S, Hogema BM, Novelletto A, Malaspina P, Salomons GS, Maropoulos GD, et al. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum Mutat. 2003;22:442–450. doi: 10.1002/humu.10288. [DOI] [PubMed] [Google Scholar]
- 16.Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12:307–309. doi: 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- 17.Baumgartner MR, Hu CA, Almashanu S, Steel G, Obie C, Aral B, et al. Hyperammonemia with reduced ornithine, citrulline, arginine and proline: a new inborn error caused by a mutation in the gene encoding delta(1)-pyrroline-5-carboxylate synthase. Hum Mol Genet. 2000;9:2853–2858. doi: 10.1093/hmg/9.19.2853. [DOI] [PubMed] [Google Scholar]
- 18.Enomoto N, Takase S, Takada N, Takada A. Alcoholic liver disease in heterozygotes of mutant and normal aldehyde dehydrogenase-2 genes. Hepatology. 1991;13:1071–1075. [PubMed] [Google Scholar]
- 19.Yokoyama A, Muramatsu T, Omori T, Yokoyama T, Matsushita S, Higuchi S, et al. Alcohol and aldehyde dehydrogenase gene polymorphisms and oropharyngolaryngeal, esophageal and stomach cancers in Japanese alcoholics. Carcinogenesis. 2001;22:433–439. doi: 10.1093/carcin/22.3.433. [DOI] [PubMed] [Google Scholar]
- 20.Kamino K, Nagasaka K, Imagawa M, Yamamoto H, Yoneda H, Ueki A, et al. Deficiency in mitochondrial aldehyde dehydrogenase increases the risk for late-onset Alzheimer’s disease in the Japanese population. Biochem Biophys Res Commun. 2000;273:192–196. doi: 10.1006/bbrc.2000.2923. [DOI] [PubMed] [Google Scholar]
- 21.Niederreither K, Subbarayan V, Dolle P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet. 1999;21:444–448. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- 22.Dupe V, Matt N, Garnier JM, Chambon P, Mark M, Ghyselinck NB. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc Natl Acad Sci U S A. 2003;100:14036–14041. doi: 10.1073/pnas.2336223100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lassen N, Bateman JB, Estey T, Kuszak JR, Nees DW, Piatigorsky J, et al. Multiple and Additive Functions of ALDH3A1 and ALDH1A1: Cataract phenotype and ocular oxidative damage in Aldh3a1(-/-)/Aldh1a1(-/-) knockout mice. J Biol Chem. 2007;282:25668–25676. doi: 10.1074/jbc.M702076200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- 25.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 27.Kapustin Y, Souvorov A, Tatusova T, Lipman D. Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct. 2008;3:20. doi: 10.1186/1745-6150-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogers GR, Markova NG, De L, V, Rizzo WB, Compton JG. Genomic organization and expression of the human fatty aldehyde dehydrogenase gene (FALDH) Genomics. 1997;39:127–135. doi: 10.1006/geno.1996.4501. [DOI] [PubMed] [Google Scholar]
- 30.Hu CA, Lin WW, Obie C, Valle D. Molecular enzymology of mammalian Delta1-pyrroline-5-carboxylate synthase. Alternative splice donor utilization generates isoforms with different sensitivity to ornithine inhibition. J Biol Chem. 1999;274:6754–6762. doi: 10.1074/jbc.274.10.6754. [DOI] [PubMed] [Google Scholar]