

Abstract
Perinatal asphyxia (PA) affects the synaptic function and morphological organization. In previous works, we have shown neuronal and synaptic changes in rat neostriatum subjected to hypoxia leading to long-term ubi-protein accumulation. Since F-actin is highly concentrated in dendritic spines, modifications in its organization could be related with alterations induced by hypoxia in the central nervous system (CNS). In the present study, we investigate the effects of PA on the actin cytoskeleton of hippocampal postsynaptic densities (PSD) in 4-month-old rats. PSD showed an increment in their thickness and in the level of ubiquitination. Correlative fluorescence-electron microscopy photooxidation showed a decrease in the number of F-actin-stained spines in hippocampal excitatory synapses subjected to PA. Although Western Blot analysis also showed a slight decrease in β-actin in PSD in PA animals, the difference was not significant. Taken together, this data suggests that long-term actin cytoskeleton might have role in PSD alterations which would be a spread phenomenon induced by PA.
1. Introduction
Dendritic spines are small protrusions that serve as a postsynaptic site for the 90% of the excitatory synapses in the CNS. Different kinds of dendritic spines were described based on their shape and their actin content in adult rat brains. Mushroom dendritic spines have stalks with a clear head differentiation, stubby spines are thick and have no neck, and thin spines are characterized as long and without neck [1]. Mushroom dendritic spines have a rich actin cytoskeleton network [1, 2], which is highly regulated by proteins that either stabilize the actin monomer (G-actin), such as thymosin or profiling, arp2/3, Rho-GTPase kinase contractin, or prevent polymerization and convert several polymers into small fragments of actin such as cofilin and gelsolin [3].
Several functions have been suggested for dendritic spines as they have been implicated in the mechanism of synaptic plasticity, learning and memory [4–7], and protein translocation [8]. Functional decline of dendritic spines is a consequence of synaptic loss in neurodegenerative disease and brain insults [9]. Furthermore, processes such as loss of dendritic spines, dendritic pruning, and loss of synaptic proteins precede neuronal death in many neurodegenerative disorders [10–12]. Moreover, activation and impaired function of the ubiquitin-proteasome pathway is thought to contribute to a number of neurodegenerative disorders [13]. Therefore, spine pathologies may be involved in different brain insults including hypoxia ischemia [14–17]. Perinatal asphyxia (PA) is a serious clinical complication with high mortality and morbidity [18]. Following PA, approximately 45% of newborn die and 25% have permanent neurological deficits including cerebral palsy, mental retardation and developmental delay, learning disabilities, visual and hearing problems, and different issues in school readiness [19–24].
In previous works, we have observed several alterations in striatum and hippocampal areas after PA, such as high level of ubiquitinization in dendritic spines, reactive gliosis, alterations in dendritic microtubular organization, and modification in cytoskeleton organization [17, 25, 26]. Given that numerous reports support the idea that dendritic spines are the main site damaged during brain ischemia [25, 27], we aimed to investigate whether dendritic spine changes are a spread feature induced by PA. For this purpose, we studied dendritic spine modifications in the Stratum radiatum of CA1 hippocampal area.
2. Material and Methods
2.1. Animals
All procedures involving animals were approved by the Institutional Animal Care and Use Committee at the University of Buenos Aires (School of Medicine) and conducted according to the principles of the Guide for the Care and Use of Laboratory Animals (NIH Publications no. 80–23, revised 1996). Sprague-Dawley female rats in the fifteenth day of pregnancy were placed in individual cages and maintained on a 12 : 12 h light/dark cycle in a controlled temperature (21 ± 2°C) and humidity (65 ± 5%) environment. The animals had access to food (Purina chow) and tap water ad libitum. One group of animals (n = 10) was used as surrogate mothers, another group (n = 10) was assigned to PA procedures.
2.2. Materials
Eosin-phalloidin and Phalloidin-Alexa568 were purchased from Invitrogen (Carlsbad, CA). Secondary antibodies against mouse were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Paraformaldehyde, EM grade glutaraldehyde, sodium cacodylate, and Durcopan ACM resin were obtained from Electron Microscopy Sciences (Fort Washington, PA); special welled tissue culture plates were obtained from MatTek (Ashland, MA). β-actin antibody was purchased from Sigma-Aldrich (cat no. A5441).
2.3. Induction of Asphyxia
Ten full-term pregnant rats on gestational day 22 were anesthetized [28], rapidly decapitated, and the uterus horns were isolated through an abdominal incision and placed in a water bath at 37°C for 19 min (subsevere PA: n = 10 full-term pregnant rats) [25, 26, 29–31]. We have used 19 min as the maximum time of PA because more than 20 minutes result in a survival rate lower than 3% [25]. Following asphyxia, the uterus horns were rapidly opened, the pups were removed, the amniotic fluid was cleaned, and the pups were stimulated to breathe by performing tactile stimulation with pieces of medical wipes for a few minutes until regular breathing was established. The umbilical cords were ligated, and the animals were left to recover for 1 hour under a heating lamp. When their physiological conditions improved, they were given to surrogate mothers who had delivered normally within the past 24 hours. The different groups of pups were marked and mixed with the surrogate mothers' normal litters (control animals (CTL) that were left undisturbed). We maintained litters of 10 pups with each surrogate mother.
2.4. Post-Asphyctic Procedure
Four-month old male rats (6 per group) were used. Briefly, an intracardiac perfusion was performed with normal rat Ringer's at 35°C followed by fixative under deep anaesthesia (containing 50 mg/kg ketamine, 1 mg/kg rhompun and 5 mg/kg acetopromazine in sterile saline). For light microscopy analysis, rats were perfused with 4% formaldehyde (freshly made from paraformaldehyde) in 0.1 M phosphate buffer, pH 7.2. The brain was removed and fixed for 2 additional hours in the same solution at 4°C. Then, sections were embedded in Durcupan ACM resin. After removing the brain from the skull, it was postfixed in the same fixative during 2 h. Coronal or sagital sections (50–80 μm) were made with a Vibratome (Leica). Some of these sections were stained with cresyl violet according to the procedures described in Capani et al. [32].
2.5. Photooxidation
Vibratome sections were washed with 50 mM glycine-PBS containing 0.5% cold water fish gelatin to block nonspecific binding. Following 30 min of washing, the sections were incubated on a shaker, in a solution of 0.05% of eosin phalloidin-0.5% cold-water fish gelatin/50 mM glycine-PBS for 2 h at 4°C. For light microscopic studies, phalloidin conjugated to Alexa488 was also used because of its superior fluorescent quantum yield compared to eosin. As a negative control, eosin-phalloidin was omitted. Tissue sections stained with eosin-phalloidin were mounted on glass-welled tissue culture dishes (Mat Tek Corp) pretreated with polyethylenimine. The slices were fixed again for 2–5 min with 2% glutaraldehyde in 0.1 M cacodylate buffer, rinsed in buffer for several minutes, and placed in 50 mM glycine and potassium cyanide in cacodylate buffer for an additional 5 min to reduce nonspecific staining. Photooxidation was performed on the Zeiss Axiovert described above, equipped with a 75-W xenon arc light source. The samples were immersed in a solution of 2.8 mM diaminobenzidine in 0.1 M sodium cacodylate at 4°C bubbled with pure O2, final pH 7.4, and then irradiated under conventional epifluorescence using a xenon lamp. After 6–8 min, a brownish reaction product began to appear in place of the fluorescence. The process was stopped by halting the excitation [1].
2.6. Electron Microscopy Procedure
Following photooxidation, tissue sections were rinsed in 0.1 M sodium cacodylate several times and incubated for 30 min with 1% osmium tetroxide in 0.1 M sodium cacodylate, pH 7.2. After several washes in double-distilled H2O, the sections were dehydrated in an ascending ethanol series, flat-embedded in Durcopan ACM resin, and polymerized for 24 h at 60°C. Serial thin sections (80–100 nm) were cut with Reichert Ultracut E ultramicrotome using glass knives and examined using a JEOL 100CX electron microscope at 80–100 keV. One set of thin sections was poststained with a combination of uranyl acetate and lead citrate. For E-PTA staining, sections were dehydrated in an ascending series of ethanol to 100% and stained for 1 h with 1% PTA stained prepared by dissolving 0.1 g of PTA in 10 mL of 100% ethanol and adding four drops of 95% ethanol [33]. Then, sections were embedded in Durcupan ACM resin.
2.7. Morphometric Analysis of Confocal Data
The volume fraction of immunoreactive material for phalloidin was estimated using the point-counting method of Weibel [34] and a grid delimiting 5000 μm2 in the hippocampus. A total area of 75,000 μm2 was evaluated in each animal. Percentage of reactive area was estimated using Image J Program (Image J 1.410, NIH, USA). For electron microscopy analysis sampling, procedures were adapted from Harris et al. [35] and Capani et al. [1]. For analysis, spines were sampled from Stratum radiatum CA1 hippocampal area. All of the synapses that have the characteristic of mushrooms dendritic spines (head larger than the neck) were used in this study since mushrooms dendritic spines are the unique F-actin positive spines [1]. Random fields of neuropil containing at least one synapse were photographed at 10000x magnification and analyzed at a total magnification of 30000x. We analyzed 643 control spines and 638 spines for tissue subjected to PA.
2.8. Quantitative Analyses of E-PTA Material
CA1 Hippocampal specimens were selected for quantitative analyses based on the quality of E-PTA staining and the degree of ultrastructural preservation, as determined from conventionally stained material from the same animals. Samples were analyzed from controls (n = 4) and 19 min PA animals (n = 8). Tissue sections were cut at thickness of 100 nm and examined and photographed at 80 keV at a magnification of 8300x with a Zeiss 109 electron microscope. For each animal, five micrographs were obtained from hippocampus. As described above, each negative was digitized into a PC computer. Using NIH Image 1.6, PSDs were first manually outlined, and then the maximal thickness, minimum thickness, length, and total area of each PSD were determined. All synapses in which the postsynaptic density, intracleft line, and presynaptic grid were clearly visible were chosen for analysis. The selection criterion resulted in the analysis of between 30 and 50 PSDs per animal for each hippocampus.
2.9. Quantitative Analysis of Dendritic Spines
For analysis, spines were sampled from hippocampus. All of the synapses that have the characteristic of mushrooms type dendritic spines (head larger than the neck) were used in this study since mushrooms spines are the unique F-actin positive spines [1]. Random fields of neuropil containing at least one synapse were photographed at 10000x magnification and analyzed at a total magnification of 30000x. We analyzed 643 control spines and 638 spines for tissue subjected to hypoxia.
2.10. Subcellular Fractionation and Preparation of PSDs
Biochemical fractionation was performed as described previously by Saraceno et al. using the whole dorsal hippocampus [17] (CTL, n = 6; PA n = 6). Dounce homogenates (H) of the pellets in ice cold TEVP buffer (10 mM Tris-HCl, pH 7.4, 5 mM NaF, 1 mM Na3VO4, 1 mM EDTA, and 1 mM EGTA, 1.25 μg/mL pepstatin A, 10 μg/mL leupeptin, 2.5 μg/mL aproptionin, 0.5 mM PMSF) containing 320 mM sucrose were centrifuged at 1000 × g to remove nuclei and large debris (P1). The supernatant (S1) was centrifuged at 10.000 × g for 10 min to obtain a crude synaptosomal fraction (P2) and subsequently was lysed hypoosmotically and centrifuged at 45.000 × g for 90 min to obtain a pellet of the synaptosomal membrane fraction (LP1). After each centrifugation, the resulting pellet was rinsed briefly with ice cold TEVP buffer before subsequent fractioning to avoid possible crossover contamination. Protein concentration was estimated by Bradford technique.
2.11. Western Blot
Western Blot analysis was carried out using LP1 fractions separated on 10% SDS-PAGE. Samples containing 50 μg of protein from each group were applied to each lane. After electrophoresis, proteins were transferred to polyvinylidene difluoride (PVDF) membrane as described previously [36–38]. The membranes were incubated with a primary antibody anti-β-actin (Sigma, 1 : 1000) overnight at 4°C. Then, after appropriate washing procedures, they were incubated with horseradish peroxidase-conjugated anti-mouse secondary antibody for 2 hours at room temperature. The blots were developed with an ECL detection kit (Amersham). The films were scanned, and the optical density of protein bands was quantified using Gel Pro Analyzer software 3.1.00.00 (Media Cybernetics, USA). We used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as load controls [17, 39–41].
2.12. Statistical Analysis
The results were expressed as the means ± SEM. Student's t-test were carried out. A probability was considered to be significant at 5% or less. Statistical analyses were performed using the GraphPad Prism 5.03 for windows statistical package (GraphPad software, Inc, San Diego, CA, USA).
3. Results and Discussion
3.1. Microscopic Analysis of Hippocampal Sections
The study of nuclear morphology by Cresyl violet staining showed that PA animals present clear nuclear condensation 4 months after injury respect to CTL animals in the Stratum radiatum CA1 hippocampal area sections (Figure 1). Statistical analyses showed alterations in pyramidal neurons of hippocampal CA1 area, showing an abundance of pyknotic nuclei in asphyctic animals as compared to control animals (Table 1). Then, we employed neuronal nuclei (NeuN) immunolabeling to determine the nature of the cells presenting condensed nuclei (Figure 1). Statistical analyses showed no significant difference in the number of NeuN+ nuclei in the CA1 hippocampal area of asphyctic animals respect to controls. When we analyzed the cellular distribution of NeuN labeling, it was determinate that asphyctic animals showed a significant increase in the number of abnormal NeuN+ nuclei and a significant decrease in the number of normal NeuN+ nuclei in the CA1 hippocampal area compared with CTL group (Table 1). To determine the morphology of these cells, we performed a conventional electron microscopy study. Morphological analyses showed that most cells presenting condensed nuclei evidence dark cytoplasm with rare vacuoles and compaction, a hypertrophic nucleolus, a nucleus with a festoon shape, and a twisted nuclear envelope, corresponding to neurons in degeneration [25, 32, 42, 43] (Figure 1).
Figure 1.
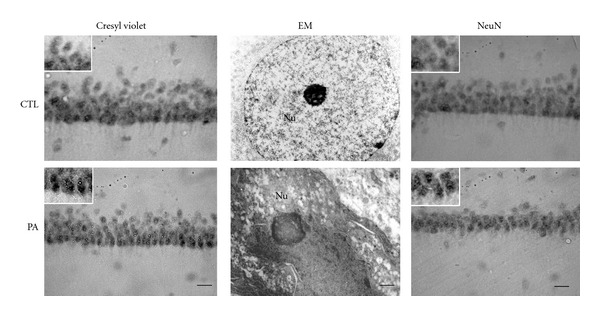
Micrographs of Stratum radiatum of CA1 hippocampal area from 4-month-old control rats and rats subjected to 19 min of PA. Vibratome sections of 50 μm were cut and stained with cresyl violet (Left), analysed by electron microscopy (EM) (Middle) and NeuN immunostaining (Right). A clear nuclear condensation was observed after 19 min of PA. Electron micrograph showed that most of the condensed cells correspond to neurons in degeneration. Abnormal NeuN+ nuclei were increased in asphyctic animals respect to control group. Scale bar: 30 μm and 0.5 μm for EM. Nu: nucleus.
Table 1.
Analysis of neuron cells in CA1 hippocampal area.
| Groups | Neurons with pyknotic nuclei | Neurons NeuN+ | Normal neurons | Abnormal neurons |
|---|---|---|---|---|
| CTL | 2.94 ± 0.2 | 75.34 ± 2.4 | 72.20 ± 0.5 | 3.04 ± 0.5 |
| PA | 6.29 ± 1.3* | 71.58 ± 5.5 | 64.12 ± 0.4* | 7.46 ± 1.2* |
Data are expressed as means ± SD. Significative differences were obtain using Student t-test. *P < 0.05.
3.2. Modification in Hippocampal PSD Stained with E-PTA
Osmium-lead-citrated staining showed no obvious alterations in the Stratum radiatum CA1 hippocampal area sections from 4-month-old CTL and PA rats (Figure 2). Presynaptic terminals, presynaptic vesicles, and ultrastructural organization of PSD were intact (Figure 2). On the other hand, E-PTA immunostaining showed clear alterations in synapses of rats subjected to PA (Figure 3). Following PA, the thickness of PSD increased as compared to controls (Figure 3). There was also a general increment in the amount of E-PTA-stained material in PSD of PA animals compared to controls. The statistical analysis performed confirmed these changes. t Student analyses for the area and length of the PSD and for the minimum and maximum thickness of the PSD were significant (P < 0.05). Post hoc tests revealed that the means of PSD area was significantly bigger as compared to the CTL group (P < 0.01) (Table 2). These inconsistencies between the osmium and E-PTA staining may be attributable to the fact that general heavy metal staining obscures the synaptic modifications occurring in post asphyctic tissue. In addition, it is possible that E-PTA stains different components in the PSD than osmium-uranium-lead methods. It has been known that PSDs stained with E-PTA are shorter and probably wider than those stained with the osmium-heavy metal method [44]. E-PTA preferentially stains protein(s) rich in basic amino acid residues, including lysine, arginine, and histidine, such as collagen or histones [45]. In contrast, conventional heavy metal staining stains a wide type of lipids and cytoskeletal and cytoplasmic elements [44]. Since both markers stain different components, this might explain why E-PTA staining is more effective to detect the PSDs alterations than heavy metal-stained sections.
Figure 2.
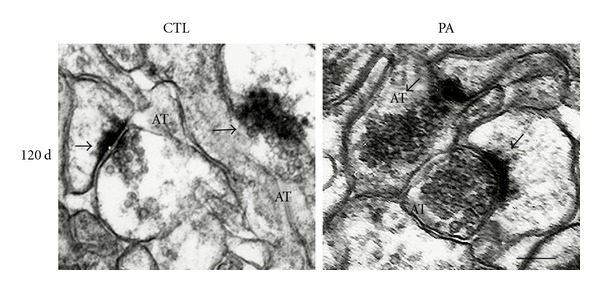
Electron micrographs of osmium-uranium-lead-stained synapses in Stratum radiatum of CA1 hippocampal area from 4-month-old control rats and rats subjected to PA. The synapses (arrows) were intact, and no obvious alterations were seen in these osmium-uranium-lead-stained synapses after PA. AT: axon terminal. Scale bar: 0.5 μm.
Figure 3.

Electron micrographs of E-PTA-stained PSDs (arrows) in Stratum radiatum of CA1 hippocampal tissue section from 4-month-old control rats and rats subjected to 19 min of PA. Note the increased thickness and dispersed appearance of the PSDs in the asphyctic brains, compared with the control. Scale bar: 0.5 μm. PRE: presynapses; PSD: postsynaptic density.
Table 2.
Analysis of PSDs features in CA1 hippocampal area.
| Groups | Área × 103 (nm2) | Length (nm) | Minimum thickness (nm) | Maximum thickness (nm) |
|---|---|---|---|---|
| CTL | 2.0 ± 0.2 | 97.1 ± 2.4 | 16.2 ± 0.5 | 43.0 ± 2.5 |
| PA | 5.7 ± 1.3** | 105.1 ± 5.5 | 35.6 ± 0.4** | 80.1 ± 2.2** |
Data are expressed as means ± SD. Significative differences were obtained using Student t-test. **P < 0.01.
Consistent with other studies in different models of ischemia [27, 33, 43] and using this long-term PA model [1], we did not observe any alterations in the subcellular organization of hippocampus material stained with osmium-heavy metals. However, we observed a marked increase in E-PTA-stained material in subsevere PA. Although not too much data is available about the mechanism of cell death during PA [25, 30], these findings suggest that the increased in the thickness could be related with the degradation of abnormal proteins probably before neurons trigger death mechanisms. Thus, we hypothesize that some early signals triggered in PSDs could induce late neuronal alterations in post asphyctic hippocampal tissue.
3.3. Ubiquitin-Protein Conjugates in Hippocampal PSDs
Since we and others demonstrated that E-PTA-stained aggregates could be composed of abnormal protein [25, 33, 46], we performed immunoelectron microscopy following the procedures previously described by Capani et al. [25] in order to detect ubiquitin (Figure 4). We observed ubiquitinated synaptic proteins after 19 min of PA in the Stratum radiatum CA1 hippocampal area sections, while negative controls, in which the primary antibody was omitted, did not show immunolabeling (data not shown). We rarely observed ubiquitin labeling in PSD of CTL animals. Taking these results into account, we could suggest that aggregates of ubi-proteins are present in PSDs of asphyctic animals, as it was observed in some neurodegenerative diseases [47].
Figure 4.

Electron micrographs of ubiquitin immunolabeling from Stratum radiatum of CA1 hippocampal tissue of 4-month-old control rats and rats subjected 19 min of PA. Strong ubiquitin stained was observed in asphyctic PSDs (arrowheads). In control animals ubiquitin staining was very weak and rare. AT: axon terminal. Scale bar: 0.5 μm.
Even though data about cell death mechanisms during PA are scarce [25, 30], these findings suggest that PSD thickening could be related to degradation of abnormal proteins, probably before cell death mechanisms are triggered in neurons. Consistent with this view, persistent ubiquitination was found in the PSD of hippocampal neurons [46, 48] after transient cerebral ischemia [43], which suggests that increased ubi-protein conjugates might produce protein damage. In addition, damage in protein can be produced by the increment in ROS production and calpain activation as consequence of a rise in Ca levels after hypoxic-ischemic insult [1, 49, 50]. On the other hand, while other heat shock proteins reversibly attach to denatured proteins and help to refold or reassemble them, ubiquitin-conjugated proteins are degraded by 26 S proteasome [47]. PA insult activates the ubiquitin pathway, which might affect neuronal survival by damaged protein accumulation. Since neurons do not have the capacity to remove it, ubi-protein accumulation leads neurons to death.
3.4. F-Actin Changes in Hippocampal Dendritic Spines Induced by PA
Brain hypoxia-ischemia triggers an early increment of glutamate in the extracellular space at synaptic level [51]. High levels of glutamate produce a cascade of events in dendritic spines that lead to cell death [25, 31, 33, 43, 48, 52–55]. Since structure and function of dendritic spines are dynamically regulated by different cellular pathways acting on the actin cytoskeleton, we used light and electron microscopic techniques that had previously been used in our laboratory [1, 56] and others [4, 8] to study F-actin modifications induced by PA. By confocal microscope analyses, we observed dendritic spines represented by punctate staining using Phalloidin-Alexa568. PA animals showed a decrease in punctate staining respect to CTL group (Figure 5 Top) (P < 0.01). The morphometric analysis confirmed these data. Since F-actin is mostly concentrated in mushroom dendritic spines [1], this decrement is tightly related with the F-actin contained in the dendritic spines.
Figure 5.

Top: confocal microscope images of Phalloidin-Alexa568 from Stratum radiatum of CA1 hippocampal tissue from 4-month-old control and asphyctic rats. A decrease in the punctate staining was observed after 19 minutes of PA (arrows). The assessment of the percentage of the reactive area from Stratum radiatum of CA1 hippocampal Phalloidin-Alexa568 staining in PA rats showed a decrement in the reactivity area staining with phalloidin. *P < 0.01. Bars and error bars represent mean + SEM. DEN: dendrites; CB: cell body. Scale bar: 10 μm. Bottom: electron micrograph of photooxidated area in the Stratum radiatum of CA1 hippocampal tissue of 4-month-old rats. Arrows point out the dendritic spines stained. A decrement in the number of the F-actin-positive spines was observed after 19 min of PA. AT: axon terminal; DEN: dendritic shaft. Scale bar: 1 μm. The graph shows the assessment of the number of spines per field from Stratum radiatum of CA1 hippocampal slices. A significant decrement in the number of positive F-actin spines was observed in the PA group in comparison with the CTL group. *P < 0.01. Bars and error bars represent mean + SEM.
Electron microscopic analyses of spine population in the photooxidated samples confirmed confocal microscopic observations. When we analyzed different dendritic spine populations, we observed that only the number of mushroom dendritic spines, the only F-actin-positive spines in control animals, showed a significant decrease after 19 min of PA (P < 0.05) (Figure 5 Bottom). In contrast, synapses did not show any sign of evident degeneration in asphyxic rats. Isolated synaptosome (LP1) fractions were analyzed by immunoblotting using anti-β-actin antibody and quantified (Figures 5 and 6). Statistical analysis showed no significant differences in mean optical densities of F-actin bands (P = n.s.) from PA and control group. However, PA animals showed a decrease in amount of β-actin with respect to CTL animals (P = 0,058). Both in vivo and in vitro studies showed high concentration of β-actin in dendritic spines are involved in the organization of the synapses in adult brains [57–60]. Although we observed a reduction of the number of spines F-actin positive in PA animals, the maintenance of β-actin concentration observed in synaptosomal fraction may represent the cytoskeletal support of a stable dendritic spines structure that maintains, thus, the potential morphological plasticity in circumstances where adaptive changes in synaptic connectivity are adequate [60]. Consistent with this point of view, disruption of receptor-scaffold proteins as NMDAR-PSD 95, which depends on actin polymerization interactions, can prevent cell death after ischemia [61].
Figure 6.
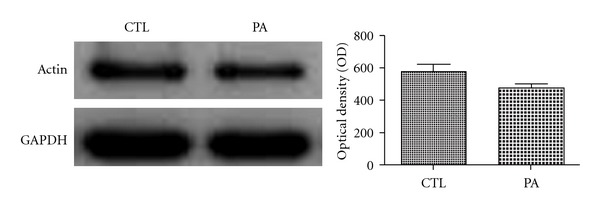
Immunoblots of hippocampal sinaptosomal fractions of 4-month-old CTL and PA rats. We used the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as loading controls. The assessment of the percentage of optical density of immunoblots from the 4-month-old CTL and PA rats showed no significant difference in the optical density with respect to control group (CTL). P = 0,058. Bars and error bars represent mean + SEM.
Actin cytoskeleton is highly regulated by several actin binding proteins (ABP) [3]. Many ABPs have been involved in the regulation of neuronal death during ischemia. Changes in spine morphology are strongly linked with some ABP such as gelsolin. Several studies using different models of neuronal cell death have demonstrated that endogenous gelsolin's antiapoptotic properties correlated to its dynamic actions in the cytoskeleton. Gelsolin-null neurons have higher rates of cell death and a rapid and sustained elevation of Ca2+ levels following glucose/oxygen deprivation, as well as augmented cytosolic Ca2+ levels in nerve terminals following in vitro depolarization [52]. Gelsolin also diminishes infarct size after ischemia, preventing neuronal death [62]. Furthermore, the increment in histone acetylation induces upregulation of gelsolin, dramatically reducing the levels of actin filaments and cell death following cerebral ischemia in mice [63]. Although a recent study by Gisselsson et al. [15] has shown that actin depolymerization prevents neuronal death, we hypothesize that the decrease in β-actin in synaptosomal fraction could also be related with cell death observed after PA insult, as this process is connected with an abnormal ubi-protein increment.
In addition, our group has previously observed learning; reference and working spatial memory impairments in the Morris water maze in 3-months-old rat, subjected to acute asphyxia immediately after birth, using the hypoxic-ischemia model described in the present manuscript [31]. It is well known that the performance in these spatial tests is disrupted after hippocampal damage [64]. Moreover, deficits were observed in the exploration of a novel environment. Therefore, synaptic modifications observed in asphyctic animals could be related with behavioral deficits previously described by our group [31].
4. Conclusions
These findings suggest that excessive protein ubiquitination in hippocampal PSD, 4 months after a subsevere PA insult, seems to be related to the increment in protein accumulation in this area. In spite of this increment, we observed a decrease in β-actin which suggests that PA is damaging the actin cytoskeleton. Moreover, the amount of β-actin in PA animals is correlated with the decrement in the number of mushroom-shaped dendritic spines. Although further studies will be necessary to determine the role of ubi-protein accumulation in PSD, we could speculate that PSD alterations might be involved in the generation of an aberrant biochemical pathway leading to long-term modifications in the brain of PA animals, as we described in a previous paper [25]. In agreement with this point of view, Alzheimer's disease has a deleterious action on the actin cytoskeleton linked with PSD, leading to dendritic spine dysfunction and synaptic degeneration [65].
Acknowledgments
This research was supported by UBATYC 20020090100118 CONICET 11420100100159. The authors also thank Ing. Elisa María Bocanegra, Ing. Roberto Francisco Domizio from IHEM–CONICET, National University of Cuyo. G. E. Saraceno is fellowship holder from CONICET (Argentina).
References
- 1.Capani F, Martone ME, Deerinck TJ, Ellisman MH. Selective localization of high concentrations of F-actin in subpopulations of dendritic spines in rat central nervous system: a three-dimensional electron microscopic study. Journal of Comparative Neurology. 2001;435(2):156–170. doi: 10.1002/cne.1199. [DOI] [PubMed] [Google Scholar]
- 2.Fifkova E, Delay RJ. Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. Journal of Cell Biology. 1982;95(1):345–350. doi: 10.1083/jcb.95.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollard TD, Borisy G. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112(4):453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 4.Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38(3):447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- 5.Hoogenraad CC, Akhmanova A. Dendritic spine plasticity: new regulatory roles of dynamic microtubules. Neuroscientist. 2010;16(6):650–661. doi: 10.1177/1073858410386357. [DOI] [PubMed] [Google Scholar]
- 6.Svitkina T, Lin WH, Webb DJ, et al. Regulation of the postsynaptic cytoskeleton: roles in development, plasticity, and disorders. Journal of Neuroscience. 2010;30(45):14937–14942. doi: 10.1523/JNEUROSCI.4276-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgado-Bernal I. Learning and memory consolidation: linking molecular and behavioral data. Neuroscience. 2011;176:12–19. doi: 10.1016/j.neuroscience.2010.12.056. [DOI] [PubMed] [Google Scholar]
- 8.Ouyang Y, Wong M, Capani F, et al. Transient decrease in F-actin may be necessary for translocation of proteins into dendritic spines. European Journal of Neuroscience. 2005;22(12):2995–3005. doi: 10.1111/j.1460-9568.2005.04521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waataja JJ, Kim HJ, Roloff AM, Thayer SA. Excitotoxic loss of post-synaptic sites is distinct temporally and mechanistically from neuronal death. Journal of Neurochemistry. 2008;104(2):364–375. doi: 10.1111/j.1471-4159.2007.04973.x. [DOI] [PubMed] [Google Scholar]
- 10.Masliah E, Terry R. The role of synaptic proteins in the pathogenesis of disorders of the central nervous system. Brain Pathology. 1993;3(1):77–85. doi: 10.1111/j.1750-3639.1993.tb00728.x. [DOI] [PubMed] [Google Scholar]
- 11.Swann JW, Al-Noori S, Jiang M, Lee CL. Spine loss and other dendritic abnormalities in epilepsy. Hippocampus. 2000;10(5):617–625. doi: 10.1002/1098-1063(2000)10:5<617::AID-HIPO13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 12.Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Research Reviews. 2002;39(1):29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- 13.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443(7113):780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 14.Gisselsson LL, Matus A, Wieloch T. Actin redistribution underlies the sparing effect of mild hypothermia on dendritic spine morphology after in vitro ischemia. Journal of Cerebral Blood Flow and Metabolism. 2005;25(10):1346–1355. doi: 10.1038/sj.jcbfm.9600131. [DOI] [PubMed] [Google Scholar]
- 15.Gisselsson L, Toresson H, Ruscher K, Wieloch T. Rho kinase inhibition protects CA1 cells in organotypic hippocampal slices during in vitro ischemia. Brain Research. 2010;1316(C):92–100. doi: 10.1016/j.brainres.2009.11.087. [DOI] [PubMed] [Google Scholar]
- 16.Luebke JI, Weaver CM, Rocher AB, et al. Dendritic vulnerability in neurodegenerative disease: insights from analyses of cortical pyramidal neurons in transgenic mouse models. Brain Structure and Function. 2010;214(2-3):181–199. doi: 10.1007/s00429-010-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saraceno GE, Ayala MV, Badorrey MS, et al. Effects of perinatal asphyxia on rat striatal cytoskeleton. Synapse. 2012;66(1):9–19. doi: 10.1002/syn.20978. [DOI] [PubMed] [Google Scholar]
- 18.van Bel F, Groenendaal F. Long-term pharmacologic neuroprotection after birth asphyxia: where do we stand? Neonatology. 2008;94(3):203–210. doi: 10.1159/000143723. [DOI] [PubMed] [Google Scholar]
- 19.Hill A, Volpe JJ. Seizures, hypoxic-ischemic brain injury, and intraventricular hemorrhage in the newborn. Annals of Neurology. 1981;10(2):109–121. doi: 10.1002/ana.410100202. [DOI] [PubMed] [Google Scholar]
- 20.Amiel-Tison C, Ellison P. Birth asphyxia in the fullterm newborn: early assessment and outcome. Developmental Medicine and Child Neurology. 1986;28(5):671–682. doi: 10.1111/j.1469-8749.1986.tb03914.x. [DOI] [PubMed] [Google Scholar]
- 21.Vannucci RC, Perlman JM. Interventions for perinatal hypoxic-ischemic encephalopathy. Pediatrics. 1997;100(6):1004–1014. doi: 10.1542/peds.100.6.1004. [DOI] [PubMed] [Google Scholar]
- 22.Gunn AJ. Cerebral hypothermia for prevention of brain injury following perinatal asphyxia. Current Opinion in Pediatrics. 2000;12(2):111–115. doi: 10.1097/00008480-200004000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Progress in Retinal and Eye Research. 2004;23(1):91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Shankaran S. Neonatal encephalopathy: treatment with hypothermia. Journal of Neurotrauma. 2009;26(3):437–443. doi: 10.1089/neu.2008.0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capani F, Saraceno GE, Botti V, et al. Protein ubiquitination in postsynaptic densities after hypoxia in rat neostriatum is blocked by hypothermia. Experimental Neurology. 2009;219(2):404–413. doi: 10.1016/j.expneurol.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Saraceno GE, Bertolino ML, Galeano P, Romero JI, Garcia-Segura LM, Capani F. Estradiol therapy in adulthood reverses glial and neuronal alterations caused by perinatal asphyxia. Experimental Neurology. 2010;223(2):615–622. doi: 10.1016/j.expneurol.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Hu BR, Park M, Martone ME, Fischer WH, Ellisman MH, Zivin JA. Assembly of proteins to postsynaptic densities after transient cerebral ischemia. Journal of Neuroscience. 1998;18(2):625–633. doi: 10.1523/JNEUROSCI.18-02-00625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorfman VB, Vega MC, Coirini H. Age-related changes of the GABA-B receptor in the lumbar spinal cord of male rats and penile erection. Life Sciences. 2006;78(14):1529–1534. doi: 10.1016/j.lfs.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 29.Bjelke B, Andersson K, Ogren SO, Bolme P. Asphyctic lesion: proliferation of tyrosine hydroxylase-immunoreactive nerve cell bodies in the rat substantia nigra and functional changes in dopamine neurotransmission. Brain Research. 1991;543(1):1–9. doi: 10.1016/0006-8993(91)91041-x. [DOI] [PubMed] [Google Scholar]
- 30.van de Berg WD, Kwaijtaal M, de Louw AJA, et al. Impact of perinatal asphyxia on the GABAergic and locomotor system. Neuroscience. 2003;117(1):83–96. doi: 10.1016/s0306-4522(02)00787-x. [DOI] [PubMed] [Google Scholar]
- 31.Galeano P, Blanco Calvo E, Madureira de Oliveira D, et al. Long-lasting effects of perinatal asphyxia on exploration, memory and incentive downshift. International Journal of Developmental Neuroscience. 2011;29(6):609–619. doi: 10.1016/j.ijdevneu.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 32.Capani F, Loidl F, Lopez-Costa JJ, Selvin-Testa A, Saavedra JP. Ultrastructural changes in nitric oxide synthase immunoreactivity in the brain of rats subjected to perinatal asphyxia: neuroprotective effects of cold treatment. Brain Research. 1997;775(1-2):11–23. doi: 10.1016/s0006-8993(97)00714-2. [DOI] [PubMed] [Google Scholar]
- 33.Martone ME, Jones YZ, Young SJ, Ellisman MH, Zivin JA, Hu BR. Modification of postsynaptic densities after transient cerebral ischemia: a quantitative and three-dimensional ultrastructural study. Journal of Neuroscience. 1999;19(6):1988–1997. doi: 10.1523/JNEUROSCI.19-06-01988.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weibel HP. The effect of noise trauma on speech discrimination in silence and under influence of party noise. Archives Of Oto-Rhino-Laryngology. 1978;219(2):413–414. doi: 10.1007/BF00463862. [DOI] [PubMed] [Google Scholar]
- 35.Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. Journal of Neuroscience. 1992;12(7):2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang P, Royer M, Houtz RL. Affinity purification of ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit N-epsilon-methyltransferase. Protein Expression and Purification. 1995;6(4):528–536. doi: 10.1006/prep.1995.1070. [DOI] [PubMed] [Google Scholar]
- 37.Dunah AW, Yasuda RP, Wang YH, et al. Regional and ontogenic expression of the NMDA receptor subunit NR2D protein in rat brain using a subunit-specific antibody. Journal of Neurochemistry. 1996;67(6):2335–2345. doi: 10.1046/j.1471-4159.1996.67062335.x. [DOI] [PubMed] [Google Scholar]
- 38.Luo L, Chen H, Zirkin BR. Are Leydig cell steroidogenic enzymes differentially regulated with aging? Journal of Andrology. 1996;17(5):509–515. [PubMed] [Google Scholar]
- 39.Murphy DD, Rueter SM, Trojanowski JQ, Lee VMY. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. Journal of Neuroscience. 2000;20(9):3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng V, Matagne V, Banine F, et al. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Human Molecular Genetics. 2007;16(6):640–650. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- 41.Wang CE, Tydlacka S, Orr AL, et al. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington’s disease. Human Molecular Genetics. 2008;17(17):2738–2751. doi: 10.1093/hmg/ddn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aggoun-Zouaoui D, Margalli I, Borrega F, et al. Ultrastructural morphology of neuronal death following reversible focal ischemia in the rat. Apoptosis. 1998;3(2):133–141. doi: 10.1023/a:1009653126347. [DOI] [PubMed] [Google Scholar]
- 43.Liu CL, Martone ME, Hu BR. Protein ubiquitination in postsynaptic densities after transient cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 2004;24(11):1219–1225. doi: 10.1097/01.WCB.0000136706.77918.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burry RW, Engel EL, Lasher RS, Wood JG. The numerical density and morphology of synaptic contacts stained either by Os-UL or by E-PTA. Brain Research. 1978;157(2):321–324. doi: 10.1016/0006-8993(78)90034-3. [DOI] [PubMed] [Google Scholar]
- 45.Bloom FE, Aghajanian GK. Fine structural and cytochemical analysis of the staining of synaptic junctions with phosphotungstic acid. Journal of Ultrasructure Research. 1968;22(5-6):361–375. doi: 10.1016/s0022-5320(68)90027-0. [DOI] [PubMed] [Google Scholar]
- 46.Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. Journal of Neuroscience. 2000;20(9):3191–3199. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korhonen L, Lindholm D. The ubiquitin proteasome system in synaptic and axonal degeneration: a new twist to an old cycle. Journal of Cell Biology. 2004;165(1):27–30. doi: 10.1083/jcb.200311091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu JJ, Zhao H, Sung JH, Sun GH, Steinberg GK. Hypothermia blocks ischemic changes in ubiquitin distribution and levels following stroke. NeuroReport. 2006;17(16):1691–1695. doi: 10.1097/01.wnr.0000236868.83970.79. [DOI] [PubMed] [Google Scholar]
- 49.Capani F, Loidl CF, Piehl LL, Facorro G, De Paoli T, Hager A. Long term production of reactive oxygen species during perinatal asphyxia in the rat central nervous system: effects of hypothermia. International Journal of Neuroscience. 2003;113(5):641–654. doi: 10.1080/00207450390200099. [DOI] [PubMed] [Google Scholar]
- 50.Dingman A, Lee SY, Derugin N, Wendland MF, Vexler ZS. Aminoguanidine inhibits caspase-3 and calpain activation without affecting microglial activation following neonatal transient cerebral ischemia. Journal of Neurochemistry. 2006;96(5):1467–1479. doi: 10.1111/j.1471-4159.2006.03672.x. [DOI] [PubMed] [Google Scholar]
- 51.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends in Neurosciences. 1995;18(2):58–60. [PubMed] [Google Scholar]
- 52.Endres M, Fink K, Zhu J, et al. Neuroprotective effects of gelsolin during murine stroke. Journal of Clinical Investigation. 1999;103(3):347–354. doi: 10.1172/JCI4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li D, Shao Z, Vanden Hoek TL, Brorson JR. Reperfusion accelerates acute neuronal death induced by simulated ischemia. Experimental Neurology. 2007;206(2):280–287. doi: 10.1016/j.expneurol.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. Journal of Neuroscience. 2009;29(42):13242–13254. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aon-Bertolino ML, Romero JI, Galeano P, et al. Thioredoxin and glutaredoxin system proteins-immunolocalization in the rat central nervous system. Biochimica et Biophysica Acta. 2011;1810(1):93–110. doi: 10.1016/j.bbagen.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 56.Capani F, Saraceno E, Boti VR, et al. A tridimensional view of the organization of actin filaments in the central nervous system by use of fluorescent photooxidation. Biocell. 2008;32(1):1–8. [PubMed] [Google Scholar]
- 57.Fagg GE, Matus A. Selective association of N-methyl aspartate and quisqualate types of L-glutamate receptor with brain postsynaptic densities. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(21 I):6876–6880. doi: 10.1073/pnas.81.21.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cohen RS, Chung SK, Pfaff DW. Immunocytochemical localization of actin in dendritic spines of the cerebral cortex using colloidal gold as a probe. Cellular and Molecular Neurobiology. 1985;5(3):271–284. doi: 10.1007/BF00711012. [DOI] [PubMed] [Google Scholar]
- 59.Wu K, Carlin R, Siekevitz P. Binding of L-[3H]glutamate to fresh or frozen synaptic membrane and postsynaptic density fractions isolated from cerebral cortex and cerebellum of fresh or frozen canine brain. Journal of Neurochemistry. 1986;46(3):831–841. doi: 10.1111/j.1471-4159.1986.tb13047.x. [DOI] [PubMed] [Google Scholar]
- 60.Matus A. Growth of dendritic spines: a continuing story. Current Opinion in Neurobiology. 2005;15(1):67–72. doi: 10.1016/j.conb.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 61.Aarts M, Liu Y, Liu L, et al. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002;298(5594):846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- 62.Harms C, Bösel J, Lautenschlager M, et al. Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Molecular and Cellular Neuroscience. 2004;25(1):69–82. doi: 10.1016/j.mcn.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 63.Yildirim F, Gertz K, Kronenberg G, et al. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Experimental Neurology. 2008;210(2):531–542. doi: 10.1016/j.expneurol.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 64.Cassel JC, Cassel S, Galani R, Kelche C, Will B, Jarrard L. Fimbria-fornix vs selective hippocampal lesions in rats: effects on locomotor activity and spatial learning and memory. Neurobiology of Learning and Memory. 1998;69(1):22–45. doi: 10.1006/nlme.1997.3807. [DOI] [PubMed] [Google Scholar]
- 65.Penzes P, Cahill ME, Jones KA, Vanleeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature Neuroscience. 2011;14(3):285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]