

Abstract
Expansion of CAG/CTG trinucleotide repeats (TNRs) in humans is associated with a number of neurological and neurodegenerative disorders including Huntington’s disease. Increasing evidence suggests that formation of a stable DNA hairpin within CAG/CTG repeats during DNA metabolism leads to TNR instability. However, the molecular mechanism by which cells recognize and repair CAG/CTG hairpins is largely unknown. Recent studies have identified a novel DNA repair pathway specifically removing (CAG)n/(CTG)n hairpins, which is considered a major mechanism responsible for TNR instability. The hairpin repair (HPR) system targets the repeat tracts for incisions in the nicked strand in an error-free manner. To determine the substrate spectrum of the HPR system and its ability to process smaller hairpins, which may be the intermediates for CAG/CTG expansions, we constructed a series of CAG/CTG hairpin heteroduplexes containing different numbers of repeats (from 5 to 25) and examined their repair in human nuclear extracts. We show here that although repair efficiencies differ slightly among these substrates, removal of the individual hairpin structures all involve endonucleolytic incisions within the repeat tracts in the nicked DNA strand. Analysis of the repair intermediates defined specific incision sites for each substrate, which were all located within the repeat regions. Mismatch repair proteins are not required for, nor do they inhibit, the processing of smaller hairpin structures. These results suggest that the HPR system ensures CAG/CTG stability primarily by removing various sizes of (CAG)n/(CTG)n hairpin structures during DNA metabolism.
Keywords: Trinucleotide repeats, Hairpin repair, Endonucleolytic incision, MutSβ
1. Introduction
Trinucleotide repeat (TNR) sequences are widely spread throughout the human genome, and can be present in both the introns and exons of a gene. Expansion of TNR sequences is associated with many neurological, neurodegenerative and neuromuscular diseases, including myotonic dystrophy, Huntington’s disease and fragile X syndrome [1-3]. For each disease, pathological symptoms are triggered and become progressively more severe when the number of repeats reaches and then exceeds a critical threshold. In the case of Huntington’s disease, for example, the threshold for CAG repeats in the HTT gene is 35, i.e., the repeat lengths from 11 to 34 are not associated with disease pathology, but the repeat lengths ≥35 result in clinical symptoms of Huntington’s disease. All these diseases exhibit strong age-dependency and the severity of the syndrome is tightly associated with an increased number of repeats. The longer the TNR sequence is, the more severe the syndrome is. Till now, the mechanism by which TNR instability occurs is not fully understood.
It has been proposed that TNR instability could result from strand slippage-caused hairpin formations within the TNR sequence in the newly synthesized strand during DNA replication or repair [2-8]. Indeed, formation of a hairpin structure within CAG/CTG repeats has been well documented by both in vitro and in vivo studies [9-11]. Despite the presence of A-A or T-T mismatches in the CAG or CTG hairpin stem, respectively, these hairpin structures are highly stable and have a melting temperature higher than the physiological temperature in mammalian cells [9,12]. Therefore, CAG/CTG hairpins are expected to persist in vivo once they form, and to require an active mechanism for removal.
Recent biochemical studies have revealed that human cells possess a nick-directed DNA hairpin repair (HPR) system that can efficiently remove DNA hairpins containing 20 or 25 CAG/CTG repeats [13,14]. The HPR system always targets the nicked (i.e., newly synthesized) DNA strand for hairpin removal, mainly using structure-specific endonucleases [13]. When a (CTG)25- or (CAG)25-containing hairpin is located in the nicked strand, it is removed either by dual incisions flanking the heterology or by a combination of nick-directed excision and flap endonucleolytic cleavage, which generates a small single-strand gap. Although the hairpin removal via dual incisions is similar to the bulky DNA adduct removal by nucleotide excision repair, the enzymes responsible for nucleotide excision repair are not essential for HPR [13]. Interestingly, a recent study shows that XPG, one of the two endonucleases required for nucleotide excision repair, greatly enhances CAG/CTG hairpin repair [15]. When such a hairpin is located in the continuous strand (template strand), incisions occur in the nicked strand opposite the hairpin, followed by hairpin unwinding, which generates a relatively large single-strand gap. In both cases, the gap is filled by a replicative DNA polymerase using the continuous strand as a template [13]. Thus, the HPR pathway plays an important role in maintaining TNR stability by targeting hairpin removal in the newly synthesized/nicked strand.
DNA mismatch repair (MMR) is another DNA repair pathway that has been implicated in TNR stability [16-18]. MMR is well known for its role in stabilizing simple repetitive sequences called microsatellites, which are prone to forming small loops or insertion/deletion (ID) mispairs. Repair of these heteroduplexes requires key mismatch proteins MutSα (MSH2–MSH6) and MutSβ (MSH2–MSH3). Interestingly, genetic studies in mice suggest that MutSβ promotes (CAG)n expansion and TNR instability. These studies show that expansion of a heterologous (CAG)n tract occurs in wild type and MSH6-/- mice, but that expansion of the (CAG)n tract is suppressed in MSH2-/- and MSH3-/- mice [16,17]. Based on the fact that MutSβ specifically interacts with (CAG)n/(CTG)n hairpins [17,19,20], it has been hypothesized that binding of (CAG)n/(CTG)n hairpins by MutSβ inhibits hairpin removal [21]. However, in vitro HPR experiments reveal that MutSβ binding does not interfere with the repair of (CAG)25 and (CTG)25 hairpins in human cell extracts [20]. Nevertheless, whether or not MutSβ promotes CAG/CTG repeat expansions by suppressing the repair of smaller hairpins (i.e., those less than 25 repeats) in human cells is unknown.
Given the fact that TNR expansion-associated diseases are age-dependent, it is possible that the disease-onset TNR expansion is a result of accumulation of many small expansions, which are due to escaped repair of small hairpins. Little is known how cells deal with various sizes of (CAG)n/(CTG)n hairpins. To address these questions, we constructed a series of DNA hairpin heteroduplexes containing different numbers of CAG or CTG repeats and performed in vitro HPR assays using cell-free nuclear extracts. We demonstrate here that human cells efficiently process these smaller hairpin DNA substrates in a manner similar to what they use for larger hairpin removal. MMR proteins, e.g., MutSβ, appear to play little role in processing these heteroduplexes, as nuclear extracts derived from MMR-deficient cells repair these heteroduplexes as actively as MMR-proficient extracts and addition of excess amount of purified MMR proteins in MMR-proficient nuclear extracts does not stimulate or inhibit the HPR reaction.
2. Materials and methods
2.1. Preparation of (CAG)n/(CTG)n hairpin substrates
Heteroduplex DNA substrates preparation is based on previous methods [13]. Briefly, we cloned oligonucleotide duplexes containing (5′-CAG-3′)35/(3′-GTC-5′)35, (5′-CAG-3′)25/(3′-GTC-5′)25, (5′-CAG-3′)15/(3′-GTC-5′)15, (5′-CAG-3′)10/(3′-GTC-5′)10 or (CAG)n/(CTG)n (5′-CTG-3′)35/(3′-GAC-5′)35, (5′-CTG-3′)25/(3′-GAC-5′)25, (5′-CTG-3′)15/(3′-GAC-5′)15, (5′-CTG-3′)10/(3′-GAC-5′)10 into the HindIII and EcoRI sites of bacteriophage M13mp18 replication form DNA. The phage derivatives were confirmed by DNA sequencing. Heteroduplex DNA substrates were prepared by combination of single strand viral DNA and double strand replication form DNA with different CAG/CTG repeat numbers as described [13] (Fig. 1). For example, V-(CAG)25 was prepared by hybridization of BglI digested (CAG)10 replication form DNA with (CAG)35 viral DNA. Different sizes of (CAG)n and (CTG)n (n = 25, 15, 10 or 5) in either the continuous strand (un-nicked strand, also called viral or V-strand because of its origin as single-stranded phage DNA) or the nicked strand (also called complementary or C-strand) were constructed. All substrates contain a nick 5′ to the hairpin structure generated from the BglI digestion of the double strand replication form DNA prior to hybridization.
Fig. 1.
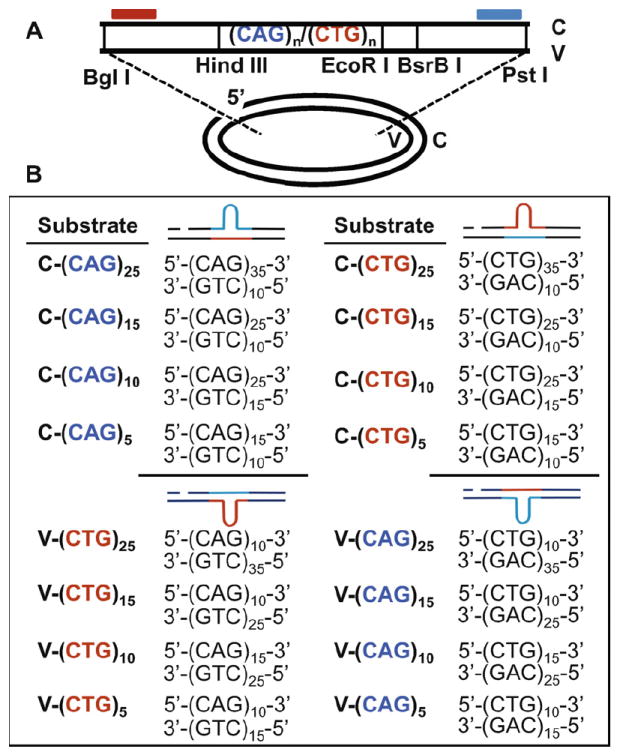
Construction of DNA heteroduplex substrates containing different sizes of CAG/CTG hairpins. (A) Oligonucleotides containing different numbers of CAG/CTG repeats were cloned into the HindIII and EcoRI restriction enzyme sites of an M13mp18 phage derivative. Heteroduplex substrates were constructed as described [13]. All substrates contain a single strand nick in the complementary strand generated by BglI digestion prior to heteroduplex formation. (B) Substrates with different sizes of CAG or CTG hairpins were constructed by heteroduplexing DNA containing different numbers of CAG and CTG repeats in the complementary and viral strands. Predicted structures are shown to the right of the sequences. Repeat sequences are indicated as red (CTG) and blue (CAG).
2.2. Cell lines and nuclear extracts
HeLa S3 cells were grown to a density of 5 × 105 cells/mL in RPMI 1640 media with 5% newborn calf serum, 5% fetal bovine serum (Hyclone) and 4 mM glutamine. NALM-6 (N6) cells were grown to a density of 5 × 105 cells/mL in RPMI 1640 media with 10% fetal bovine serum (Hyclone) and 4 mM glutamine. Nuclear extracts were prepared as described [22].
2.3. Protein purification
MutSα and MutSβ proteins were expressed in insect cells and purified to near homogeneity and examined for MMR activity as described [23].
2.4. DNA hairpin repair assay
Hairpin repair assays were performed by Southern blot analysis as described [13,20,23,24] unless otherwise mentioned. Briefly, 42 fmol of DNA hairpin substrate were incubated with 100 μg of HeLa or N6 nuclear extracts in a 40-μL reaction containing 20 mM Tris–HCl (pH 7.6), 110 mM KCl, 5 mM MgCl2, 1.5 mM ATP, 1 mM glutathione and 0.1 mM each of the four dNTPs in the absence or presence of 400 ng of MutSα or MutSβ. After incubating at 37 °C for 30 min, repair reactions were terminated with the addition of proteinase K to a final concentration of 30 μg/mL and incubating for 20 min at 37 °C. DNA products were recovered by phenol extraction and ethanol precipitation. DNA samples were digested with BglI and BsrBI before loading on a 6% denaturing polyacrylamide gel. After electrophoresis, the DNA products were electrophoretically transferred to a nylon membrane. The nicked strand was probed with a 32P-end labeled oligonucleotide that specifically anneals near the BglI site (Fig. 2) to score the change in the size of the repeats sequence. Repaired molecules and un-repaired molecules were visualized by exposing the probed membrane to X-ray film. Repair efficiency was quantified using a Kodak Image Station 2000.
Fig. 2.
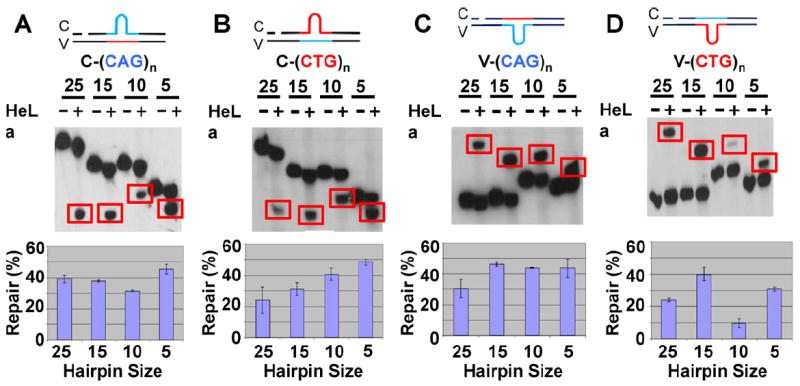
Repair of (CAG)n and (CTG)n substrates in HeLa cell extracts. (A–D, top) The indicated (CAG)n and (CTG)n hairpin substrates (200 ng) were incubated with (+) or without (−) 100 μg of HeLa nuclear extracts under the normal HPR conditions (see Section 2. Repaired products were analyzed by Southern blotting using a probe close to the BglI site of the nicked strand (red bar on the substrate structure). Repair products are highlighted by red boxes. (A–D, bottom) Repair efficiencies of the different (CAG)n and (CTG)n substrates. Repair efficiencies are quantified from at least three individual assays and error bars represent the standard error of the mean.
2.5. DNA hairpin incision/excision assay
The incision/excision assay was performed using the same reaction conditions as the repair assay except without adding exogenous dNTPs to prevent resynthesis (repair) of DNA. DNA products were digested with BglI and PstI and the repair intermediates were scored by Southern blot analysis. 32P-end labeled oligonucleotide annealing to the BglI site (BglI probe) was used for detecting incision products while 32P-end labeled oligonucleotide annealing to PstI site (PstI probe) was used for detecting both incision and excision products.
2.6. Creation of size markers for determining locations of incisions
DNA sequencing reactions were used to read the DNA TNR sequences in the nicked strand of the substrates to create size markers for determining the locations of hairpin incisions. Sequence markers were produced with a primer that anneals to the single strand phage DNA (viral-strand DNA) at the BglI digestion site and extended from the nick site towards the repeats region by sequenase. α-32P-dATP was incorporated into the newly synthesized strand. ddA, ddG, ddC and ddT termination mixes were used to terminate the primer extension reactions at specific nucleotides (A, G, C and T, respectively). Reactions were terminated by the addition of stop solution. Sequence markers were loaded to a 6% denaturing polyacrylamide gel together with incision/excision DNA samples. For the C-(CTG)25 and C-(CTG)15 substrates, V-(CAG)35 and V-(CAG)15 phage viral DNA were used to generate the markers, respectively. For the V-(CAG)n substrates, V-(CAG)10 and V-(CAG)15 phage viral DNA were used to generate the markers. For the V-(CTG)n substrates, V-(CTG)10 and V-(CTG)15 phage viral DNA were used to generate the markers. These are corresponding to the repeat sequences in the complementary strands of the substrates (Fig. 1).
3. Results
3.1. Repair of various (CAG)n/(CTG)n hairpins in human nuclear extracts
To understand potential DNA repair mechanisms that contribute to TNR stability, we constructed a series of heteroduplexes containing hairpins of 5-, 10-, 15-, or 25-CAG or CTG repeats either in the nicked (complementary or C-strand) or continuous strand (viral or V-strand) (Fig. 1). The individual substrates were assayed for repair by HeLa nuclear extracts. Because DNA hairpin repair (HPR) is targeted to the nicked DNA strand resulting in either the deletion of the hairpin if it is in the nicked strand or addition the TNR sequence of the hairpin if the hairpin is in the template strand, HPR in this study was scored by monitoring any change in the length of the nicked strand using Southern blot analysis as described. The results show that HeLa nuclear extracts were capable of processing all the substrates in a nick-dependent manner. For all the substrates that contain a hairpin in the nicked strand, i.e., C-(CAG)n and C-(CTG)n (n = 5, 10, 15, or 25, see Fig. 1), incubation with HeLa nuclear extracts resulted in conversion of the original longer nicked strand to a relatively shorter one (Fig. 2A and B, red rectangles). This indicates that the hairpin is removed from the nicked strand and the gap produced by the removal is filled in, producing the shorter product. Based on the size of the repeat sequence in the continuous strand of these substrates (10 TNRs for n = 5, 15 or 25 and 15 TNRs for n = 10), the repair products for the C-(CAG)n and C-(CTG)n, where n = 5, 15 or 25 should be the same size, while the repair product for the n = 10 substrates should be larger since the size of the repeat sequence in the continuous strand of this substrate is larger (15 TNRs). This is what is seen in Fig. 2A and B. For all the substrates with a hairpin in the continuous strand, i.e., V-(CAG)n and V-(CTG)n (see Fig. 1), a product that is longer than the original substrate was produced after incubation with HeLa nuclear extracts (Fig. 2C and D). These results indicate that like processing CAG/CTG hairpins with 20- or 25-repeats, human cells repair smaller CAG/CTG hairpins in the continuous strand by targeting and removing nucleotides from the nicked strand, followed by repair DNA synthesis using the continuous strand, including the unwound hairpin, as a template. As with the C-(CTG)n and C-(CAG)n substrates, the sizes of the repair products seen (Fig. 2C and D) with the different V-(CAG)n and V-(CTG)n substrates is consistent with the number of TNRs in the continuous strands of the substrates.
Quantification of the repair products revealed that the repair efficiency for these hairpin substrates was between 30 and 40% (see Fig. 2), except for V-(CTG)10, which showed a repair efficiency of only ~10% (Fig. 2D).
3.2. Influence of MutSα and MutSβ on small hairpin repair
Mismatch repair proteins, particularly MutSβ, have been implicated in promoting TNR instability [14,16,17,21,25], possibly by binding to the hairpin structure and inhibiting HPR [17,26]. To determine the role that MutSβ may play during the processing of CAG/CTG hairpins containing a relatively small number (5, 10, or 15) of repeats, we compared HPR activity of nuclear extracts derived from MMR-proficient HeLa and MMR-deficient NALM-6 cells. NALM-6 (N6) cells harbor deletions in the MSH2 gene [27], and are therefore defective in both MutSα and MutSβ. We first assayed their repair activity with the V-(CTG)10 substrate. As shown in Fig. 3A (lanes 6–10) with the V-(CTG)10 substrate, N6 extracts possess a repair activity at least twice as high as that in HeLa extracts (compare lanes 7 and 8). This result suggests that MutSα and/or MutSβ might be responsible for the low HPR activity in HeLa extracts since these proteins are not present in N6 cells. However, addition of purified MutSα or MutSβ to the assays did not inhibit but slightly stimulated V-(CTG)10 repair in N6 extracts (Fig. 3A, lanes 9 and 10). Adding MutSα and MutSβ together also fail to inhibit the repair activity in N6 extracts (data not shown). We further compared HeLa and N6 extracts for the processing of other hairpin substrates, and the results show that other than with the V-(CTG)10 substrate, these two extracts exhibited similar activities in repair of the remaining hairpin heteroduplexes examined (Fig. 3A and B). Consistently, exogenous MutSα or MutSβ did not inhibit repair activity but had either no effect or slightly stimulated HPR in almost every case. We therefore conclude that MutSα or MutSβ has no inhibitory role in vitro during the processing of CAG/CTG hairpins with 5, 10, or 15 repeats.
Fig. 3.
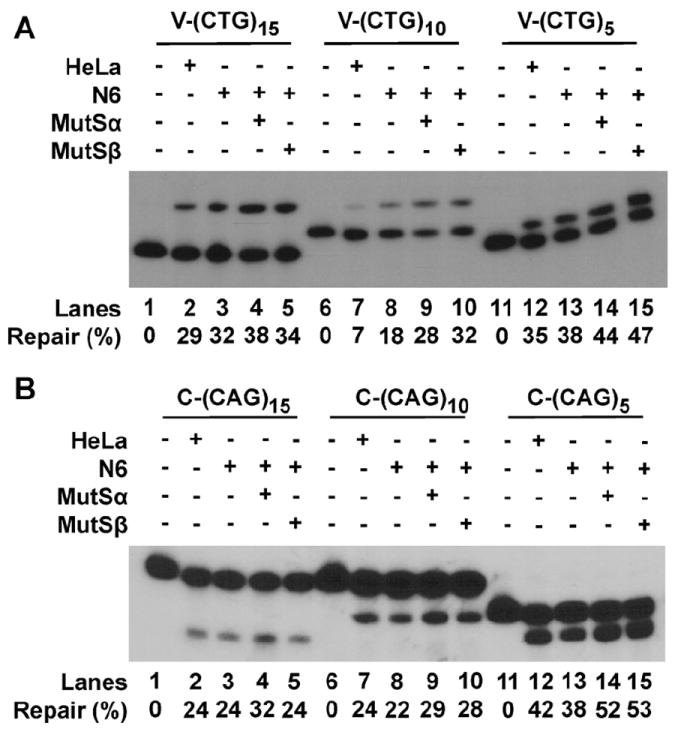
Influence of MutSα and MutSβ on smaller hairpin repair. HPR assays were performed as described in Fig. 2 legend, but in the presence or absence of purified MutSα or MutSβ. (A) V-(CTG)n substrates. The upper bands are repair products, and lower bands are substrates. (B) C-(CAG)n substrates. The lower bands are repair products, while the upper bands are substrates.
3.3. Excision and flap endonuclease activity together remove (CAG)n hairpins in the nicked strand
Previous studies have shown that endonucleotic activities were involved in the repair of (CAG)25 repeat hairpins located in the nicked DNA strand [13]. To determine the repair intermediates for different sizes of C-(CAG)n hairpins, repair assays were performed under conditions that support incision/excision but not DNA synthesis (see materials and Methods). Radiolabeled probes annealing to the nicked strand close to the BglI site (BglI probe) or PstI site (PstI probe) were used. The BglI probe can only detect intermediates generated by incision(s) because the probe’s target sequence is quickly removed by excision starting from the nick, while the PstI probe detects both incision and excision products since its target sequence is on the other side of the hairpin from the nick where excision begins (Fig. 1).
Incision/excision time course assays were performed for the C-(CAG)n substrates. It was shown previously that the removal of the hairpin in the C-(CAG)25 substrate includes an incision within the CAG hairpin region and a 5′–3′ excision starting from the original nick [13]. For the C-(CAG)15, C-(CAG)10 and C-(CAG)5 substrates, however, we detected no specific incision signals even at 30 min using the BglI probe (data not shown). When using the PstI probe, we could clearly see excision products at about 5 min for all three substrates. Excision for these three substrates started from the original nick and stopped somewhere within the repeats area (Fig. 4A–C, red rectangles). At the same time, a band close to the end of the repeats was detected (Fig. 4A–C, black arrows). This indicates that excision starting from the original nick has removed the BglI probe recognition site so that the BglI probe produced no signal. The excision may be blocked by the hairpin structure and stops, leaving a 5′ flap structure. The remaining flap structure, which is close to the end of the repeat sequence, could then be removed by a flap endonuclease such as FEN1 [13]. Taken together, our results support the idea that excision started from the original nick and went 5′–3′ to remove the complementary strand sequence until encountering the hairpin structure, which is subsequently cleaved a flap endonuclease, leaving a gap for repair DNA synthesis.
Fig. 4.
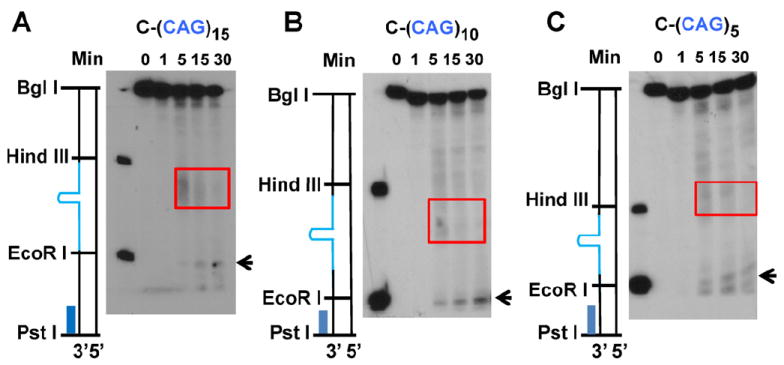
Time course analysis of repair intermediates of C-(CAG)n substrates. The incision/excision time course assays were performed under the HPR conditions, but in the absence of dNTPs. DNA samples were recovered and subjected to Southern blot analysis using the PstI probe, which detects both incisions and excisions. (A) Substrate C-(CAG)15. (B) Substrate C-(CAG)10. (C) Substrate C-(CAG)5. Red rectangles indicate excision intermediates. Black arrows indicate the products generated by a flap endonuclease.
3.4. Dual incisions remove the C-(CTG)n hairpin from the nicked strand
It is proposed that dual incisions occur at the two ends of the C-(CTG)25 substrate and remove the hairpin structure from the nicked strand [13]. Our results show that for all the C-(CTG)n substrates, incision(s) occur within the repeats, while excision also participates in removal of the hairpin structure. For C-(CTG)5, one incision occurred close to the EcoRI site within 1 min of the start of the assay. With a longer incubation time (~5 min), another incision closer to the Hind III site was observed. However, further incubation showed that the both of the two incision products diminished over time (compare 5, 15 and 30 min, Fig. 5A, red arrows). This may be due to degradation of the intermediates or the BglI probe recognition site, or both. When the PstI probe (which can detect incision as well as excision products) was used to reprobe the same blot (Fig. 5B) excision starting from the original nick was detected. This confirmed that the excision removed the BglI probe recognition sequence so that the incision signals seen with the probe became weaker with longer incubation. However, the excision was terminated as it encountered the repeat sequence and the repeat sequences were not removed by the excision (Fig. 5B, rectangle). Therefore, the excision did not directly remove the hairpin structure. The PstI probe also detected two dominant bands within the repeats region. These two products corresponded to the two incision products detected by the BglI probe (Fig. 5B, red arrows). These results again strongly support the idea that the C-(CTG)5 hairpin is removed by incision, not excision.
Fig. 5.
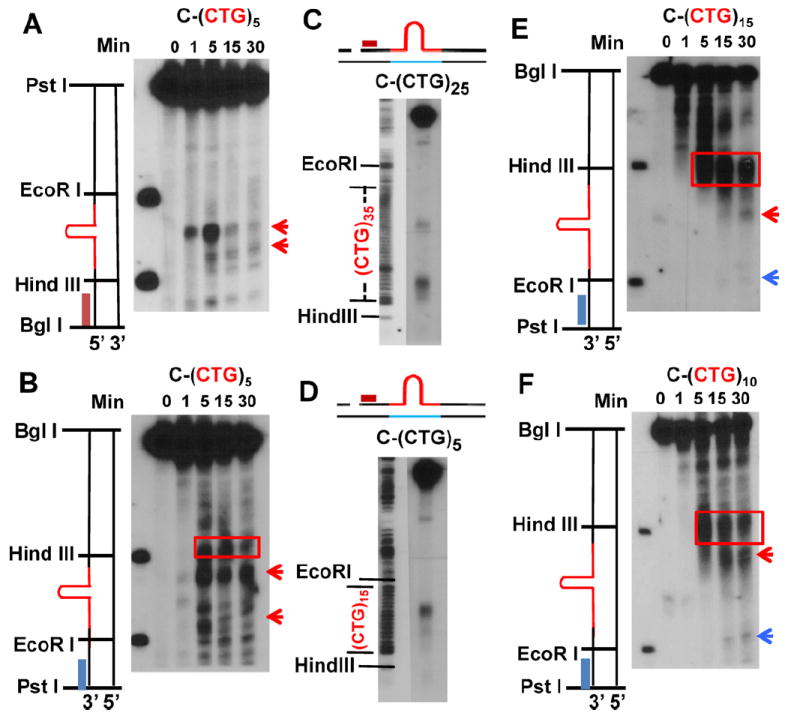
Analysis of repair intermediates of C-(CTG)n substrates. (A) C-(CTG)5 incision/excision time course assay probed with the BglI probe. (B) C-(CTG)5 incision/excision time course assay probed with the PstI probe. (C) Determination of incision sites for substrate C-(CTG)25. The left panel shows the size markers generated from (CTG)35 viral DNA by DNA sequencing analysis (only the T reaction shown). The right panel shows the incision intermediates derived from substrate C-(CTG)25 after incubation with HeLa nuclear extracts for 30 min. (D) Determination of incision sites for substrate C-(CTG)5. The left panel shows the size markers generated from (CTG)15 viral DNA by DNA sequencing analysis (only the T reaction shown). The right panel shows the incision intermediates derived from substrate C-(CTG)5 after incubation with HeLa nuclear extracts for 5 min. (E) C-(CTG)15 incision/excision time course assay probed with PstI prode. (F) C-(CTG)10 incision/excision time course assay probed with PstI probe. Red rectangles indicate excision products; red arrows indicate endonuclease products; and blue arrows represent flap endonuclease products.
The C-(CTG)25 substrate contains 35 CTG repeats in the continuous strand while C-(CTG)5 contains 15 CTG repeats in the continuous strand (Fig. 1). To determine the precise sites of the incisions for substrates C-(CTG)25 and C-(CTG)5, HPR intermediates were analyzed with DNA size markers generated by sequencing using the viral strand DNA of M13mp18-(CAG)35 or M13mp18-(CAG)15 as a template and a primer located at the BglI cutting site in the complementary strand. These markers and the incision/excision reaction intermediates were loaded on the same gel and probed with the BglI probe. As shown in Fig. 5C and D, there are about 20 CTG repeats between the two incisions of for C-(CTG)25; for C-(CTG)5, there are about 5 CTG repeats between the two incisions. This has confirmed the previously proposed model that dual incisions happen close to the two ends of the C-(CTG)n hairpin and remove the hairpin structure from the DNA backbone [13].
In the study of C-(CTG)15 and C-(CTG)10, in contrast, we detected no signal with the BglI probe suggesting that excision of the BglI probe site has occurred (data not shown). We then probed these membranes with the PstI probe (Fig. 5E and F) and at early times during the assay we detected similar excision products for both substrates originating from the nick. The size of these products decreases with time, suggesting that these are excision products. However, the excision appeared to stop at the beginning of the repeat area (Fig. 5E and F, red rectangles). Another product slightly smaller than the excision product, which may be the result of an incision in the hairpin, began to appear around 5 min (Fig. 5E and F, red arrow). At 15–30 min, a third product closer to the end of the repeat sequence appears (Fig. 5E and F, blue arrow). This product resembles that detected for substrates C-(CAG)25 and C-(CAG)5 (see Fig. 4 and Ref. [14]), indicating flap endonuclease activity may be involved in the repair. Therefore, these results suggest that with the C-(CTG)15 and C-(CTG)10 substrates excision occurs starting from the original nick and stops at the hairpin and then an incision occurs within the repeat sequences, which may generate a 5′ flap structure that is further removed by a flap endonuclease.
In conclusion, different C-(CTG)n substrates are repaired by a mechanism involving both endonuclease and exonuclease activities. For C-(CTG)25 and C-(CTG)5, dual incisions cut at the two ends of the CTG hairpin structure and remove it from double stranded DNA. Together with the incisions, excision also helps to remove the nucleotides beginning from the original nick and generates a gap, which can be filled by DNA polymerase in the resynthesis step. For C-(CTG)15 and C-(CTG)10, excision starts first and stops when encountering the repeats. A nick is then generated within the hairpin, which may lead to a 5′ flap structure and further incisions by a flap endonuclease.
3.5. Incision(s) occur in the nicked strand of V-(CAG)n and V-(CTG)n
Southern blot analysis was used to determine the mechanism(s) by which V-(CAG)15, V-(CAG)10 and V-(CAG)5 hairpin substrates are processed. As shown in Fig. 6, the BglI probe detected two incisions in the repeat region of the complementary strand for these substrates. The larger incision product was by far the major product (much stronger band) compared with the smaller product (Fig. 6A–C, red arrows). This supports a previous study showing that incisions remove the (CAG)25 hairpin in the continuous strand [13].
Fig. 6.
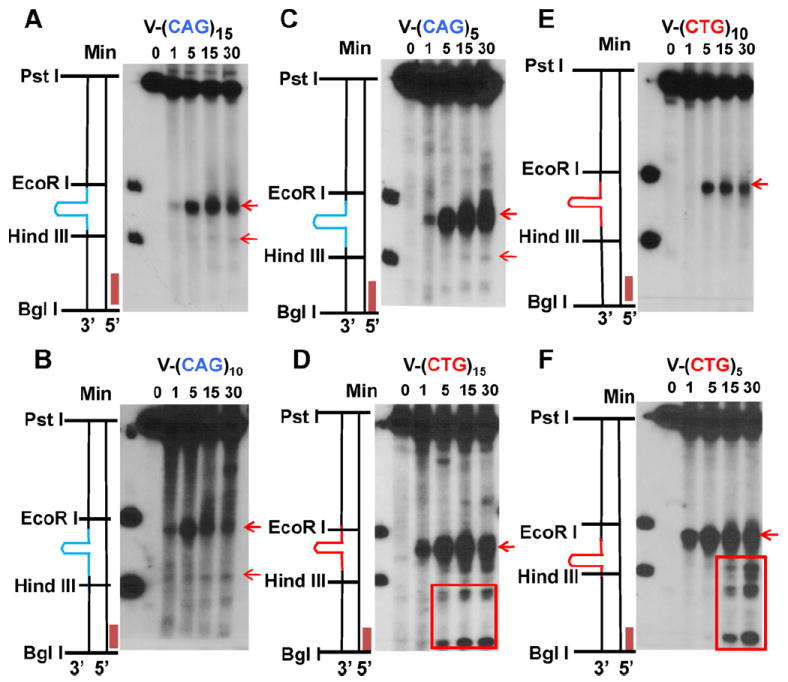
Analysis of repair intermediates of V-(CAG)n and V-(CTG)n substrates. (A–C) Incision/excision time course assays for all V-(CAG)n substrates. (D–F) Incision/excision time course assays for all V-(CTG)n substrates. All assays were probed with the BglI probe. Red arrows indicate endonuclease products. Red rectangles indicate further degradation of the incision products.
In contrast, a single incision was detected for all V-(CTG)n substrates (Fig. 6D–F, red arrow). In consistent with its lowest repair efficiency, substrate V-(CTG)10 also shows the weakest incision signal (Fig. 6, compare D–F). These results suggest that, despite differences in hairpin size, repair of V-(CTG)n hairpins is initiated by a single incision opposite the hairpin structure. It was also noted that for V-(CTG)15 and V-(CTG)5 substrates, some further degradation occurs after the initial incision (Fig. 6D and F, red rectangle), but this phenomenon is not observed in substrate V-(CTG)10. This difference could be related to the stability and/or conformation of the hairpin structure, which may dictate the accessibility of endonucleases involved in HPR (see Section 4 below).
DNA sequencing reactions were used to generate size markers for determining the precise incision site(s) in these hairpin substrates (Fig. 7). For the V-(CAG)25, V-(CAG)15 and V-(CAG)5 substrates, the nicked strand contains 10 CTG repeats. However, for the V-(CAG)10, the nicked strand contains 15 CTG repeats (Fig. 1). In the sequencing reactions, we used viral strand DNA derived from M13mp18-(CAG)10 and M13mp18-(CAG)15 as templates to generate sequence markers for the V-(CAG)n. substrates. Similarly, viral strand DNA derived from M13mp18-(CTG)10 and M13mp18-(CTG)15 was used as templates to generate sequence markers for the V-(CTG)n substrates.
Fig. 7.
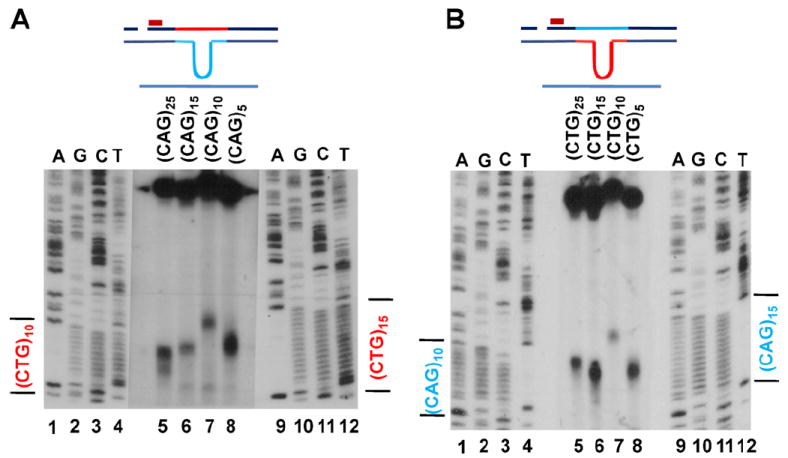
Incision locations for V-(CAG)n and V-(CTG)n substrates. (A) Size markers were generated from (CAG)15 and (CAG)10 viral DNAs. V-(CAG)n substrates were incubated with HeLa extracts for 15 min. (B) Sequence markers were generated from (CTG)15 and (CTG)10 viral DNAs. V-(CTG)n substrates were incubated with HeLa extracts for 15 min. The assays were probed with the BglI probe.
This analysis revealed that with V-(CAG)25, two incisions occurred close to the middle of the CTG repeats, one close to the fourth CTG and the other close to the eighth CTG (Fig. 7A); for the V-(CAG)15, V-(CAG)10 and V-(CAG)5 substrates, two incisions also occurred, one within the CTG repeats nearer to the EcoRI site and the other weaker incision close to the HindIII site of the CTG repeats. It appears that the pattern for V-(CAG)n hairpins is independent of the hairpin size and the repeat number in the nicked strand (Fig. 7A, compare lanes 5–8).
For all the V-(CTG)n substrates, a single incision occurs close to the end of the repeat sequence near the EcoRI site. This is also independent of the repeat numbers in the nicked strand (Fig. 7B, compare lanes 5, 6, 8 and 7). Taken together, the results above suggest that there may be structural specific endonuclease(s) that recognize (CAG)n/(CTG)n hairpin structures in the continuous strand and incise at specific positions in the repeat sequences of the nicked strand that help to repair the hairpins.
4. Discussion
In this study, we examined the in vitro repair of CAG/CTG hairpins of different sizes, which are believed to be intermediates in the processes that result in the increase or decrease in the size of CAG/CTG trinucleotide repeat regions [2-8]. Previous studies using hairpins containing 25 CAG or CTG trinucleotide repeats showed nick-dependant repair of these hairpins involving more than one mechanism. Which mechanism is used depends upon the strand location, sequence specificity (whether it is CAG or CTG) and secondary structure of the hairpin. Analysis of repair intermediates of (CAG)25 and (CTG)25 hairpin substrates revealed that the HPR system involves multiple endonucleolytic activities [13]. However, it is unknown whether or not the HPR pathway can process smaller CAG/CTG hairpins that are thought to be the transition molecules leading to TNR instability. In this study, we studied the repair of DNA hairpin heteroduplexes containing various numbers of CAG/CTG repeats, and we demonstrated that despite the differences in hairpin sizes, HeLa cell extracts can repair all of the (CAG)n and (CTG)n substrates examined in this study (i.e., hairpins of 5, 10, 15, and 25 TNRs) in a similar manner.
One of the mismatch recognition proteins, MutSβ, is responsible for repairing small insertion/deletion heteroduplexes [28], including small loops derived from random DNA sequence [24] or CAG/CTG repeats [14]. However, MutSβ is not required for the repair of CAG/CTG hairpin structures consisting of 25 TNRs [20]. Our results using mismatch repair deficient cell extracts indicates that for the repair of the smaller hairpins, MutSβ does not play a critical role, consistent with previous results for the repair of larger hairpins.
Studies by Hou et al. [13] suggest structure-specific endonucleases play important role in removing hairpins containing 25 TNRs. The results shown in this study reveal that removal of smaller hairpins also involves structure-specific endonucleases. In the case of (CAG)n hairpins containing 5, 10 or 15 repeats in the continuous strand, two incisions occur within the repeat area in the nicked strand, but only one incision for the V-(CTG)n substrates. These results are consistent with those obtained for the V-(CAG)25 and V-(CTG)25 substrates [13].
It is worth mentioning that among all V-(CTG)n substrates tested, V-(CTG)10 is the only one that shows no further degradation following the initial incision (Fig. 6). In addition, the hairpin substrate demonstrates the lowest incision and repair efficiencies. A possible explanation is that the V-(CTG)10 hairpin is more stable than other V-(CTG)n hairpins so that it is not easily accessed by an endonuclease, which leads to the initial poor incision and eventually the poor repair. We believe that the degradation observed in other V-(CTG)n substrates following the initial incision is due to the unwinding of the hairpin in the continuous strand. The gap generated by unwinding of the hairpin allows nuclease(s) access to attack the complementary strand. However, the stable V-(CTG)10 hairpin does not unwind as easily. As a result, there is no further degradation of the incision product. Poor accessibility for endonuclease and low efficiency for helicase may be the reason that V-(CTG)10 has the lowest repair efficiency. It is also possible that the low repair of V-(CTG)10 in HeLa extracts represents cell specificity for this substrate, as a better repair the substrate was observed in N6 extracts. Further investigations are required to resolve these issues.
When a smaller hairpin (5, 10, or 15 TNRs) forms in the nicked strand, we observed products of both endonuclease and exonucleases. These results are similar to what has been reported for the processing of the C-(CAG)25 hairpin [15]. It appears that the excision starts at the original nick and stops when it reaches the hairpin structure. The removal of the 5′ immediate sequences of the hairpin allows the hairpin structure “float” around, leading to the formation of a 5′ flap structure. This structure may be further removed by a flap endonuclease. Thus, the complete removal of a C-(CAG)n hairpin requires coordinating actions from both exonucleases and endonucleases.
Unlike the C-(CAG)n substrates, which appear to be processed by a common mechanism, the C-(CTG)n hairpins are removed differently. Whereas the hairpins in C-(CTG)25 and C-(CTG)5 are repaired by two incisions flanking the hairpin structure (Fig. 5A–D), the hairpins in C-(CTG)15 and C-(CTG)10 heteroduplexes are removed by the combined action of exonuclease and endonucleases in a manner similar to mechanism by which the C-(CAG)n substrates are processed.
In conclusion, human cell extracts process corresponding larger and smaller (CAG)n or (CTG)n hairpins essentially in a similar manner. Exclusive incisions are involved in the removal of the hairpins (both CAG and CTG hairpins) that are located in the continuous strand, but both excisions and incisions are required to remove the hairpins located in the nicked strand. In the later case, an exonuclease may initiate the reaction at the strand break, but it stops when encountering the hairpin; the remaining job, i.e., the actual hairpin removal, is likely conducted by an endonucleases. However, there are exceptions. For example, the processing of substrates C-(CTG)25 and C-(CTG)5 appears to only involve dual incisions. However, the endonuclease(s) involved in the hairpin removal remain unknown. A recent study revealed that although XPG, an essential endonuclease that make an incision 3′ to a bulky DNA adduct during nucleotide excision repair, is not required for HPR, the enzyme stimulates the incision 5′ to the hairpin structure [15]. The molecular mechanism by which XPG promotes 5′ incision in HPR is unknown. Therefore, understanding the molecular basis of HPR awaits future investigations.
Acknowledgments
We would like to thank Drs. Charles M. Ensor, Caixia Hou, Lei Tian and other members of the Li Laboratory for valuable comments and technique assistance in hairpin repair assay and protein expression and purification. This work was supported partially by National Institutes of Health grants GM089684 (to GML) and CA104333 (to LG) and a grant (No. 30740420548 to JH) from National Natural Science Foundation of China.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
References
- 1.Lopez Castel A, Cleary JD, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. 2010;11:165–170. doi: 10.1038/nrm2854. [DOI] [PubMed] [Google Scholar]
- 2.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 3.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 4.Gordenin DA, Kunkel TA, Resnick MA. Repeat expansion—all in a flap? Nat Genet. 1997;16:116–118. doi: 10.1038/ng0697-116. [DOI] [PubMed] [Google Scholar]
- 5.Kang S, Jaworski A, Ohshima K, Wells RD. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat Genet. 1995;10:213–218. doi: 10.1038/ng0695-213. [DOI] [PubMed] [Google Scholar]
- 6.Miret JJ, Pessoa-Brandao L, Lahue RS. Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1998;95:12438–12443. doi: 10.1073/pnas.95.21.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards RI, Sutherland GR. Simple repeat DNA is not replicated simply. Nat Genet. 1994;6:114–116. doi: 10.1038/ng0294-114. [DOI] [PubMed] [Google Scholar]
- 9.Gacy AM, Goellner G, Juranic N, Macura S, McMurray CT. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell. 1995;81:533–540. doi: 10.1016/0092-8674(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 10.Liu G, Chen X, Bissler JJ, Sinden RR, Leffak M. Replication-dependent instability at (CTG) × (CAG) repeat hairpins in human cells. Nat Chem Biol. 2010;6:652–659. doi: 10.1038/nchembio.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearson CE, Tam M, Wang YH, Montgomery SE, Dar AC, Cleary JD, Nichol K. Slipped-strand DNAs formed by long (CAG)*(CTG) repeats: slipped-out repeats and slip-out junctions. Nucl Acids Res. 2002;30:4534–4547. doi: 10.1093/nar/gkf572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petruska J, Arnheim N, Goodman MF. Stability of intrastrand hairpin structures formed by the CAG/CTG class of DNA triplet repeats associated with neurological diseases. Nucl Acids Res. 1996;24:1992–1998. doi: 10.1093/nar/24.11.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou C, Chan NL, Gu L, Li GM. Incision-dependent and error-free repair of (CAG)(n)/(CTG)(n) hairpins in human cell extracts. Nat Struct Mol Biol. 2009;16:869–875. doi: 10.1038/nsmb.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panigrahi GB, Lau R, Montgomery SE, Leonard MR, Pearson CE. Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat Struct Mol Biol. 2005;12:654–662. doi: 10.1038/nsmb959. [DOI] [PubMed] [Google Scholar]
- 15.Hou C, Zhang T, Tian L, Huang J, Gu L, Li GM. The role of XPG in processing (CAG)n/(CTG)n DNA hairpins. Cell Biosci. 2011;1:11. doi: 10.1186/2045-3701-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. 1999;23:471–473. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- 17.Owen BA, Yang Z, Lai M, Gajek M, Badger JD, 2nd, Hayes JJ, Edelmann W, Kucherlapati R, Wilson TM, McMurray CT. (CAG)(n)-hairpin DNA binds to Msh2–Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol. 2005;12:663–670. doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- 18.Panigrahi GB, Slean MM, Simard JP, Gileadi O, Pearson CE. Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSbeta, but clustered slip-outs are poorly repaired. Proc Natl Acad Sci U S A. 2010;107:12593–12598. doi: 10.1073/pnas.0909087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owen BA, McMurray HLWCT. The nucleotide binding dynamics of human MSH2–MSH3 are lesion dependent. Nat Struct Mol Biol. 2009;16:550–557. doi: 10.1038/nsmb.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian L, Hou C, Tian K, Holcomb NC, Gu L, Li GM. Mismatch recognition protein MutSbeta does not hijack (CAG)n hairpin repair in vitro. J Biol Chem. 2009;284:20452–20456. doi: 10.1074/jbc.C109.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McMurray CT. Hijacking of the mismatch repair system to cause CAG expansion and cell death in neurodegenerative disease. DNA Repair (Amst) 2008;7:1121–1134. doi: 10.1016/j.dnarep.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes J, Jr, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci U S A. 1990;87:5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 24.McCulloch SD, Gu L, Li GM. Bi-directional processing of DNA loops by mismatch repair-dependent and -independent pathways in human cells. J Biol Chem. 2003;278:3891–3896. doi: 10.1074/jbc.M210687200. [DOI] [PubMed] [Google Scholar]
- 25.Gomes-Pereira M, Fortune MT, Ingram L, McAbney JP, Monckton DG. Pms2 is a genetic enhancer of trinucleotide CAG·CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum Mol Genet. 2004;13:1815–1825. doi: 10.1093/hmg/ddh186. [DOI] [PubMed] [Google Scholar]
- 26.Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo. Cell Res. 2008;18:198–213. doi: 10.1038/cr.2008.5. [DOI] [PubMed] [Google Scholar]
- 27.Gu L, Cline-Brown B, Zhang F, Qiu L, Li GM. Mismatch repair deficiency in hematological malignancies with microsatellite instability. Oncogene. 2002;21:5758–5764. doi: 10.1038/sj.onc.1205695. [DOI] [PubMed] [Google Scholar]
- 28.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]