

Abstract
Background
In humans, there is a 4:1 male:female ratio in the incidence of abdominal aortic aneurysms (AAAs). c-Jun-N-terminal kinase (JNK) is an important upstream regulator of several enzymes involved in AAA formation, including the matrix metalloproteinases (MMPs). The purpose of this study was to determine if there is a gender difference between males and females in JNK during AAA formation.
Materials and Methods
Male and female C57/B6 mice underwent aortic perfusion with elastase or heat inactivated elastase with aortas harvested at day 3 and 14 for phenotype determination, RT-PCR, Western blot, and zymography. Additionally, in vitro experiments using siRNA were conducted to define JNK regulation of matrix metalloproteinases (MMPs). A t-test was used to compare between groups.
Results
Males formed larger AAAs at day 14 compared to females (p<0.001), with significantly higher levels of JNK1 protein, proMMP9, proMMP2, and active MMP2. At day 3, males had more JNK1 mRNA, protein, and MMP activity. Knockdown of JNK 1 or 2 in vitro decreased MMP activity, while knockdown of JNK 1 and 2 together blocked all MMP activity.
Conclusion
Alterations in JNK between genders is partially responsible for the differential rates of experimental AAA formation, likely through differential regulation of MMPs.
Keywords: Abdominal aortic aneurysm, JNK, gender, rodent, elastase model
Introduction
Abdominal aortic aneurysm (AAA) is a common medical problem, with over 35,000 patients undergoing aneurysm repair in the United States each year. There is a gender disparity in the incidence of AAAs, with men being affected four times as often as women (1). The molecular basis underlying this gender disparity is unknown. However, if protective factors present in women could be identified, these may serve as potential therapeutic targets to prevent aneurysm expansion in men with small aortic aneurysms, prevent aneurysm formation in patients at high risk, or possibly even cause aneurysm regression.
The mitogen activated protein (MAP) kinases are a family of intracellular signaling molecules that transduce signals via phosphrylation. There are three main proteins in this family: p38, extracellular signal-regulated kinase (ERK), and c-Jun-N-terminal kinase (JNK). These proteins have many roles in cellular signaling, including regulation of the matrix metalloprotinases (MMPs). Prior work has demonstrated differential regulation of ERK between male and female aortic smooth muscle cells, leading to differential regulation of MMP2 (2). However this system of MAP kinases is redundant and these molecules often play redundant and paradoxical roles in regulating inflammation, including the MMPs. In humans, JNK has been shown to be correlated with increased size and rupture risk in intracranial aneurysms (3). JNK has also been demonstrated to be elevated in patients with AAAs compared to those who have a normal aortic diameter. Additionally, pharmacologic inhibition of JNK has been shown to prevent aneurysm formation, as well as lead to regression of existing AAAs in male mice (4). To date, there are no studies examining gender differences in JNK and AAAs.
Given the promising therapeutic potential of JNK inhibition in causing aneurysm regression and the known gender difference in the incidence of AAAs in humans, we sought to determine if there is a gender difference in JNK between male and female mice in experimental AAA formation.
Materials and Methods
Rodent Elastase Perfusion
Male and female wild type C57/B6 mice (Jackson Labs, Bar Harbor, ME) (N=10–15 per group) were anesthetized with 2% isofluorane with 98% oxygen. A ventral vertical midline incision was made and the abdominal aorta was exposed from the renal vein to the iliac bifurcation. All side branches were cauterized or ligated with 10–0 nylon suture (Surgical Specialties Corporation, Reading, PA) to ensure pressurization of the aorta. Baseline aortic photos and measurements were taken using a video micrometer and NIS Elements software attached to the microscope (Nikon, Melville, NY). Measurements were taken at the proximal, mid, and distal aorta. Temporary proximal and distal control was obtained with 4–0 silk suture (Ethicon Inc., Somerville, NJ) and a 30 gauge needle was used to make an aortotomy just proximal to the iliac bifurcation. A custom polyethelyne catheter (Braintree Scientific, Braintree, MA) was inserted through the aortotomy. A dilute porcine pancreatic elastase (0.4units/mL, Sigma, St. Louis, MO) for experimental animals, or heat inactivated elastase (90°C for 30 minutes) for control animals, was infused to ensure a doubling in the aortic diameter for 5 minutes. The catheter was then removed and the aortotomy closed with a single 10–0 nylon suture. Proximal and distal ligatures were then removed restoring blood flow to the lower extremities. The abdomen was closed in two layers with running 5–0 Vicryl suture (Ethicon Inc., Somerville, NJ) and the animal allowed to emerge from anesthesia. All animal experiments were approved by the University of Michigan Universal Committee on the Use and Care of Animals (UCUCA #09679).
Rodent Sacrifice
At time of sacrifice, animals were again anesthetized with 2% isofluorane and 98% oxygen and the vertical ventral laparotomy was reopened. The aorta was dissected free from the surrounding tissues and repeat intraoperative photos and measurements were taken, again at the proximal, mid, and distal aorta while the animal was still alive. Blood was then collected from the inferior vena cava using a 25 gauge needle and 1cc syringe (Becton Dickinson, Franklin Lakes, NJ). The aorta was then removed from the level of the left renal vein to the iliac bifurcation and processed.
Aortic Measurements
The increase in aortic diameter was determined by taking photographs and measurements of the aorta of each animal using a video micrometer and NIS Elements software attached to the microscope (Nikon, Melville, NY) just prior to aortic perfusion and again just prior to sacrifice. Photos were taken just below the left renal vein (proximal), at the level of the ileolumbar vessels (mid) and just above the iliac bifurcation (distal) at both time points. The diameter at baseline prior to aortic perfusion was subtracted from the diameter at harvest at each location. The largest difference (typically in the mid aorta) was then used as the increase in aortic diameter for that animal.
Messenger RNA (mRNA) Extraction, Reverse Transcription, and Real-time PCR
Established techniques using TRIzol reagent (Invitrogen, Carlsbad, CA) were used to extract mRNA for real-time reverse transcription polymerase chain reaction (RT-PCR). In brief, explanted aortic tissue was added to 1.5mL TRIzol reagent, and the tissue was homogenized. Chloroform (+99%) was then added to the homogenized tissue, vortexed, and centrifuged. The clear supernatant was pipetted into Eppendorf tubes. RNA precipitation was performed with isopropanol (+99%) and 7.5ug glycogen. The resulting solution was centrifuged. The supernatant was poured off and the remaining mRNA pellet was washed by adding 70% ethanol in diethylpyrocarbonate (DEPC) water and centrifuged. The supernatant was then aspirated off, and the pellet dried at room temperature. The pellet was then redissolved in DEPC water at 58° C. RNA concentration was then measured on the Nanodrop 1000 Spectrophotometer (ThermoScientific, Pittsburgh,PA). For producing cDNA, 5ug RNA was used in the reverse transcription reaction with standard reagents in a GeneAmp 2400 PCR System (Perkin Elmer-Applied Biosystems, Norwalk, CT).
cDNA concentration was measured using the Nanodrop 1000 Spectrophotometer (Thermo Scientific, Wilmington, DE) to calculate the amount needed to obtain 22 ng/mL for the RT-PCR reaction. mRNA expression of JNK1(Mapk8), and JNK2(Mapk9) were compared with that of β-actin, a housekeeping gene. Primers as well as the SYBR Green Master Mix used for RT-PCR were obtained from SABiosciences (Qiagen, Frederick, MD). The RotorGene 6000 Series Software 1.7 (Corbett Research, Qiagen, Frederick, MD) was used to amplify the target DNA and obtain the take-off values and melt curves for analysis. The following program was used on the Rotor Gene: 95° C, 10 min; 40 cycles of (95° C, 15 sec; 60° C, 60 sec).
Western Blot
For Western blot analysis, the tissues or cells were lysed by sonication and overnight incubation in ice-cold RIPA buffer (Sigma, St. Louis, MO) containing protease and phosphatase inhibitors (Roche, Basel, Switzerland). Protein concentration in the lysate was determined with the BCA protein assay kit (Pierce, Rockford, IL). Equal amounts of protein were loaded into each well and resolved by SDS-PAGE with 10% gels (Novex, Invitrogen, Carlsbad, CA) and electroblotted onto polyvinylidene membranes (Immobilon-P, Millipore, Billerica, MA) by semidry transfer blot (Biorad, Hercules, CA) according to the manufacturer's instructions. The membranes were incubated in StartBlock TBS (Pierce, Rockford, IL) for one hour and then incubated with anti-total-JNK or anti-phosporylated-JNK (Cell Signaling Technology, Danvers, MA) in the StartBlock solution at 1:500 dilution for overnight with gentle shaking. The membranes were washed in 25 mM Tris, 150 mM NaCl, 0.1% Tween-20, pH 7.4 (TBST) for one hour at room temperature and then incubated with HRP conjugated goat anti-rabbit secondary antibodies (1:2000) (Santa Cruz Biotechnology, Santa Cruz, CA) for one hour and again washed in TBST. For normalization of proteins on the western blots, the membranes were stripped and probed with anti-actin antibodies conjugated with HRP (Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were developed with the West-Pico ECL kit (Pierce, Rockford, IL).
Gel Zymography
Gelatin substrate zymograms were run in pre-cast 10% SDS-PAGE gels containing 1 mg/mL of gelatin (Invitrogen, Carlsbad, CA). Equal volumes of experimental media or equal amounts of proteins were diluted into 2× tris-glycine SDS sample buffer and electrophoretically separated under non-reducing conditions. Proteins were renatured for 30 min in Renaturing Buffer (Invitrogen, Carlsbad, CA) and then the gels were incubated in the developing buffer (Invitrogen, Carlsbad, CA) for 30 min and again in the same buffer overnight at 37°C. The gel was stained in SimplyBlue SafeStain (Invitrogen, Carlsbad, CA) and the gelatinase activity was observed by clear bands against the blue background.
Immunohistochemistry
Three animals from each group were randomly selected for histologic analysis. Aortic tissue was fixed in 10% buffered formaldehyde for 2 hours, transferred to 70% ethanol, and subsequently embedded in paraffin for immunohistochemistry. Sections were prepared with Hematoxylin and Eosin stain. Blank sections were then stained for macrophages using the following procedure. Aortic sections were deparaffinized in xylene and rehydrated in graded alcohols. Heat-induced antigen retrieval using 10 mM sodium citrate, pH 6.0, was performed in a microwave. Sections were subsequently incubated with 3% hydrogen peroxide in methanol to block endogenous peroxidase activity, followed by a blocking buffer to prevent nonspecific binding. Purified Anti-Mouse MAC-2 Monoclonal Antibody (1:200, Cedarlane) specific for macrophages was used for staining, followed by an Anti-Rat IgG biotinylated secondary antibody (1:500) and an avidin-biotin-HRP complex, available in the Rat IgG Elite Vectastain ABC Kit (Vector Laboratories, Burlingame, CA). Sections were then visualized using a DAB Peroxidase Substrate Kit (Vector Laboratories) and counterstained with Hematoxylin QS (Vector Laboratories). Digital photographs were then taken using a Nikon Eclipse Ti-U microscope with Nikon Elements software (Nikon, Melville, NY). The photos were then imported into Image J (National Institutes of Health, Bethesda, MA), separated into red-green-blue components, and the percent area stained was measured after appropriate thresholds were established using the green image.
Cell Culture
Reagents were obtained from Sigma Chemical Co. (St. Louis, MO) unless otherwise indicated. Rat aortic smooth muscle cells were cultured from the abdominal aortas of young (190–210 g) male and female Sprague-Dawley rats (Charles River Labs, Wilmington, MA). After animal sacrifice and aortic explanation under general inhalational anesthesia, aortic tissue was cut into 2 mm2 pieces and placed in 60mm diameter plastic tissue culture dishes. Basement membrane Matrigel (Collaborative Research, Bedford, MA) was applied to each section of explanted tissue to prevent floating. Cultures were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (HyClone Laboratories, Logan, UT), 100 units/mL penicillin, 100 mg/mL streptomycin, and glutamine 292 mcg/mL. Tissue culture media and antibiotics were obtained from Gibco (Rockville, MD). Tissues were incubated at 37°C in a humidified, 5% CO2 atmosphere for 4 to 7 days, until spindle shaped SMCs were observed extending from the tissue. After removing the explant, cells were dispersed by treatment with trypsin (Gibco), centrifuged, resuspended in complete medium, and placed into 75 cm2 culture flasks. Post confluent cultures assumed a hill and valley topography characteristic of SMCs grown in vitro. RASMCs were confirmed by staining with a monoclonal antibody against SMC-specific α-actin and determined to nearly 100% SMCs.
siRNA Transfection
Cells were plated in six-well tissue-culture plates at 60–80% confluency. One day later, the cells were transfected with 200 pmol of siRNAs for JNK-1, JNK-2 or a mixture of JNK-1 and 2 (all siRNAs were from Santa Cruz Biotechnology, Santa Cruz, CA) using the siRNA transfection reagent and medium (Santa Cruz Biotechnology, Santa Cruz, CA) according to the instructions provided by the manufacturer. Seventy-two hours after transfection, RASMCs were washed with phosphate-buffered saline (PBS), starved in serum free medium overnight, washed in PBS and then treated with 5μg/mL elastase (Sigma, St. Louis, MO) containing media or media alone for 24 hours. Media was then collected for zymography.
Statistical Analysis
Various data points were collected and entered into a database (Microsoft Excel 2007, Microsoft Corp., Redmond, WA). Comparison statistics between groups were determined using unpaired student's t-test in PRISM software (GraphPad Software Inc., La Jolla, CA). Data are presented as means ± standard deviation along with significance values.
Results
Increased JNK Levels in Males Compared to Females at Day 14
There was not a significant difference in aortic diameter between male and female mice at baseline (data not shown). An aneurysm was defined as a >50% increase compared to baseline. Additionally, there was no significant difference in JNK1 or JNK2 mRNA expression between males and females at baseline (data not shown).
At day 14, the endpoint of this model, elastase treated males (n=11) experienced an increase in aortic diameter of 130±29% compared to baseline, while elastase treated females (n=14) only had an increase of 35±18% (p<0.001) (Figure 1). All of the males formed an aneurysm compared to only 14% of females at this time point. Control males (n=14) had an average increase of 29±7% at day 14, significantly smaller than the elastase treated males (p<0.001). Control females (n=15) had an average increase of 14.5±3%, significantly less than the elastase treated females at day 14 (p=0.0008). There was not a statistical difference in aortic diameter between the male and female controls at day 14.
Figure 1.
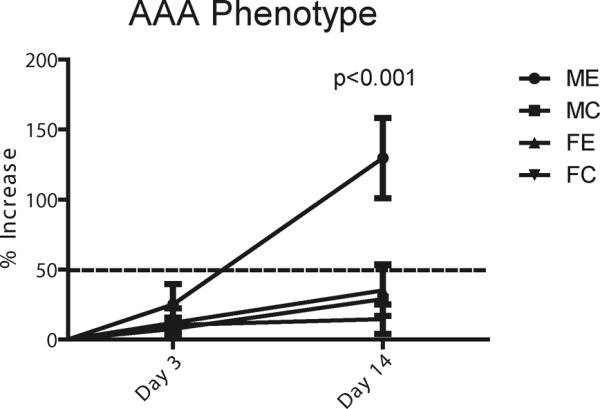
Percent increase in aortic diameter compared to baseline at 3 and 14 days for male elastase (ME), male control (MC), female elastase (FE) and female control (FC) animals. ME have an average increase of 130% at 14 days compared to 35% for FE (p<0.001)
Hematoxalin and eosin staining of male and female elastase treated samples harvested at day 14 (n=3 per group) revealed a dense inflammatory response and breakdown of the internal elastic lamina in the male, but not the female (Figure 2A), consistent with aneurysm formation in the male. Immunohistochemical staining for MAC-2 at day 14 (n=3 per group) revealed 46-fold more macrophages in the elastase treated male than the elastase treated female (p=0.017) (Figure 2B). Male elastase treated mice also had significantly more macrophages than the male controls (p=0.0173). There was not a significant difference between the male and female controls, or between the female controls and female elastase treated animals.
Figure 2.

A: Hematoxalin and eosin staining of aortic tissue samples from male and female elastase perfused mice harvested at day 14. Breakdown of the internal elastic lamina (arrows) and a dense inflammatory reaction (arrow head) in the male compared to female. B: Immunohistochemistry for MAC-2 demonstrating macrophages (brown stained cells) in male and female aortic tissue sections collected at day 14. There are 46× more macrophages in the male (arrows) compared to the female (p=0.017).
Western blot performed on aorta samples from animals treated with elastase harvested at day 14 (n=9 each group) revealed that total JNK1 was increased 47% in males compared to females (p=0.033) (Figure 3A). There was no significant difference in total JNK2 protein between the genders at day 14. Additionally, male elastase treated animals had significantly more total JNK1 and total JNK 2 than control males (p=0.0035 and p=0.0328, respectively). Western blot for phosphorylated JNK1 and JNK2 did not reveal any significant differences between the genders (data not shown). Gelatin zymography, performed on day 14 (n=9 each group) to assess for MMP 9 and 2 activity, revealed that there were significant differences in proMMP9, proMMP2, and active MMP2 (p=0.03, p=0.0009, and p=0.0112, respectively), all of which were higher in the male than female (Figure 3B).
Figure 3.

A: Western blot at day 14 demonstrates significantly more JNK1 protein in male elastase (ME) compared to female elastase (FE) (p=0.033). Also significantly more JNK1 and JNK2 protein in male elastase treated animals (ME) compared to control (MC) (p=0.0035 and p=0.0328, respectively). B: Zymography reveals significantly more proMMP9 (p=0.03), proMMP2 (p=0.0009), and active MMP2 (p=0.0112) in the elastase treated males compared to females at day 14.
Increased JNK in Males at Day 3
Given these differences at day 14 and in order to determine what happens early during aneurysm formation in association with peak inflammation (5), we examined animals three days after aortic elastase perfusion. Neither elastase treated males nor females formed aneurysms by day 3; however, males (n=13) had an average increase in aortic diameter of 25±4% compared to baseline, while females (n=11) had an average increase of 12±3% (p=0.021) (Figure 1). Real-time PCR on day 3 samples revealed significantly higher JNK1 mRNA expression in males treated with elastase (n=5) compared to elastase treated females (n=9) (p=0.0067) (Figure 4A). JNK1 expression was also significantly higher in the male elastase treated animals compared to male controls (n=8) (p=0.0031) (Figure 4A). There was a significant difference between males and females (n=9 each group) treated with elastase in both total JNK1 and total JNK2 protein levels (p=0.0022 and p=0.023, respectively), with higher levels of both in males compared to females (Figure 4B). Again, there were not any significant differences in phosphorylated JNK1 or JNK2 between the males and females (data not shown). On day 3, there was 97% more proMMP9 activity in the elastase perfused males compared to elastase perfused females (p=0.002) (n=9 each group), as well as 67% more active MMP2 (p=0.022) (Figure 4C).
Figure 4.

A: Significantly more JNK1 mRNA expression in male elastase (ME) treated animals than male controls (MC) (p=0.0031) or female elastase (FE) (p=0.0067) treated at day 3. B: Western blot reveals significantly more JNK1 (p=0.0022) and JNK2 (p=0.023) protein in the males (ME) compared to females (FE) 3 days following elastase perfusion. C: Significantly more proMMP9 (p=0.002) and active MMP2 (p=0.022) in males compared to females at day 3 following elastase perfusion by zymography
siRNA Inhibition of JNK Decreases MMP Activity in Aortic Smooth Muscle Cells
Media from rat aortic smooth muscle cells (SMCs) in culture stimulated with elastase were collected and analyzed. Western blot for total JNK was also performed on the cell lysate. Media from male control cells revealed proMMP2 and active MMP2 activity only (Figure 5). Male SMCs stimulated with elastase revealed an increase in proMMP2 (p=0.0017) and active MMP2 (p<0.0001), as well as induced proMMP9 activity compared to control cells. Knock-down of JNK1 with siRNA eliminated proMMP9, and caused decreases in both proMMP2 (p<0.0001) and active MMP2 (p=0.0014) (Figure 5). Knock down of JNK2 with siRNA revealed similar results, with elimination of proMMP9, and decreases in proMMP2 (p=0.0003) and activeMMP2 (p=0.0019) when compared with non-manipulated elastase treated cells (Figure 5). When both JNK1 and JNK2 were knocked down, MMP activity was eliminated. However, the cells appeared sick and died within 48 hours. This correlates with the lethal nature of JNK1+2 knock out in vivo. Scramble control cells did not demonstrate any significant non-specific effects of the siRNA transfection (data not shown). These changes in MMP activity correlate with decreases in JNK1 and JNK2 protein in the cells as demonstrated by Western blot (Figure 5). Similar experiments were performed in female cells, but there were not any significant differences in MMP activity with knock down of JNK.
Figure 5.

Zymogram on media from male aortic smooth muscle cells transfected with siRNA for JNK1, JNK2, or both, then stimulated with elastase. Significantly less proMMP2 and activeMMP2 activity with knock down of JNK1 (p<0.0001 and p=0.0014, respectively) or JNK2 (p=0.0003 and p=0.0019, respectively). All MMP activity is eliminated with knock down of both JNK1 and 2 simultaneously. A decrease in JNK1 and JNK2 protein in the cells is demonstrated by Western blot.
Discussion
This study demonstrates that there are indeed differences between males and females in the incidence and prevalence of experimental abdominal aortic aneurysm formation in the mouse elastase perfusion model, recapitulating the human phenotype. These differences are seen histologically with increased inflammation and macrophage influx in males compared to females. Differences between males and females in total JNK1 were observed both early (day 3) and late (day 14) in aneurysm formation, with higher levels in the male at both time points. Gender related differences also were observed in MMP activity, with more proMMP9 and active MMP2 activity at day 3, and more proMMP9, proMMP2, and active MMP2 activity at day 14 in elastase treated males compared to females. In vitro studies confirmed that knock down of JNK1 or JNK2 leads to decreases in MMP9 and 2, suggesting a possible mechanism for linking decreased levels of JNK with decreased aneurysm formation. The fact that there were differences in JNK2 at day 3 but not at day 14 are not fully explained by these experiments and require further investigation. These results also speak to the redundancy that is inherent in the JNK and overall MAP kinase system. Additionally, while the differences in phosphorylated JNK between genders did not reach statistical significance in our study, others have demonstrated that differences in both total and phosphorylated JNK are important in AAA formation in a mouse model (4).
Other investigators have demonstrated that adensosine triphosphate (ATP) stimulates MMP2 production in human aortic smooth muscle cells via the JNK pathway both in the presence and absence of interluken-1β (IL-1β) (6). Pharmacologic inhibition of the JNK pathway significantly reduced the amount of MMP2 produced from the cells, similar to our siRNA knockdown experiments. However, to our knowledge, this is the first report of differences in JNK expression and levels between males and females in an experimental model of aneurysm formation. Given these results, it appears that JNK is not only involved in AAA formation, but it plays an important role in gender-associated differences in AAA development. One of the most obvious differences between males and females is in the gonadal hormones.
There are many studies in the literature that support the linkage between estrogen and decreases in JNK in tissues other than the aorta. One example is in endometrial cells, both in vitro and in vivo. These investigators noted that the highest level of JNK in endometrial cells in normal women was during the early proliferative and later secretory phases of the menstrual cycle, the times when estrogen is at the lowest levels in the body (7). These findings were verified in vitro by subjecting endometrial stromal cells in culture to estrogen withdrawal, which led to significant increases in JNK compared to the control and non-withdrawal groups. These investigators also noted that pharmacologic inhibition of JNK in vitro led to decreased interluken-8 (IL-8) production. This finding serves as a link to prior work demonstrating that endometrial cells secrete significantly more MMP9 and MMP2 in response to IL-8 stimulation in a dose-dependent fashion, thus linking increases in JNK to increases in MMPs 9 and 2 (8).
Another tissue where the link between estrogen and JNK has been examined is in the breast, particularly in relation to breast cancer. Using estrogen receptor positive (ER+) MCF7 and estrogen receptor negative (ER−) SkBr3 breast cancer cell lines, investigators were able to demonstrate that treatment with estrogen inhibited mixed lineage kinase 3 (MLK3) in the ER+ cells, but not the ER− cells (9). JNK is downstream of MLK3 and was also inhibited by treatment with estrogen in the ER+ cells. When the estrogen receptor was pharmacologically blocked, there was no inhibition of MLK3 or JNK in the ER+ cells.
JNK is also known to play a role in the apoptosis signaling pathway, while estrogen can have cell-protective effects, especially under oxidative stress. One example of this is in hepatocytes in cell culture, where hypoxia led to upregulation of both p38 and JNK, eventually leading to increases in NF-κB (10). This upregulation was attenuated with pharmacologic inhibition of JNK, and blocked by treatment with estrogen. Another study examined the effects of estrogen on the modulation of oxidative stress in pancreatic islet cells in relation to JNK (11). These investigators found that islet cells subjected to hypoxia that were treated with 17β-estradiol had a higher viability compared to control cells. Additionally, there was a dose-dependent reduction in JNK activation for increasing concentrations of 17β-estradiol, and that this effect was partially reversed by addition of an estrogen antagonist. A similar effect was also seen in cardiac myocytes, where treatment with estrogen led to decreases in JNK and p38 activation (12).
Finally, the link between estrogen and downregulation of JNK has also been described in the context of the bone sparing effect of estrogen. In HeLa cells transfected with estrogen receptor-α, investigators noted that exposure to estrogen led to downregulation of activator protein -1 (AP-1), a downstream target of the MAP kinases including JNK. Further inhibition experiments isolated this effect to decreases in JNK (13). Other experiments in RAW 264.7 cells stimulated with IL-1β or tumor necrosis factor-α (TNFα) demonstrated that estrogen decreased JNK protein levels by 3-fold compared to cells without estrogen present (14). Additional studies demonstrated that estrogen ultimately led to decreased TNFα gene expression and protein levels mediated by decreases in JNK.
In addition to the evidence supporting downregulation of JNK by estrogen, there is also some evidence of upregulation of JNK activity by testosterone. In one study using human umbilical vein endothelial cells, treatment with testosterone increased the activation of JNK, p38 and ERK and subsequent increases in tissue factor pathway inhibitor induction (15).
Given the diverse role of JNK in many tissues and disease processes in addition to AAA formation, work examining JNK inhibitors has also been conducted. Inhibition of JNK1 can help with insulin improve insulin resistance, while inhibition of JNK2 can help prevent foam cell formation and prevent matrix degradation in the intestine in inflammatory bowel disease (16). However the development of inhibitors is somewhat difficult, as there are three different types of JNK protein (JNK1, JNK2, and JNK3) with a total of 10 different splice forms, in addition to a role in several different organs (16). Additionally, problems with drug delivery and rapid metabolism of the inhibitors must be overcome. Finally, total inhibition of JNK is not possible as it would lead to many unintended side effects such as increased susceptibility to infection due to poor bacterial clearance (17).
There are three main types of JNK inhibitors: ATP competitive inhibitors, substrate site binding inhibitors, and allosteric regulatory site blockers (18). SP600125 is an example of an ATP competitive inhibitor of JNK and is often used in laboratory experiments. It has been shown to have a protective effect on the nervous system and in inhibiting AAA formation (4, 16). AS601245 is another ATP competitive inhibitor of JNK that has been shown to protect against ischemic apoptosis in the heart of rats (16). CNI-1493 is an inhibitor that blocks the phosphorylation of JNK as well as p38. It has also been shown to suppress macrophage activation and block the production of several inflammatory cytokines (17). The immunosuppressant FK506 has been shown to inhibit JNK and p38 activation by LPS in microglial cells. Finally, CEP-1347 is an inhibitor of mixed lineage kinases and can block the activation of the JNK apoptotic pathway in neurons. It can also attenuate the neural damage in models of Parkinson's disease (17). Unfortunately, the use of many of these inhibitors is limited to the laboratory and none have been widely studied in humans due to their adverse side effect profile from a lack of specificity.
Some limitations do apply to this investigation. JNK is an upstream intracellular stress molecule that plays a role in many processes, including inflammation and cell death. Given that JNK is upstream, there are many possible opportunities for biologic compensation by alternative pathways with loss of JNK, making it difficult to draw a direct cause and effect between JNK and aneurysm formation in males and females. However, in vitro experiments performed serve to mechanistically link JNK and MMP 2 and 9 regulation. Additionally, the biology of the MAP kinases is extremely complex, with several different isoforms of each family. Each isoform has its own unique function and role in normal physiology and pathophysiology, yet also able to cross pathways and compensate for a loss of another isoform.
Despite these limitations, the present study is the first to document JNK modulation of MMP dependent vessel remodeling associated with gender differences in aneurysm phenotype. Further studies examining the effects of manipulation of the hormone environment and the subsequent effects on JNK and aneurysm formation will help to further delineate the role of estrogen and testosterone in this system. Additionally, study of the downstream molecules in the JNK pathway, as well as into the other MAP kinase pathways in relation to abdominal aortic aneurysm disease, will need to be conducted.
Acknowledgements
The authors would like to thank Mr. Chris Hedly for graphic design assistance in creating the figures for this manuscript.
This work was supported by NIH R01 HL081629-01 (GRU), NIH R01 HL081629-03S1 (GRU), Coller Society Research Fellowship (PDD), and the University of Michigan Aortic Research Fellowship (PDD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest to disclose.
References
- 1.Singh K, Bonaa K, Jacobsen B, Bjork L. Stolberg S Prevalence of and risk factors for abdominal aortic aneurysms in a population-based study: The Tromoso Study. Am J Epidemiol. 2001;154:236–244. doi: 10.1093/aje/154.3.236. [DOI] [PubMed] [Google Scholar]
- 2.Ehrlichman L, Ford J, Roelofs K, Tedeschi-Filho W, Futchko J, et al. Gender-Dependent Differential Phosphorylation in the ERK Signalinig Pathway is Associated with Increased MMP2 Activity in Rat Aortic Smooth Muscle Cells. J Surg Res. 2010;160:18–24. doi: 10.1016/j.jss.2009.03.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laaksamo E, Tulamo R, Baumann M, Dashti R, Hernesniemi J, et al. Involvement of Mitogen-Activated Protein Kinase Signaling in Growth and Rupture of Human Intracranial Aneurysms. Stroke. 2008;39:886–892. doi: 10.1161/STROKEAHA.107.497875. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 5.Hannawa K, Eliason J, Woodrum D, Pearce C, Roelofs K, et al. L-Selectin mediated neutrophil recruitment in experimental rodent aneurysm formation. Circulation. 2005;112:241–247. doi: 10.1161/CIRCULATIONAHA.105.535625. [DOI] [PubMed] [Google Scholar]
- 6.Robinson W, Douillet C, Milano P, Boucher R, Patterson C, et al. ATP stiumulates MMP-2 release from human aortic smooth muscle cells via JNK signaling pathway. Am J Physiol Heart Circ Physiol. 2006;290:H1988–H1996. doi: 10.1152/ajpheart.00344.2005. [DOI] [PubMed] [Google Scholar]
- 7.Kizilay G, Cakmak H, Yen C, Atabekoglu C, Arici A, et al. Expresion and regulation of c-Jun N-terminal kinase (JNK) in endometrial cells in vivo and in vitro. Histochem Cell Biol. 2008;130:761–771. doi: 10.1007/s00418-008-0421-z. [DOI] [PubMed] [Google Scholar]
- 8.Mulayim N, Savlu A, Guzeloglu-Kayisli O, Kayisli U. Arici A Regulation of endometrial stromal cell matrix metalloproteinase activity and invasiveness by interleukin-8. Fertil Steril. 2004;81:904–911. doi: 10.1016/j.fertnstert.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Rangasamy V, Mishra R, Mehrotra S, GSondarva G, Ray R, et al. Estrogen suppresses MLK3-mediated apoptosis sensitivity in ER+ breast cancer cells. Cancer Res. 2010;70:1731–1740. doi: 10.1158/0008-5472.CAN-09-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee M, Jung S, Lee J. Han H Estradiol-17B protects against hypoxia-induced hepatocyte injury through ER-mediated upregulation of Bcl-2 as well as ER-independent antioxidant effects. Cell Research. 2008;18:491–499. doi: 10.1038/cr.2008.42. [DOI] [PubMed] [Google Scholar]
- 11.Eckhoff D, Smyth C, Eckstein C, Bilabo G, Young C, et al. Suppression of the c-Jun N-terminal kinase pathway by 17B-estradiol can preserve human islet functional mass from proinflammatory cytokine-induced destruction. Surgery. 2003;134:169–179. doi: 10.1067/msy.2003.219. [DOI] [PubMed] [Google Scholar]
- 12.Satoh M, Matter C, Ogita H, Takeshita K, Wang C, et al. Inhibition of apoptosis-regulated signaling kinase-1 and prevention of congestive heart failure by estrogen. Circulation. 2007;115:3197–3204. doi: 10.1161/CIRCULATIONAHA.106.657981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kousteni S, Han L, Chen J, Almeida M, Plotkin L, et al. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. 2003;111:1651–1664. doi: 10.1172/JCI17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava S, Weitzmann M, Cenci S, Ross F, Adler S, et al. Estrogen decreases TNF gene exression by blocking JNK activity and the resulting production of c-Jun and JunD. J Clin Invest. 1999;104:503–513. doi: 10.1172/JCI7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin H, Wang D, Mei Y, Qiu W, Zhou Y, et al. Mitogen activated protein kinases pathway is involved in physiological testosterone-induced tissue factor pathway inhibitor expession in endothelial cells. Blood Coagul Fibrinolysis. 2010;21:420–424. doi: 10.1097/MBC.0b013e328337b475. [DOI] [PubMed] [Google Scholar]
- 16.Waetzig V. HT Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage. Trends Pharmacol Sci. 2005;26:455–461. doi: 10.1016/j.tips.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy - from molecular mechanisms to therapeutic benefits. Biochimica et Biophysica Acta. 2005;1754:253–262. doi: 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 18.Cui J ZM, Zhang Y, Xu Z. JNK pathway: diseases and theraputic potential. Acta Pharmacol Sin. 2007;28:601–608. doi: 10.1111/j.1745-7254.2007.00579.x. [DOI] [PubMed] [Google Scholar]