

Abstract
Topoisomerase II (Topo II) that decatenates newly synthesized DNA is targeted by many anticancer drugs. Some of these drugs stabilize intermediate complexes of DNA with Topo II and others act as catalytic inhibitors of Topo II. Simultaneous depletion of Topo IIα and Topo IIβ, the two isoforms of mammalian Topo II, prevents cell growth and normal mitosis, but the role of Topo II in other phases of mammalian cell cycle has not yet been elucidated. We have developed a derivative of p53-suppressed human cells with constitutive depletion of Topo IIβ and doxycycline-regulated conditional depletion of Topo IIα. The effects of Topo II depletion on cell cycle progression were analyzed by time-lapse video microscopy, pulse-chase flow cytometry and mitotic morphology. Topo II depletion increased the duration of the cell cycle and mitosis, interfered with chromosome condensation and sister chromatid segregation and led to frequent failure of cell division, ending in either cell death or restitution of polyploid cells. Topo II depletion did not change the rate of DNA replication but increased the duration of G2. These results define the effects of decreased Topo II activity, rather than intermediate complex stabilization, on the mammalian cell cycle.
Key words: topoisomerase II, mitosis, G2, conditional knockdown, S phase, mitotic catastrophe
Introduction
Topoisomerase II (Topo II), an enzyme required for decatenation of supercoiled DNA, has been implicated in key cellular processes.1 Topo II plays several essential roles in mitosis,2–4 where it is required for chromosome structure, condensation and segregation.1,5,6 Topo II has also been implicated in the activation of transcription.1,7 While Topo II is needed to decatenate newly replicated DNA, it is not required to complete DNA replication.1 Expression of a phosphorylation-deficient form of Topo II induces S-phase arrest8 but it is unknown whether Topo II activity affects the rate of DNA replication. Topo II inhibition in mammalian cells has been shown to trigger G2 checkpoint arrest, which is distinct from the checkpoint induced by DNA damage.9–11 This G2 checkpoint was initially believed to be signaled by non-decatenated DNA, but more recent studies indicate that it requires the expression and phosphorylation of Topo II protein12–14 rather than just a failure of DNA decatenation.
Much of our knowledge on the biological functions of Topo II comes from the studies utilizing small-molecule Topo II inhibitors, several of which have been developed as anticancer drugs.15–17 Topo II-inhibiting drugs can be divided into two groups. The first group is comprised of Topo II “poisons,” such as doxorubicin, etoposide or mitoxantrone, which stabilize an intermediate complex formed by Topo II bound to cleaved DNA, leading to the formation of double-stranded DNA breaks and activation of DNA damage checkpoints. The second group comprises catalytic inhibitors of Topo II, defined as compounds that exert their effect on the cells by interfering with DNA decatenation by Topo II rather than by stabilizing the “cleavable complexes.” However, the specificity of catalytic inhibitors of Topo II has been questioned due to conflicting reports on the ability of such compounds to induce DNA damage18,19 or to their off-target effects on other cellular proteins.15 Since there is considerable interest in developing “pure” catalytic inhibitors for cancer therapy, understanding the cell cycle effects of a decrease in the Topo II function (as opposed to the presence of a non-functional or partly functional protein) can help in evaluating such compounds and their antiproliferative activity.
The effects of catalytic inhibitors of Topo II, which act on both Topo IIα and Topo IIβ, the two Topo II isoforms in mammalian cells, can be modeled by simultaneous depletion of Topo IIα and Topo IIβ. Depletion of Topo IIα alone through conditional knockout20 or short interfering RNA (siRNA) knockdown of both Topo IIα and Topo IIβ14,21,22 has led to the development of mitotic abnormalities, increased duration of mitosis and failure to proliferate. Longer-term analysis of cell cycle effects of Topo II depletion, however, is complicated by the fact that double knockdown of Topo IIα and Topo IIβ is lethal to the cells. Johnson et al.13 have recently reported a derivative of chicken DT40 cell line with stable knockdown of Topo IIβ and inducible expression of short hairpin RNA (shRNA) targeting Topo IIα, which allows one to monitor the effects of Topo II depletion upon the addition of the inducer. Topo II depletion in these cells induced mitotic abnormalities similar to those observed in mammalian cells upon transfection of Topo IIα and Topo IIβ siRNA. Topo II depletion showed no effects on the fraction of cells replicating their DNA (the S phase) or on the fraction of cells that arrest in mitosis upon nocodazole treatment (a measure of G2).13 However, the steady-state measurements used in the latter study are not sensitive enough to detect more subtle kinetic effects on the cellular entry and exit in different phases of the cell cycle. In the present study, we have developed what is, to the best of our knowledge, the first mammalian cell line with regulated Topo II depletion. We then used sensitive cell cycle kinetics assays, including time-lapse video microscopy and DNA pulse-chase flow cytometric analysis, to investigate the effects of Topo II depletion on cell cycle progression in mammalian cells. Our results confirm the role of Topo II in mitosis, demonstrate that Topo II activity does not affect the rate of DNA replication and reveal that Topo II depletion delays the completion of G2.
Results
Generation of TOP73 cell line with stable Topo IIβ and inducible Topo IIα knockdown.
As the starting cell line, we used human HT1080-GSE56 fibrosarcoma cells that had been previously transduced with GSE56, a transdominant peptide inhibitor of p53.23–26 This p53-suppressed cell line was chosen to minimize the effects of p53-mediated damage-responsive cell cycle checkpoints in the analysis of cell cycle effects of Topo II depletion. In preliminary experiments, cells were co-transduced with lentiviral vectors carrying different selectable markers and constitutively expressing shRNAs targeting Topo IIβ and Topo IIα. We were unable to select any cells transduced with both vectors, indicating that simultaneous depletion of both Topo II isoforms is lethal to this cell line. We then set out to generate a cell line with conditional Topo II depletion, using the scheme shown in Figure 1. HT1080-GSE56 cells were first transduced with a lentiviral vector that constitutively expresses Topo IIβ-targeting shRNA and the DsRed fluorescent marker; the transduced cells were selected for DsRed fluorescence by flow sorting. These cells (the Topo IIβ-knockdown population) were then sequentially transduced with a lentiviral vector expressing the tTR-KRAB tetracycline/doxycyline-sensitive repressor27 and then with Topo IIα-targeting shRNA cloned into a tTR-KRAB-regulated lentiviral vector pLLCEP TU6X that expresses the EGFP-Puro fusion marker.28,29 The transduced cells were subcloned, and individual clonal cell lines were tested for growth inhibition by doxycycline, the expected effect of inducible Topo II depletion. A cell line showing the strongest growth inhibition by doxycycline was designated TOP73.
Figure 1.

Scheme of generation of HT1080-derived TOP73 cell line with regulated Topo II depletion.
Figure 2A compares the levels of Topo IIβ (left) and Topo IIα (right) mRNA, measured by quantitative reverse-transcription PCR (QPCR) in the parental HT1080-GSE56 cells in the Topo IIβ-knockdown population and in the TOP73 cell line before and at different time points after the addition of doxycycline. Figure 2B shows the results of immunoblotting analysis of Topo IIβ and Topo IIα proteins in the same cells. Topo IIβ mRNA and protein were essentially undetectable in the Topo IIβ-knockdown population and in TOP73 cells in the absence or in the presence of doxycycline. Remarkably, the levels of Topo IIα mRNA and protein, while unchanged in the Topo IIβ-knockdown population relative to HT1080-GSE56, were increased ∼4-fold in untreated TOP73 cells (Fig. 2A and B). This increase can be explained by our observations that expression from tTR-KRAB-regulated vectors is not immediately repressed upon transduction into tTR-KRAB-expressing cells. GFP expression from the pLLCEP TU6X vector was detectable by fluorescence microscopy during the first two days after transduction and disappeared only on the third day. Since Topo IIα shRNA was also expressed during this period, its detrimental effect on cell growth should have favored the selection of epigenetic variants with elevated basal expression of Topo IIα, which are relatively resistant to Topo IIα shRNA. TOP73 appears to represent such a variant.
Figure 2.
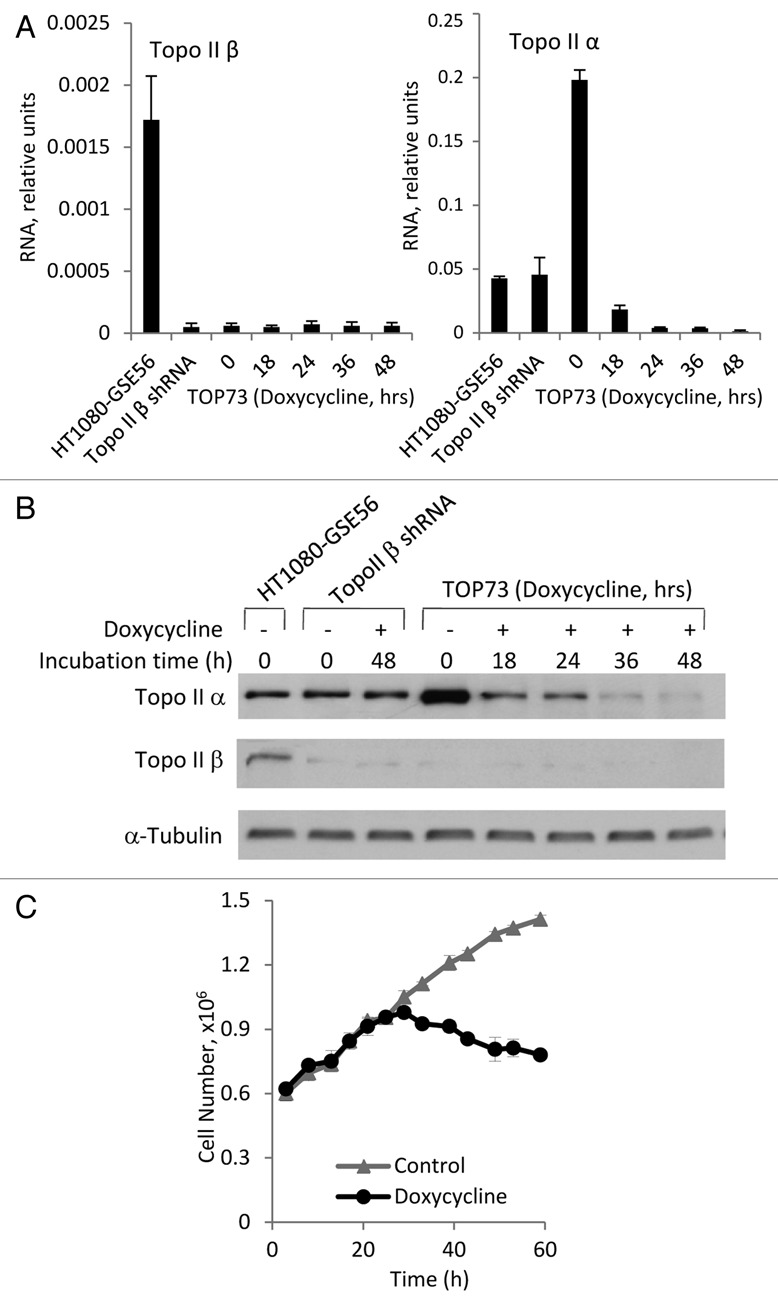
Topo II-knockdown analysis. (A) mRNA levels of Topo IIβ (left) and Topo IIα (right) in the parental HT1080-GSE56 cells, Topo IIβ-knockdown population and TOP73 cell line before and at the indicated time points after the addition of doxycycline. Means and standard deviations of QPCR measurements conducted in triplicates. (B) Immunoblotting analysis of Topo IIα (above) and Topo IIβ (below) in the same cell populations as in (A). (C) Kinetics of TOP73 cell growth in the absence and in the presence of doxycycline.
Both Topo IIα mRNA and protein levels were strongly decreased 18 h after the addition of doxycycline to TOP73 cells relative to the same cells in the absence of doxycycline; Topo II protein levels became greatly decreased even relative to the parental HT1080-GSE56 cells by 36 h of doxycycline treatment (Fig. 2A and B). Topo II depletion in doxycycline-treated TOP73 cells was associated with the inhibition of cell proliferation, the cell numbers ceasing to increase ∼36 h after the addition of doxycycline (Fig. 2C), i.e., at the time of the maximal Topo IIα protein depletion.
Effects of Topo II depletion on cell cycle distribution and mitosis.
Figure 3A shows flow cytometric analysis of cell cycle distribution as assayed by cellular DNA content in the parental HT1080-GSE56 cells and in TOP73 cell line before and 24 h or 48 h after the addition of doxycycline. TOP73 cells, even without doxycycline, show a lower fraction of cells with G1 DNA content and a higher fraction of cells with G2/M DNA content relative to HT1080-GSE56. TOP73 also showed a readily detectable fraction of cells with higher than G2 DNA content; this polyploid fraction was negligible in HT1080-GSE56 (Fig. 3A). Since the Topo IIβ-knockdown population did not differ from HT1080-GSE56 in this analysis (data not shown), the change in cell cycle distribution of TOP73 cells is most likely due to the elevated levels of Topo IIα in TOP73 (Fig. 2A and B). Upon the addition of doxycycline, the fraction of cells with G1 DNA content decreased, and the fraction with G2/M DNA content increased relative to control TOP73 cells (Fig. 3A). Concurrent determination of the fraction of mitotic cells showed no significant differences between these samples, indicating that the accumulation of cells with G2/M DNA content was not due to mitotic arrest.
Figure 3.

Flow cytometric analysis of changes in DNA content of TOP73 cells upon Topo II depletion. (A) Flow cytometric analysis of DNA content in the parental HT1080-GSE56 cells and in TOP73 cell line before and at the indicated time points after the addition of doxycycline. The percentages of cells with G1 and G2/M DNA content are shown. (B) Flow cytometric analysis of changes in DNA content of TOP73 cells that were pulse-labeled with BrdU 40 h after culture in the absence (above) or in the presence of doxycycline (below) at the indicated time points after the removal of BrdU. Only BrdU-positive cells are shown.
The accumulation of cells with G2 DNA content upon Topo II depletion was also indicated by a flow cytometric bromodeoxyuridine (BrdU) pulse-chase analysis of cellular DNA. In this experiment, TOP73 cells were cultured for 40 h in the absence or in the presence of doxycycline, pulse-labeled with BrdU for 30 min and analyzed at different chase time points by staining for cellular DNA content and immunostaining for BrdU. Figure 3B shows the course of changes in the DNA content of BrdU pulse-labeled TOP73 cells. BrdU-labeled cells first increased their DNA content to the G2 level, and by 6–8 h of chase, some of these cells appeared in the G1 fraction, indicating completion of mitosis. The most prominent difference between doxycycline-treated and untreated cells was observed after 8 h of chase, at which time 54.9% of BrdU-labeled cells have acquired G1 DNA content, as compared with only 34.8% of doxycycline-treated cells (Fig. 3B), indicating a delay in G2 or M upon Topo II depletion.
The increased fraction of cells with G2 DNA content upon Topo II depletion could result either from a failure of cells to enter mitosis or from re-entry of cells with fully replicated DNA into the interphase without cell division (restitution). We have investigated the effects of Topo II depletion on the course of mitosis in TOP73 cells by time-lapse phase-contrast video microscopy. The filming was conducted for a period of 72 h in the absence or in the presence of doxycycline. Cells were analyzed for the time of entry and exit from mitosis and the outcome of mitosis. The addition of doxycycline increased the length of the cell cycle (the time from the onset of the first mitosis after the start of filming to the onset of the second mitosis) from 26.9 ± 1.9 h to 33.9 ± 8.3 h (31 cells in each group). The duration of mitosis in Topo II-depleted cells increased more than 3-fold among cells that entered mitosis after 24 h of doxycycline treatment from 1.2 ± 0.6 h to 3.7 ± 1.0 h (41 cells in each group). Such a delay is indicative of difficulties in the mitotic process. This was corroborated by the analysis of mitotic outcomes, which were determined for 54 doxycycline-treated and 39 untreated cells starting 24 h after the addition of doxycycline. Only 51.9% of mitotic events among doxycycline-treated cells ended in cell division (Movie S1), as compared with 92.3% of control TOP73 cells. On the other hand, 42.6% of doxycycline-treated cells but only 5.1% of control cells underwent restitution (Movies S2 and S3), whereas 5.6% of mitoses in doxycycline-treated cells and 2.6% of mitoses in control cells ended in cell death through apoptosis (Movie S4). Cells that underwent restitution became micronucleated, and some of the micronucleated cells died through apoptosis (Movie S5). The frequent restitution events indicate that the increase in the fraction of cells with G2 DNA content upon Topo II depletion is due, at least in part, to the failure of mitosis and accumulation of polyploid cells.
We have also analyzed the effects of Topo II depletion on mitosis by microscopic assays, including morphological analysis of chromosome spreads and immunofluorescence analysis of mitotic figures. Examination of chromosome spreads showed several types of chromosomal morphology, classified as normal, shortened, entangled, severely entangled or amorphous (see examples in Fig. 4A). 100 chromosome spreads were classified for each of HT1080-GSE56 cells, the Topo IIβ-knockdown cells, TOP73 control cells and doxycycline-treated TOP73 cells. As shown in Figure 4B, the first three cell populations showed predominantly normal chromosomes, but 60% of doxycycline-treated TOP73 cells displayed abnormal chromosome morphology, with the most common categories being amorphous chromosomes (suggesting poor chromosome condensation) and severely entangled chromosomes (suggesting failure to separate the daughter chromatids).
Figure 4.

Morphological analysis of changes in mitosis of TOP73 cells upon Topo II depletion. (A) Mitotic spread images of different types of chromosomal morphology. Arrows indicate entangled chromosomes. (B) Distribution of different types of chromosomal morphology in TOP73 cells, untreated or treated for 48 h with doxycycline (107 cells in each group). Means and standard deviations for three independent experiments. (C) Distribution of mitotic phases among mitotic figures of TOP73 cells, untreated (114 cells) or treated for 48 h with doxycycline (111 cells), as determined by immunofluorescence microscopy. Means and standard deviations for three independent experiments. (D) Immunofluorescent images of normal metaphases (control cells), metaphase plates with poorly condensed DNA and multipolar mitoses (doxycycline-treated TOP73 cells). Mitotic cells were stained with DAPI (DNA staining, blue) and with antibodies against α-tubulin (mitotic spindle, green) and γ-tubulin (centrosomes, red). (E) Immunofluorescence analysis of DNA damage in mitotic chromosomes of TOP73 cells, untreated or treated with doxycycline for 24 h or 48 h and in HT1080-GSE56 cells treated with 100 nM doxorubicin for 9 h. Cells were stained with PI (DNA, red) and an antibody against γH2AX, a marker of DNA damage (green).
Immunofluorescence analysis of mitotic figures was conducted by staining of DNA, α-tubulin (mitotic spindle) and γ-tubulin (centrosomes). Figure 4C shows the distribution of mitotic phases in TOP73 cells grown in the absence or in the presence of doxycycline (because of the difficulty of distinguishing metaphase and prometaphase in Topo II-depleted cells, these phases were grouped together). The main effects of Topo II depletion were a decrease in the prometaphase/metaphase fraction and an increase in the prophase fraction (Fig. 4C), suggesting a difficulty in reaching metaphase in Topo II-depleted cells. TOP73 control cells exhibited predominantly normal mitotic morphology, but doxycycline-treated cells showed a peculiar combination of apparently normal metaphase-type mitotic spindles with diffuse, poorly condensed DNA that failed to form a metaphase plate (Fig. 4D). In addition, we have observed an increased incidence of multipolar mitotic figures upon Topo II depletion (Fig. 4D). We have also analyzed the effects of Topo II on the staining of mitotic figures with the antibody against γH2AX, a marker of DNA breaks,30 with DNA counterstaining. The images in Figure 4E show the lack of γH2AX-positive foci (yellow) in mitotic chromosomes of cells treated with doxycycline for 24 h or 48 h, in contrast to strong γH2AX staining of the chromosomes in cells treated with a Topo II “poison” doxorubicin (Fig. 4E).
Effects of Topo II depletion on the duration of S and G2 phases.
The time course of DNA replication (the S phase) and of the transition between DNA replication and mitosis (the G2 phase) was analyzed by a flow cytometric pulse-chase assay similar to the one shown in Figure 3B, but utilizing 5-ethynyl-2′-deoxyuridine (EdU) incorporation, followed by a chemical reaction for fluorescent labeling31 instead of BrdU immunostaining. The use of EdU allowed us to combine the analysis of EdU pulse-labeling with DNA staining by 4′,6-diamidino-2-phenylindole (DAPI) and mitotic cell staining with GF-7, an antibody that selectively stains mitotic cells32 and that produced, in our hands, the strongest immunofluorescence signal for mitotic cells among several mitotic cell-specific antibodies. GF-7 reactivity with cells in different phases of mitosis was verified by immunoperoxidase staining (Fig. 5A). TOP73 cells were cultured for 40 h in doxycycline-containing or control media, and after EdU pulse-labeling for 30 min, chased for 1, 2, 3, 4 or 5 h, followed by staining and flow cytometric analysis. Figure 5B shows the time course of changes in DNA content for EdU pulse-labeled TOP73 cells during the 5 h pulse. The DNA content of pulse-labeled cells (a representation of DNA replication) was indistinguishable between doxycycline-treated and untreated cells at any time point (Fig. 5B). Similarly, no difference in the rate of increase in the DNA content of S-phase cells was detectable in the BrdU pulse-chase experiment in Figure 3B. Hence, Topo II depletion does not affect the rate of DNA replication.
Figure 5.

Pulse-chase analysis of the effects of Topo II depletion on DNA replication and G2 phase of TOP73 cells. (A) Immunoperoxidase staining of the interphase and different mitotic phases of HT1080 cells with GF-7 antibody (above); DAPI counterstaining of DNA (below). (B) Flow cytometric analysis of changes in DNA content of TOP73 cells that were pulse-labeled with EdU 40 h after culture in the absence (above) or in the presence of doxycycline (below) at the indicated time points after the removal of EdU. Only EdU-positive cells are shown. (C) Flow cytometric analysis of EdU and GF-7 labeling of TOP73 cells that were pulse-labeled with EdU 40 h after culture in the absence (above) or in the presence of doxycycline (below), at the indicated time points after the removal of EdU. Percentages of EdU+ GF-7− and EdU+ GF-7+ cells are shown. (D) Time course of mitotic entry in TOP73 cells, untreated or treated with doxycycline. Percentages of EdU+ GF-7+ cells from (C) are plotted.
The duration of G2 was measured by the kinetics of the appearance of EdU pulse-labeled cells that express the mitotic marker GF-7. GF-7 staining was not only specific for mitotic vs. interphase cells, but also strongly associated with cells in pro-phase (Fig. 5A), indicating that the acquisition of GF-7 positivity coincides with the entry in mitosis. The distribution of EdU and GF-7-labeled cells in each sample is shown in Figure 5C, and the time course of the appearance of cells positive for both EdU and GF-7 is plotted in Figure 5D. This analysis demonstrates that doxycycline-treated TOP73 cells display slower progression through G2 relative to control cells, indicating that Topo II depletion causes a delay in G2.
Discussion
The TOP73 human cell line with inducible Topo II depletion developed in the present study offers the first cellular model for monitoring long-term consequences of Topo II depletion in mammalian cells. Such a model has been previously developed in avian DT40 cell line,13 but accomplishing this goal in mammalian cells proved to be very difficult due to the lethal cellular effects of Topo II depletion. We believe that we have succeeded in developing a Topo IIβ-depleted cell line that also showed conditional Topo IIa knockdown due to the selection of a rare variant with a highly elevated basal level of Topo IIα, a likely prerequisite for conditional Topo IIα depletion in mammalian cells. The TOP73 line can be used to study the role of Topo II in cell cycle progression and other biological processes, such as transcription or DNA repair. The effects of Topo II depletion observed in TOP73 cells allow one to differentiate the effects of decreased DNA decatenation expected from “pure” catalytic inhibition of Topo II from the consequences of stabilization of intermediate Topo II complexes or other non-functional forms of Topo II and from off-target effects of Topo II inhibitors.
Previous studies of Topo II knockdown in mammalian or avian cells revealed severe perturbations of mitosis, in agreement with the well-known role of Topo II in chromosome condensation and segregation. The impairment of mitosis has also been observed upon treatment with ICRF-193, generally believed to be a catalytic inhibitor of Topo II.33,34 Our study indicates that disruption of mitosis is the principal cell cycle effect of Topo II depletion and offers a more detailed description of the effects of Topo II depletion on the outcomes of mitosis and chromosome morphology. As a result of impaired mitosis, Topo II depletion leads to a failure of cell division, with the ensuing cell death (mitotic catastrophe) or restitution. The latter outcome leads to an increase in DNA ploidy, which has also been observed in ICRF-193-treated cells33 and which can be misinterpreted as “G2 arrest” in flow cytometric analysis of DNA content. The increase in the chromosome number upon restitution can lead to multipolar mitosis, which is suppressed by p53,35 and we have, indeed, observed multipolar mitosis upon Topo II depletion in our p53-suppressed cellular system.
We have used flow cytometric analysis of pulse-chase DNA labeling to investigate the effects of Topo II depletion on the kinetics of DNA replication and G2. Topo II depletion had no detectable effect on the DNA content of cells with pulse-labeled DNA at any time point until the completion of DNA replication, arguing that DNA decatenation by Topo II does not affect S-phase progression. Similarly, no effect on DNA replication was observed upon knockdown of both Topo II isoforms in chicken cells, albeit with a less sensitive approach.13 Topo II depletion also did not affect DNA replication in budding yeast, whereas the completion of DNA replication in this system was prevented by the expression of catalytically inactive Topo II.7 On the other hand, we observed a delay in the entry of DNA pulse-labeled cells into mitosis, a measure of G2 in Topo II-depleted cells. The G2 checkpoint induced by Topo II inhibitors is presently believed to require the presence of the Topo II protein,12–14 and previous Topo II-knockdown studies did not detect apparent changes in the length of G2.13 Those studies, however, did not utilize the sensitive pulse-chase approach used in the present work. The delay in mitotic entry (Fig. 5D) was moderate and gradual, suggesting that extension of G2 upon Topo II depletion is not due to the activation of a checkpoint.
Importantly, Topo II depletion induced mitotic catastrophe, i.e., abnormal mitosis that eventually leads to cell death or permanent cessation of cell proliferation.36 This was evidenced by video microscopy observations of cell death that occurred either during mitosis or after restitution that followed unsuccessful mitosis as well as by the formation of multiple micronuclei, the most common marker of mitotic catastrophe, upon Topo II depletion. Mitotic catastrophe is the principal cause of tumor cell death induced by Topo II “poisons” and other anticancer agents,24,36,37 which argues for the potential utility of catalytic inhibitors of Topo II as cytotoxic anticancer agents.
Materials and Methods
Development of TOP73 cell line.
Lentiviral transduction was performed as described in reference 29. HT1080-GSE56 cells38 were transduced with a lentiviral vector expressing shRNA against Topo IIβ (target sequence GGA TTA TGT GGT AGA TCA A), cloned into a modified version of pLL3.7 vector,39 where EGFP marker was replaced with DsRed. Transduced cells were selected for DsRed fluorescence using FACS Aria (BD Biosciences). These cells were then consecutively transduced with lentiviral vector pLV-tTR-KRAB-red expressing doxycycline-regulated tTR-KRAB repressor (a gift of D. Trono, University of Geneva)27 and with shRNA against Topo IIα (target sequence GGA AAC AGC CAG TAG AGA ATA) in tTR-KRAB-regulated pLLCEP TU6X vector.28,29 The infected cells were selected for GFP fluorescence and subcloned. Individual clones were analyzed for growth inhibition by 200 ng/ml doxycycline; the best-responding clone was designated TOP73. Growth kinetics were determined by plating 2 × 105 cells per P100, adding doxycycline or vehicle 12 h later and counting cells using Z1 cell counter (Beckman Coulter) every 5–6 h, starting 3 h after the addition of doxycycline.
Topo II RNA and protein expression assays.
Topo II mRNA expression was analyzed by QPCR, as described in reference 29, using the following pairs of primers: forward 5′-TGG CTG AGG TTT TGC CTT CT-3′ and reverse 5′-GGC CTT CTA GTT CCA CAC CA-3′ (Topo IIα) and forward 5′-AGG AAA GCA TCT GGC TCT GA-3′ and reverse 5′-TCT GAG GGG AAG ATG TCC AC-3′ (Topo IIβ). Immunoblotting analysis was performed by standard procedures, using rabbit polyclonal antibody against Topo IIβ (SC-13059, Santa Cruz Biotechnology, Inc.) and mouse monoclonal antibody against Topo IIα (Novocastra Laboratories Ltd., 3F6), HRP-conjugated anti-rabbit antibody (Amersham Biosciences, NA934V) and R-Phycoerythrin-conjugated goat anti-mouse IgG (Sigma-Aldrich).
Flow cytometric assays.
Flow cytometric analysis was performed using LSRII flow cytometer (BD Biosciences) with FACS Diva acquisition and analysis software. Ethanol-fixed cells were stained for DNA using either 1 µg/ml DAPI or 5 µg/ml propidium iodide (PI). Mitotic cells were detected by staining with mouse monoclonal antibody GF-7 (a gift of Dr. Peter Davies, Albert Einstein College of Medicine) at 1:1,000 dilution, followed by 1:200 dilution of R-Phycoerythrin-conjugated goat anti-mouse IgG (Sigma-Aldrich). For BrdU assays, cells labeled in vivo with 10 µM BrdU for 30 min were fixed with ethanol and stained with anti-BrdU antibody (Bu20a, DAKO Corp.), at 1:100 dilution, followed by Alexa Fluor 488-labeled goat antimouse IgG (Molecular Probes/Invitrogen) at 1:200 dilution. For EdU assays, cells were labeled for 30 min in vivo with 5 µM EdU from Click-iT EdU Alexa Fluor Cell Proliferation Assay kit (Invitrogen). Cells were permeabilized and labeled with Click-iT reaction mixture per the manufacturer's instructions. The labeled cells were stained with DAPI and with GF-7 antibody, followed by Alexa Fluor 633-labeled goat anti-mouse IgG (Invitrogen).
Microscopic assays.
Confocal fluorescence microscopy was performed using Zeiss Axiovert 200 with the LSM 510 META detector and LSM 510 software. For immunofluorescence assays, methanol-fixed cells were stained with α-tubulin mouse monoclonal antibody (Sigma-Aldrich, B-5-1-2) (1:200) and Alexa Fluor 488-labeled goat anti-mouse antibody (Invitrogen) (1:200), tetramethyl rhodamine isothiocyanate (TRITC)-conjugated γ-tubulin goat antibody (Santa Cruz Biotechnology, SC-7396) and DAPI. For γH2AX labeling, cells were incubated with 1:100 γH2AX rabbit polyclonal antibody (Sigma-Aldrich, H5912 or Trevigen, 4411pc-100) (1:100) and Alexa Fluor 594-conjugated goat anti-rabbit antibody (Invitrogen) (1:200). Chromosome spreads were prepared by collecting mitotic cells through mitotic shake-off and fixing the cells in cold methanol:glacial acetic acid (4:1). Pellets were collected by centrifugation, washed, resuspended in methanol:acetic acid and dropped onto microscope slides from 10″ height. Slides were dried and treated with mounting media Vectashield (Vector Laboratories) containing 0.05 µg/µl PI and 1 mg/ml RNase A. GF-7 monoclonal antibody staining was detected using an HRP-linked horse anti-mouse IgG (Cell Signaling Technology, 7076) with DAPI counterstaining. Time-lapse video microscopy of live cells was done using Leica DM IRE2 inverted microscopes in a climate-controlled 37°C room using GIBCO™ CO2-independent culture medium (Invitrogen, 18045). The microscopes interfaced with personal computers running Leica's FW4000 program, which controlled the camera (Leica DC350FX) as well as focus position, filter cube, light source and transmitted light intensity. The FW4000 program was used to run acquisition protocols over at least three days and images were taken every 3 min.
Acknowledgments
We thank Dr. John Nitiss for his review of this manuscript and valuable advice; Gregory Hurteau and Ordway Functional Genomics Facility for assistance with QPCR analysis; Swathi Kalurupalle for assistance with some experiments; Dr. Peter Davies for GF-7 antibody, Dr. Didier Trono for tTR-KRAB vector and Dr. Michael Shtutman for helpful discussions. Supported by NIH grants RO1 CA95727 and R01 AG028687 (I.B.R.).
Supplementary Material
References
- 1.Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer. 2009;9:327–337. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holm C, Goto T, Wang JC, Botstein D. DNA topoisomerase II is required at the time of mitosis in yeast. Cell. 1985;41:553–563. doi: 10.1016/S0092-8674(85)80028-3. [DOI] [PubMed] [Google Scholar]
- 3.Uemura T, Ohkura H, Adachi Y, Morino K, Shiozaki K, Yanagida M. DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell. 1987;50:917–925. doi: 10.1016/0092-8674(87)90518-6. [DOI] [PubMed] [Google Scholar]
- 4.Gasser SM, Laroche T, Falquet J. Boy dlT, Laemmli UK. Metaphase chromosome structure. Involvement of topoisomerase II. J Mol Biol. 1986;188:613–629. doi: 10.1016/S0022-2836(86)80010-9. [DOI] [PubMed] [Google Scholar]
- 5.Giménez-Abián JF, Clarke DJ, Devlin J, Giménez-Abián MI, De la Torre C, Johnson RT, et al. Premitotic chromosome individualization in mammalian cells depends on topoisomerase II activity. Chromosoma. 2000;109:235–244. doi: 10.1007/s004120000065. [DOI] [PubMed] [Google Scholar]
- 6.Tapia-Alveal C, Outwin EA, Trempolec N, Dziadkowiec D, Murray JM, O'Connell MJ. SMC complexes and topoisomerase II work together so that sister chromatids can work apart. Cell Cycle. 2010;9:2065–2070. doi: 10.4161/cc.9.11.11734. [DOI] [PubMed] [Google Scholar]
- 7.Baxter J, Diffley JF. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell. 2008;30:790–802. doi: 10.1016/j.molcel.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Wang Y, Liu X. Plk1-dependent phosphorylation regulates functions of DNA topoisomerase IIalpha in cell cycle progression. J Biol Chem. 2008;283:6209–6221. doi: 10.1074/jbc.M709007200. [DOI] [PubMed] [Google Scholar]
- 9.Downes CS, Clarke DJ, Mullinger AM, Gimenez-Abian JF, Creighton AM, Johnson RT. A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature. 1994;372:467–470. doi: 10.1038/372467a0. [DOI] [PubMed] [Google Scholar]
- 10.Deming PB, Cistulli CA, Zhao H, Graves PR, Piwnica-Worms H, Paules RS, et al. The human decatenation checkpoint. Proc Natl Acad Sci USA. 2001;98:12044–12049. doi: 10.1073/pnas.221430898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skoufias DA, Lacroix FB, Andreassen PR, Wilson L, Margolis RL. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol Cell. 2004;15:977–990. doi: 10.1016/j.molcel.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 12.Luo K, Yuan J, Chen J, Lou Z. Topoisomerase IIalpha controls the decatenation checkpoint. Nat Cell Biol. 2009;11:204–210. doi: 10.1038/ncb1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson M, Phua HH, Bennett SC, Spence JM, Farr CJ. Studying vertebrate topoisomerase 2 function using a conditional knockdown system in DT40 cells. Nucleic Acids Res. 2009;37:98. doi: 10.1093/nar/gkp480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bower JJ, Karaca GF, Zhou Y, Simpson DA, Cordeiro-Stone M, Kaufmann WK. Topoisomerase IIalpha maintains genomic stability through decatenation G(2) checkpoint signaling. Oncogene. 2010;29:4787–4799. doi: 10.1038/onc.2010.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen AK, Escargueil AE, Skladanowski A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther. 2003;99:167–181. doi: 10.1016/S0163-7258(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 17.Hawtin RE, Stockett DE, Wong OK, Lundin C, Helleday T, Fox JA. Homologous recombination repair is essential for repair of vosaroxin-induced DNA double-strand breaks. Oncotarget. 2010;1:606–619. doi: 10.18632/oncotarget.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang KC, Gao H, Yamasaki EF, Grabowski DR, Liu S, Shen LL, et al. Topoisomerase II poisoning by ICRF-193. J Biol Chem. 2001;276:44488–44494. doi: 10.1074/jbc.M104383200. [DOI] [PubMed] [Google Scholar]
- 19.Mikhailov A, Shinohara M, Rieder CL. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J Cell Biol. 2004;166:517–526. doi: 10.1083/jcb.200405167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carpenter AJ, Porter AC. Construction, characterization and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol Biol Cell. 2004;15:5700–5711. doi: 10.1091/mbc.E04-08-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakaguchi A, Kikuchi A. Functional compatibility between isoformalpha and beta of type II DNA topoisomerase. J Cell Sci. 2004;117:1047–1054. doi: 10.1242/jcs.00977. [DOI] [PubMed] [Google Scholar]
- 22.Toyoda Y, Yanagida M. Coordinated requirements of human topo II and cohesin for metaphase centromere alignment under Mad2-dependent spindle checkpoint surveillance. Mol Biol Cell. 2006;17:2287–2302. doi: 10.1091/mbc.E05-11-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ossovskaya VS, Mazo IA, Chernov MV, Chernova OB, Strezoska Z, Kondratov R, et al. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc Natl Acad Sci USA. 1996;93:10309–10314. doi: 10.1073/pnas.93.19.10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–3767. [PubMed] [Google Scholar]
- 25.Broude EV, Swift ME, Vivo C, Chang BD, Davis BM, Kalurupalle S, et al. p21(Waf1/Cip1/Sdi1) mediates retinoblastoma protein degradation. Oncogene. 2007;26:6954–6958. doi: 10.1038/sj.onc.1210516. [DOI] [PubMed] [Google Scholar]
- 26.Broude EV, Demidenko ZN, Vivo C, Swift ME, Davis BM, Blagosklonny MV, et al. p21 (CDKN1A) is a negative regulator of p53 stability. Cell Cycle. 2007;6:1468–1471. doi: 10.4161/cc.6.12.4313. [DOI] [PubMed] [Google Scholar]
- 27.Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated druginducible RNA interference. J Virol. 2003;77:8957–8961. doi: 10.1128/JVI.77.16.8957-1.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maliyekkel A, Davis BA, Roninson IB. Cell cycle arrest drastically extends the duration of gene silencing after transient expression of short hairpin RNA. Cell Cycle. 2006;5:2390–2395. doi: 10.4161/cc.5.20.3363. [DOI] [PubMed] [Google Scholar]
- 29.Shtutman M, Maliyekkel A, Shao Y, Carmack CS, Baig M, Warholic N, et al. Function-based gene identification using enzymatically generated normalized shRNA library and massive parallel sequencing. Proc Natl Acad Sci USA. 2010;107:7377–7382. doi: 10.1073/pnas.1003055107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buck JW, Dong W, Mueller DS. Effect of light exposure on in vitro germination and germ tube growth of eight species of rust fungi. Mycologia. 2010;102:1134–1140. doi: 10.3852/09-283. [DOI] [PubMed] [Google Scholar]
- 32.Rundle NT, Xu L, Andersen RJ, Roberge M. G2 DNA damage checkpoint inhibition and antimitotic activity of 13-hydroxy-15-oxozoapatlin. J Biol Chem. 2001;276:48231–48236. doi: 10.1074/jbc.M103990200. [DOI] [PubMed] [Google Scholar]
- 33.Ishida R, Sato M, Narita T, Utsumi KR, Nishimoto T, Morita T, et al. Inhibition of DNA topoisomerase II by ICRF-193 induces polyploidization by uncoupling chromosome dynamics from other cell cycle events. J Cell Biol. 1994;126:1341–1351. doi: 10.1083/jcb.126.6.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwai M, Hara A, Andoh T, Ishida R. ICRF-193, a catalytic inhibitor of DNA topoisomerase II, delays the cell cycle progression from metaphase, but not from anaphase to the G1 phase in mammalian cells. FEBS Lett. 1997;406:267–270. doi: 10.1016/S0014-5793(97)00282-2. [DOI] [PubMed] [Google Scholar]
- 35.Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J. 2010;29:1272–1284. doi: 10.1038/emboj.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Broude EV, Loncarek J, Wada I, Cole K, Hanko C, Swift M, et al. Mitotic catastrophe in cancer therapy in Beyond Apoptosis: Cellular outcomes of cancer therapy. In: Roninson IB, Brown JM, Bredesen DE, editors. Informa Healthcare. 2008. pp. 307–320. [Google Scholar]
- 37.Tao Y, Leteur C, Calderaro J, Girdler F, Zhang P, Frascogna V, et al. The aurora B kinase inhibitor AZD1152 sensitizes cancer cells to fractionated irradiation and induces mitotic catastrophe. Cell Cycle. 2009;8:3172–3181. doi: 10.4161/cc.8.19.9729. [DOI] [PubMed] [Google Scholar]
- 38.Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, et al. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–4818. doi: 10.1038/sj.onc.1203078. [DOI] [PubMed] [Google Scholar]
- 39.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.