

Abstract
The ability of cells to respond and repair DNA damage is fundamental for the maintenance of genomic integrity. Ex vivo culturing of surgery-derived human tissues has provided a significant advancement to assess DNA damage response (DDR) in the context of normal cytoarchitecture in a non-proliferating tissue. Here, we assess the dependency of prostate epithelium DDR on ATM and DNA-PKcs, the major kinases responsible for damage detection and repair by nonhomologous end-joining (NHEJ), respectively. DNA damage was caused by ionizing radiation (IR) and cytotoxic drugs, cultured tissues were treated with ATM and DNA-PK inhibitors, and DDR was assessed by phosphorylation of ATM and its targets H2AX and KAP1, a heterochromatin binding protein. Phosphorylation of H2AX and KAP1 was fast, transient and fully dependent on ATM, but these responses were moderate in luminal cells. In contrast, DNA-PKcs was phosphorylated in both luminal and basal cells, suggesting that DNA-PK-dependent repair was also activated in the luminal cells despite the diminished H2AX and KAP1 responses. These results indicate that prostate epithelial cell types have constitutively dissimilar responses to DNA damage. We correlate the altered damage response to the differential chromatin state of the cells. These findings are relevant in understanding how the epithelium senses and responds to DNA damage.
Key words: DNA damage, prostate, γH2AX, ATM, DNA-PK
Introduction
Preservation of genomic integrity is vital for multicellular organisms. Extrinsic and intrinsic DNA damage constantly challenge the cellular environment. These insults activate DNA damage signaling and repair cascades that are highly conserved and couple with other major DNA metabolic activities, transcription, replication and chromatin organization.1,2 Defects in DNA damage response (DDR) and repair contribute to aging, developmental disorders, neurodegenerative diseases and cancer.1,3 Failures of DDR and repair lead to accumulation of DNA lesions, predisposition to cancer and are the underlying cause in most hereditary cancer syndromes.4,5
DDR is a signal transduction pathway initiated by damage detection by phosphatidylinositol-3-kinase-like protein kinases (PIKK), ATM and DNA-dependent protein kinase catalytic subunit (DNA-PKcs).2,6 Both are activated by autophosphorylation, and whereas ATM has hundreds of downstream targets,7 DNA-PKcs has a more limited number of proposed substrates, including damage signaling molecules and proteins involved in nonhomologous end-joining (NHEJ).6 ATM, through its kinase activity, orchestrates the assembly of key downstream targets, among those, H2AX and MDC1, that are responsible for the DNA damage foci formation, and it increases chromatin accessibility and recruitment of repair complexes.8,9 Phosphorylation of H2AX on serine 139 (γH2AX) is one of the most rapid and sensitive mark for the presence of DNA double-strand (ds) breaks.10 γH2AX serves as a platform for the signal amplification and is essential for the maintenance of genomic integrity and repair.11 In cultured cells, γH2AX is phosphorylated within minutes, peaks within an hour and declines thereafter, presumably at the rate of repair, i.e., dissolution of the breaks. ATM, DNA-PKcs as well as ATR, as a consequence of replication stress, phosphorylate H2AX.11–14 In addition to phosphorylation and dephosphorylation,15–17 H2AX is subject to acetylation and ubiquitination to multiple sites, both constitutively and in response to DNA damage.10,17 In particular, γH2AX is a sensitive and feasible marker for the presence of genotoxic stress.14,18
DNA breaks cause extensive local and global chromatin changes. These include chromatin unwinding to increase accessibility in the immediate vicinity of the broken DNA ends to allow recruitment of the large repair complexes, resolution of the potential transcriptional and replication machineries potentially colliding with the damaged area and restoration of the chromatin following successful repair.8,19 Therefore, the higher order of chromatin structure and condensation will likely impact how the damage is detected, repaired and resolved.20 In fact, ATM substrates include heterochromatin proteins (HP) 21,22 and KAP1 that bind to histone repressive marks and maintain condensed chromatin states.23,24 Their damage-dependent release from chromatin is considered a feature relevant in facilitating repair.21,25 The heterochromatin marks also include histone marks such as histone 3 (H3) lysine 9 di- and trimethylation (K9me2, K9me3, respectively) and H3 lysine 27 methylation (K27me).26 The chromatin marks vary within single cells and between cell types. A high level of chromatin condensation is associated with cell differentiation, whereas actively replicating cells maintain open chromatin. This distinction is relevant, as the degree of chromatin condensation relates to damage foci formation27,28 and the strength of the response.20
DNA damage may be sensed and repaired differently in post-mitotic differentiated cells. Nucleotide excision repair is attenuated in neurons, adipocytes and keratinocytes,29 and base excision repair is impaired in myotubes as compared with myoblasts,30 whereas NHEJ is upregulated during adipocyte differentiation.31 In this regard, the prostate represents a relevant model to address DDR responses, as it has a high frequency of malignant conversion and a tendency to form multifocal tumors.32,33 The prostate gland consists of two epithelial cell layers, basal and luminal, surrounded by fibromuscular stroma and occasional neuroendocrine cells.32 The basal and luminal cells are distinct in terms of their expression of differentiation markers, response to hormonal and growth stimuli and function.32 While controversy still exists as to the cell type origin of prostate cancer, ample evidence ascribes this to the luminal cells.34 We have earlier shown that DDR of non-proliferating human prostate epithelium is exceptional due decreased responsiveness of the ubiquitous DNA damage mark, γH2AX, to ds DNA breaks.35 Specifically, its absence was evident in the prostate luminal cells and correlated with low expression of the H2AX histone subtype.35 However, the basal cell responses were consistent with a fully active DDR cascade. Here, we have analyzed the upstream pathways that regulate γH2AX activity and addressed the correlation of the DDR response to chromatin marks. We show that the H2AX and KAP1 phosphorylation responses to ds DNA breaks are rapid, transient and fully dependent on ATM activity. Consistent with the attenuated H2AX response in the luminal cells, we find that the phosphorylation of KAP1 is also lower in the luminal than in the basal cells. We show that basal and luminal cells differ in their staining of certain chromatin condensation and histone modification marks. These findings indicate that differentiated human prostate cells have distinct DDR responses and chromatin states, which may impact how they respond to damage.
Results
DNA damage response to IR is rapid, ATM-dependent and epithelial cell type-specific. We have earlier shown that human prostate tissues respond to IR and cytotoxic drugs by activating DDR within 4 h of the damage, but that the luminal compartment displays attenuated γH2AX response.35 To assess the rapid kinetic response, we radiated ex vivo cultured prostate tissues and measured the imminent activation and consequent resolution of DDR based on phosphorylation of DDR proteins. Tissues were irradiated with 4 Gy and stained for Ser1981-phosphorylated ATM (phospho-ATM), γH2AX and Ser824-phosphorylated KAP1 (phospho-KAP1). KAP1 is a heterochromatin-binding protein and a direct ATM target, the phosphorylation of which affects chromatin condensation and repair.23,25 Signals were quantified using FrIDA image analysis software.36 Peak numbers of cells with phosphorylated ATM, H2AX and KAP1 were detected within 1–2 h and declined rapidly (Fig. 1A–C). By 18 h, these damage signals had ceased. Strikingly, and in accordance to our previous observations,35 γH2AX response was attenuated in the luminal epithelium even at its expected peak induction time despite robust activation of phospho-ATM (Fig. 1B). Here, we also show that phosphorylation of KAP1 was less prominent in the luminal cells as compared with basal cells (Fig. 1C). That the highly intense phospho-KAP1-positive cells were predominantly basal cell type was confirmed by dual staining for phospho-KAP1 and basal cell maker p63 (Fig. 1E). Quantitative image analysis of PKAP1 intensities in p63-positive (basal) cells and p63-negative (luminal and stromal) cells indicated higher phospho-KAP1 staining intensity in the p63-expressing basal cells. It should be noted that the analysis of p63-negative cells also includes stromal cells, which are abundantly positive for phospho-KAP1, thus underestimating the difference between the epithelial cell types (Fig. 1E). Whereas the difference in the luminal γH2AX response can be attributed to lower levels of total H2AX protein, as shown by our previous work in reference 35, KAP1 was equally expressed in all cells (Fig. 1D). This indicated that the activation of two ATM-regulated DDR proteins is attenuated in the luminal cells.
Figure 1.

Kinetics of prostate epithelium DNA damage response. Human prostate ex vivo cultured tissues were radiated with 4 Gy and incubated for the indicated times. Tissues were stained for (A) phospho-ATM (PATM), (B) γH2AX, (C) phospho-KAP1 (PKAP1) and (D) total KAP1. Basal (B) and luminal (L) compartments are indicated. Percentages of positive cells are shown to the right. (e) Confocal imaging for phospho-KAP1 (green) and p63 (red) of tissues either mock-treated (ctrl) or treated with Ir and incubated for 1 h. Merge, merged image for green, red and blue (DNA) channels. Scale bars, 50 µm. Mean nuclear intensities in p63-negative and p63-positive cells are shown to the right.
ATM, ATR and DNA-PKcs are all kinases that phosphorylate H2AX.11–14 To address whether the DDR response is dependent on the activation of ATM kinase, we used a potent and selective ATM kinase inhibitor (ATMi) KU5593337 to pretreat the tissues prior to radiation. As shown in Figure 2A–C and quantitative image analysis, pretreatment with ATMi reduced phospho-ATM activation by IR and distinctly inhibited the phosphorylation of H2AX and KAP1. Interestingly, even at later timepoints (up to 24 h), there was no compensatory activation of H2AX or KAP1 phosphorylation by other kinases (not shown).
Figure 2.

ATM-dependency of Ir-activated DNA damage responses. Tissues were pretreated with KU55933 (ATMi, 20 µM) for 1 h followed by radiation (4 Gy) and incubation of the tissues for 1 h. Tissues were stained for (A) phospho-ATM (PATM), (B) γH2AX and (C) phospho-KAP1 (PKAP1). Basal (B) and luminal (L) compartments are indicated. Percentages of positive cells are shown to the right. Error bars, SD. Scale bar, 50 µm.
Topoisomerase II (TOP2) inhibitor-activated DNA damage response is dependent on ATM.
We have previously shown that TOP2 poisons do not effectively activate luminal cell γH2AX response.35 We hence detected the prostate phospho-KAP1 responses and assayed for the dependency of γH2AX and phospho-KAP1 responses on ATM. Prostate tissues were treated with TOP2 poisons daunomycin and etoposide. As shown in Figure 3, daunomycin and etoposide activated phospho-ATM, γH2AX and phospho-KAP1 responses. These responses were dependent on ATM activity, as shown by absence of their activation in the ATMi-pretreated tissues and quantitative image analysis (Fig. 3). However, the luminal cell H2AX and KAP1 responses were largely absent (Fig. 3B and C). This indicated that TOP2 poisons, which lead to formation of covalent TOP2-DNA complexes, DNA strand breaks and formation of protein-DNA adducts, also act selectively through activation of ATM-dependent DDR in the prostate tissue.
Figure 3.

TOP2 inhibitors activate ATM-dependent DNA damage responses. Tissues were pretreated with KU55933 (ATMi, 20 µM) for 1 h followed by treatment of the tissues with daunomycin (DAU, 2 µM) and etoposide (ETO, 40 µm) for 6 h. Tissues were stained for (A) phospho-ATM (PATM), (B) γH2AX and (C) phospho-KAP1 (PKAP1). Basal (B) and luminal (L) compartments are indicated. Percentages of positive cells are shown to the right. Error bars, SD. Scale bar, 50 µm.
Inhibition of DNA-PKcs activity prolongs DDR signaling.
IR-induced damage repair in non-proliferative cells takes place predominantly by NHEJ and depends on DNA-PK activity.38 DNA-PKcs is autophosphorylated to multiple sites in response to DNA damage and is a prerequisite for NHEJ.6,38 To address whether the absence of the luminal DDR response affects the activation of repair, we assessed phospho-DNA-PKcs response to IR. DNA-PKcs was phosphorylated in the epithelial cells within 6 h of IR (10 Gy) (Fig. 4A). Interestingly, although the phospho-ATM response had ceased by 18 h, the phospho-DNA-PKcs response lasted for at least 30 h, suggesting either continuous activity or a very slow switch-off of the phosphorylation signals (Fig. 4A). Similar results were obtained with 4 Gy IR (not shown).
Figure 4.

DNA-PK activation by irradiation is inhibited by NU7441. (A) Prostate ex vivo tissues were radiated with 10 Gy and incubated for the indicated times. Tissues were stained for phospho-DNA-PK. (B) tissues were pretreated with NU7441 (DNAPKi, 10 µM) for 1 h followed by radiation (10 Gy) and incubation of the tissues for 6 h. Tissues were stained for phospho-DNA-PK (P-DNAPK) and DNA-PK. Percentages of positive cells are shown to the right. Error bars, SD. Scale bars, 50 µm.
We next used a specific inhibitor of DNA-PK, NU7441 to block DNA-PK-dependent damage sensing and repair.39 This allowed us to test whether any of DDR markers depend on DNA-PK. On the other hand, quenching of DNA repair is predicted to lead to persistent DNA damage signaling.40 Therefore, this approach facilitated testing whether the DDR markers are sustained and whether luminal cell DDR signaling could be amplified if DNA-PK activity is switched off. We first confirmed that pretreatment of the tissues with NU7441 attenuated the phospho-DNA-PKcs response to IR, as both the number of positive cells and signal intensity decreased (Fig. 4B).41 However, there was no change in total DNA-PK (Fig. 4B). We then followed the IR-induced DDR responses over time. At 6 h after IR (10 Gy), the phosphorylation of ATM, H2AX and KAP1 in the NU7441-preteated cells was largely unaffected indicating that NU7441 does not affect the ATM-mediated responses. However, strikingly, NU7441 caused substantial activation of all DDR responses at 24 h (Fig. 5A–C). The sustained phosphorylation of ATM, H2AX and KAP1 was indicative of presence of persistent damage signaling.
Figure 5.
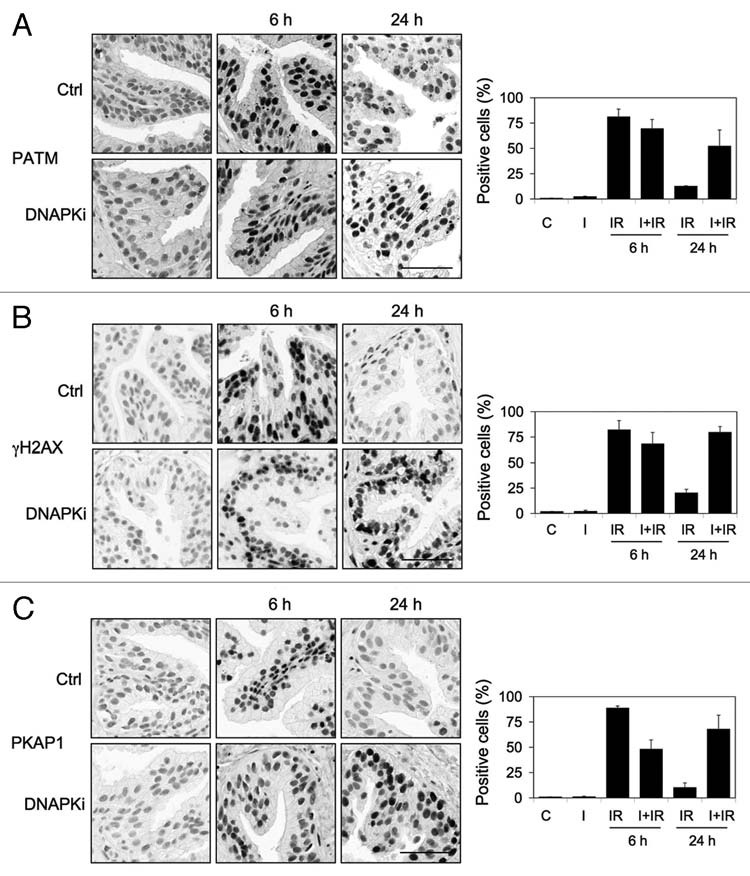
Inhibition of DNA-PK leads to sustained DNA damage signaling following irradiation. Tissues were pretreated with NU7441 (DNAPKi; I, 10 µM) for 1 h followed by radiation (10 Gy) and incubation of the tissues for 6 and 24 h. Tissues were stained for (A) phospho-ATM (PATM), (B) γH2AX and (C) phospho-KAP1 (PKAP1). Percentages of positive cells are shown to the right. Error bars, SD. Scale bars, 50 µm.
Luminal and basal prostate epithelial cells differ in their chromatin marks.
DDR and DNA repair involve extensive remodeling of the chromatin. To address whether the differential responses observed in the luminal and basal compartments could be ascribed to differences in chromatin marks, such as his-tone modifications or expression of heterochromatin proteins, we detected markers of these in the prostate tissue. H3K9 methylation has been associated with differentiated cells,42 constitutive and facultative heterochromatin43,44 but is also a mark for active transcription.45 HP1γ is enriched in heterochromatin but also localizes to euchromatin.46 As shown in Figure 6A and B, basal but not luminal cells generally showed higher levels of staining for H3K9me3, whereas H3K9me2 was present in both. On the other hand, luminal cells, but not basal cells, highly expressed H3K27me3 (Fig. 6C). In addition, staining for HP1γ was more prominent in p27-expressing luminal cells (Fig. 6D). On the other hand, HP1β staining, tested in parallel, was weak and did not suggest distinctive differences between the basal and luminal compartments (not shown). These findings suggested that the basal and luminal cell types differed in their nature of chromatin marks relevant in chromatin architecture.
Figure 6.

Chromatin states of basal and luminal cells are different. Prostate tissues were stained for (A) dimethylated H3K9 (H3K9me2) and (B) trimethylated H3K9 (H3K9me3), (C) H3K27me3 and (D) HP1γ (green), p27 (red) and merge (HP1γ, p27, DNA). B and L, basal and luminal epithelial layers, respectively. Scale bar, 100 µm. Insets, 1.7- and 2-fold enlarged sections for (A–C) and (D), respectively.
Discussion
Despite the extensive number of studies of DNA damage signaling and repair in cell and animal models, few models exist that address how human tissues in the physiological context and normal cytoarchitecture respond to external DNA damage. Ex vivo cultures of surgery-derived prostate tissue are amenable for experimentation with drug treatment and radiation and assessment of the damage responses, e.g., by the use of marker proteins. A specific feature of the prostate tissue is that the histologically normal sections used here and in our previous study contain cells with very low proliferative capacity. The cells express distinct differentiation markers specific for the basal and luminal (secretory) epithelium, and therefore, compartment-specific responses can be identified.35 Hence, the prostate epithelium represents highly differentiated, largely postmitotic cells, which, by default, are expected to undergo repair by the NHEJ pathway.
We show here that the DDR response, as measured by phosphorylation of ATM, H2AX and KAP1, is fast and is resolved with kinetics consistent with proficient repair. These responses are abrogated by a specific inhibitor of ATM, indicating that in normal epithelium, ATM represents the main PIKK responsible for DDR activation. However, the luminal cell responses differed. We have shown that while ATM is activated in both cell types, the H2AX response is negligible in the luminal cells.35 Here, we recapitulate similar findings at the peak activation of ATM and show also that phosphorylation of KAP1 in response to IR is less prominent, despite that the protein was expressed at comparable levels in all epithelial layers. We also found that all prostate compartments express high levels of DNA-PKcs, and that it undergoes rapid and sustained phosphorylation consequent to IR. Inhibition of DNA-PK led to prolonged expression of DDR markers, suggesting that inhibition of repair maintains the damage signaling. However, inhibition of DNA-PK did not activate γH2AX and phospho-KAP1 responses in the luminal compartment to a similar degree as in the basal compartment. This indicated that the luminal and basal cells differ in their capacity to activate certain DDR responses.
Only a few examples exist of cell types that do not display a profound γH2AX response to DNA damage by IR. These include neutrophils and differentiated layers of artificial skin cells.47 In general, many differentiated cells, like neurons, adipocytes and erythrocytes, have decreased repair,29,30 suggesting that in cell types with short lifespan or cells incapable of entering proliferative cycles, the preservation of genomic integrity is no longer a priority, or that the pathways to do so are distinct. The latter may be the case in the prostate. It can be argued that damage repair is a non-essential function in terminally differentiated cells, as the progeny that would distribute errors is non-existent. However, in terms of preserving the functionality of transcriptionally active programs, the integrity of such genes still predisposes a challenge and stresses the need for adequate damage detection and repair. Moreover, under constant genomic damage, the risk of accruing multiple lesions causing dedifferentiation and replicative re-entry increases. Hence, the ability of cancer-prone organs, such as the human prostate, to respond to genetic damage is of considerable interest.
We find that the luminal and basal epithelia contain distinct chromatin marks. Basal cells showed higher levels of staining for trimethylated H3K9, while luminal cells showed higher levels of staining for HP1γ and H3K27me3. These are considered heterochromatin marks, suggesting that such marks may also be specific to cells at different differentiation stages. HP1 proteins are expressed during prostate development in adult prostates but generally decreased in prostate cancer.48 Chromatin condensation is relevant for the DDR. Recent studies have shown that phosphorylation of H2AX is affected by gene transcription49 and is more restricted in heterochromatin areas, occurring preferentially in open chromatin (euchromatin, reviewed in ref. 28).20,27 This indicates that assembly of DDR foci and signaling to repair complexes is dependent on chromatin relaxation and more restricted in compacted chromatin areas. The finding that the luminal cells display less prominent δH2AX and KAP1 phosphorylation is consistent with this notion and suggests that cells with high expression of certain heterochromatin marks may not be as amenable for DDR signal propagation, or that the signals are selective. High expression of HP1? in the luminal cells is indicative that their chromatin is condensed and therefore may be less accessible to modification and repair.
Capability to complete successful repair depends on seamless upstream signaling, but also on the levels of the signaling and repair molecules. Variations in their levels with respect to cell cycle, cell type or genetic alterations may severely compromise the repair capability of the cells. Here, we show that these variations relate also to differentiated cells and may depend on the chromatin condensation marks. Although this model does not allow us to assay conclusively whether appropriate repair has been accomplished, the finding that following IR the rate of resolution of ATM phosphorylation in the basal and luminal cells is equal suggests that breaks are being repaired at similar kinetics. Also, luminal cells are proficient in formation of 53BP1 foci, indicating that at least some ATM downstream targets are activated.35 In addition, DNA-PKcs phosphorylation is similar in both compartments, suggesting that relevant repair complexes may be activated irrespective of the unusual low activation of DDR markers.
Our findings suggest that differentiated cells may have less stringent requirements for a fully proficient DDR as compared with replicating cells challenged with checkpoint control and management of the replicative processes. In other words, the requirement of the full set of ATM responses may be lower in differentiated as compared with replicating cells and may depend on the extent of damage. Ultimately, whether these are sufficient to promote preservation of genomic integrity will determine the susceptibility of the differentiated cells to cancer.
Materials and Methods
Ex vivo tissue culture.
Prostate tissues were obtained with approval of the Johns Hopkins Institutional Review Board (#NA00015481). Radical prostatectomy specimens were cored at the unaffected lobe and sliced at 300 µm with a Krumdieck precision tissue slicer (Alabama Research and Development Corporation). The tissue slices were placed onto titanium grids containing RPMF-4A medium supplemented with growth factors and R1881 androgen analog and rotated on an inclined plane in a humidified tissue culture incubator at 37°C.35,50 Ionizing radiation was conducted using Gammacell 40 137Cs irradiator at 0.67 Gy/min (Atomic Energy Commission of Canada Ltd.). Daunomycin and etoposide were obtained from Sigma-Aldrich. KU55933 and NU7441 were from Tocris. Following treatments, tissue slices were fixed with 10% formalin, embedded in paraffin and cut to 4 µm.
Immunohistochemistry.
Tissue sections were deparaffinized, rehydrated, and antigen retrieval was conducted in citrate buffer at pH 6.1 (Dako) or EDTA (H3K27me3) using a steamer for 45 min. Slides were blocked with 3% hydrogen peroxide and incubated with primary antibodies for 45 min at room temperature or overnight at 4°C. The slides were stained with PowerVision detection system (Leica Biosystems) for 30 min at room temperature, followed by development with DAB solution (Invitrogen). Nuclei were counterstained with hemotoxylin, and slides were visualized under brightfield microscope (Nikon Eclipse 50i) and imaged using Nikon DS-Fi1 color CCD camera. The following primary antibodies and dilutions were used: phospho-ATM (1:8,000, Cell Signaling Technology), γH2AX (1:4,000, Millipore), KAP1 (1:8,000, BD Transduction Laboratories), phospho-KAP1 (1:2,000, Bethyl Laboratories), DNA-PKcs (1:800, Epitomics), phospho-DNA-PKcs (1:800, Abcam), H3K9me2 and H3K9me3 (1:500, Cell Signaling Technology), H3K27me3 (1:200, Abcam). Immunofluorescence staining was conducted using anti-p27 (1:100, Santa Cruz Biotechnology), anti-p63 (1:100, Neomarkers) and anti-HP1γ (Millipore) antibodies. Secondary anti-mouse and anti-rabbit antibodies used were Alexa-488 and Alexa-594 (Molecular Probes, Invitrogen) and counterstaining was with Hoechst 33,342 (Molecular Probes). Confocal images were acquired using Zeiss LSM 510 META confocal microscope and LSM software release 3.2.
Image analysis.
Image analysis of IHC specimens was conducted using FrIDA designed for the analysis of RGB color image data sets.36 Hue saturation and brightness ranges for DAB (brown) and hematoxylin alone (blue) were defined for each image set. The fraction of positive cells was defined as the DAB area divided by the sum of the DAB area and the hematoxylin area. Thresholds for automated image analysis were set such that any brown staining within nuclei detected visually was included. An average of 900 cells was quantified from three fields. Colocalization analysis for immunofluorescence images was conducted using Cell Image Classification and Segmentation analysis program as in reference 35.
Acknowledgments
We thank Helen Fedor and others from the Brady Urological Research Institute Prostate Specimen Repository, which is funded in part by the NIH/NCI SPORE in prostate cancer (P50 CA58236) for specimen collection and members of Laiho labs in Helsinki and Hopkins for discussion. Source of funding: Patrick C. Walsh Prostate Cancer Research Fund (M.L.), Academy of Finland (grant no. 129699, M.L.; 126827, T.Ma.H.). S.J. has received support by Finnish Cancer Organization and Helsinki Biomedical Graduate School.
Abbreviations
- DDR
DNA damage response
- HP
heterochromatin protein
- IR
ionizing radiation
- NHEJ
nonhomologous end-joining
- PIKK
phosphatidylinositol-3-kinase-like protein kinase
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seviour EG, Lin SY. The DNA damage response: Balancing the scale between cancer and ageing. Aging. 2010;2:900–907. doi: 10.18632/aging.100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 5.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 6.Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 7.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 8.Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol. 2009;10:243–254. doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 11.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, et al. gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010;9:662–669. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 15.Moon SH, Nguyen TA, Darlington Y, Lu X, Donehower LA. Dephosphorylation of γH2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control. Cell Cycle. 2010;9:2092–2096. doi: 10.4161/cc.9.11.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mac rek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010;29:2281–2291. doi: 10.1038/onc.2009.501. [DOI] [PubMed] [Google Scholar]
- 17.Xie A, Odate S, Chandramouly G, Scully R. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle. 2010;9:3602–3610. doi: 10.4161/cc.9.17.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Redon CE, Nakamura AJ, Martin OA, Parekh PR, Weyemi US, Bonner WM. Recent developments in the use of γH2AX as a quantitative DNA double- strand break biomarker. Aging. 2011;3:168–174. doi: 10.18632/aging.100284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 20.Murga M, Jaco I, Fan Y, Soria R, Martinez-Pastor B, Cuadrado M, et al. Global chromatin compaction limits the strength of the DNA damage response. J Cell Biol. 2007;178:1101–1108. doi: 10.1083/jcb.200704140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- 22.Dinant C, Luijsterburg MS. The emerging role of HP1 in the DNA damage response. Mol Cell Biol. 2009;29:6335–6340. doi: 10.1128/MCB.01048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 24.Noon AT, Shibata A, Rief N, Löbrich M, Stewart GS, Jeggo PA, et al. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol. 2010;12:177–184. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- 25.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Löbrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 26.Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- 27.Cowell IG, Sunter NJ, Singh PB, Austin CA, Durkacz BW, Tilby MJ. gammaH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS ONE. 2007;2:1057. doi: 10.1371/journal.pone.0001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodarzi AA, Jeggo P, Lobrich M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair (Amst) 2010;9:1273–1282. doi: 10.1016/j.dnarep.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Nouspikel T. DNA repair in differentiated cells: some new answers to old questions. Neuroscience. 2007;145:1213–1221. doi: 10.1016/j.neuroscience.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Fortini P, Dogliotti E. Mechanisms of dealing with DNA damage in terminally differentiated cells. Mutat Res. 2010;685:38–44. doi: 10.1016/j.mrfmmm.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Meulle A, Salles B, Daviaud D, Valet P, Muller C. Positive regulation of DNA double strand break repair activity during differentiation of long life span cells: the example of adipogenesis. PLoS ONE. 2008;3:3345. doi: 10.1371/journal.pone.0003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14:2410–2434. doi: 10.1101/gad.819500. [DOI] [PubMed] [Google Scholar]
- 33.Kiviharju-af Hällström TM, Laiho M. Genetic changes and DNA damage responses in the prostate. Prostate. 2008;68:902–918. doi: 10.1002/pros.20746. [DOI] [PubMed] [Google Scholar]
- 34.De Marzo AM, Nelson WG, Bieberich CJ, Yegnasubramanian S. Prostate cancer: New answers prompt new questions regarding cell of origin. Nat Rev Urol. 2010;7:650–652. doi: 10.1038/nrurol.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jäämaa S, Af Hällström TM, Sankila A, Rantanen V, Koistinen H, Stenman UH, et al. DNA damage recognition via activated ATM and p53 pathway in non-proliferating human prostate tissue. Cancer Res. 2010;70:8630–8641. doi: 10.1158/00085472.CAN-10-0937. [DOI] [PubMed] [Google Scholar]
- 36.Cornish T, Morgan J, Gurel B, De Marzo AM. FrIDA: An open source framework for image dataset analysis. Arch Pathol Lab Med. 2008;132:856. [Google Scholar]
- 37.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN04-2727. [DOI] [PubMed] [Google Scholar]
- 38.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by nonhomologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, et al. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66:5354–5362. doi: 10.1158/0008-5472.CAN05-4275. [DOI] [PubMed] [Google Scholar]
- 40.O'Connor MJ, Martin NM, Smith GC. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene. 2007;26:7816–7824. doi: 10.1038/sj.onc.1210879. [DOI] [PubMed] [Google Scholar]
- 41.White JS, Choi S, Bakkenist CJ. Transient ATM kinase inhibition disrupts DNA damage-induced sister chromatid exchange. Sci Signal. 2010;3:44. doi: 10.1126/scisignal.2000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL. Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell. 2001;107:727–738. doi: 10.1016/S0092-8674(01)00598-0. [DOI] [PubMed] [Google Scholar]
- 44.Peters AH, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AA, Perez-Burgos L, et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell. 2003;12:1577–1589. doi: 10.1016/S10972765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 45.Wiencke JK, Zheng S, Morrison Z, Yeh RF. Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene. 2008;27:2412–2421. doi: 10.1038/sj.onc.1210895. [DOI] [PubMed] [Google Scholar]
- 46.Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol. 2004;5:296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- 47.Redon CE, Dickey JS, Bonner WM, Sedelnikova OA. gamma-H2AX as a biomarker of DNA damage induced by ionizing radiation in human peripheral blood lymphocytes and artificial skin. Adv Space Res. 2009;43:1171–1178. doi: 10.1016/j.asr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shapiro E, Huang H, Ruoff R, Lee P, Tanese N, Logan SK. The heterochromatin protein 1 family is regulated in prostate development and cancer. J Urol. 2008;179:2435–9. doi: 10.1016/j.juro.2008.01.091. [DOI] [PubMed] [Google Scholar]
- 49.Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, et al. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiviharju-af Hällström TM, Jäämaa S, Mönkkönen M, Peltonen K, Andersson LC, Medema RH, et al. Human prostate epithelium lacks Wee1A-mediated DNA damage-induced checkpoint enforcement. Proc Natl Acad Sci USA. 2007;104:7211–7216. doi: 10.1073/pnas.0609299104. [DOI] [PMC free article] [PubMed] [Google Scholar]