

Abstract
Summary: The differences between observed and predicted 13Cα chemical shifts can be used as a sensitive probe with which to detect possible local flaws in protein structures. For this reason, we previously introduced CheShift, a Web server for protein structure validation. Now, we present CheShift-2 in which a graphical user interface is implemented to render such local flaws easily visible. A series of applications to 15 ensembles of conformations illustrate the ability of CheShift-2 to locate the main structural flaws rapidly and accurately on a per-residue basis. Since accuracy plays a central role in CheShift predictions, the treatment of histidine (His) is investigated here by exploring which form of His should be used in CheShift-2.
Availability: CheShift-2 is free of charge for academic use and can be accessed from www.cheshift.com
Contact: has5@cornell.edu; jv84@cornell.edu
Supplementary information: Supplementary data are available at the Bioinformatics online.
1 INTRODUCTION
Chemical shifts provide important information about the conformations of proteins in solution (see, for example, Wishart, 2011, and references therein). For this reason, we developed CheShift-2, a Web server for protein structure validation based on a quantum mechanics database of 13Cα chemical shifts (Vila et al., 2009). CheShift was originally developed to return a list of predicted values of 13Cα chemical shifts. It was the user's responsibility to compare the predicted with the observed 13Cα chemical shifts to assess the global quality of a protein. However, it is a highly desirable goal of any accurate validation method (Nabuurs et al., 2006; Vila and Scheraga, 2009) to identify the existence of local flaws, in addition to the global quality; see analysis of local versus global chemical shift validation of Dynein light chain 2A protein in Supplementary Material. In order to automate and facilitate the validation process on a per-residue basis, we added a GUI to CheShift. The GUI displays the differences between observed and predicted 13Cα chemical shifts by using a four-color code mapped onto a 3D protein model.
A set of 15 proteins was used to test the ability of CheShift-2 to detect local flaws. This set was selected from the Protein Data Bank [(PDB), Berman et al., 2000] and corresponds to obsolete and superseded NMR protein structures. Released PDB data (coordinates and experimental data) are rendered obsolete when the authors have collected new data or had re-refined the structure. The obsolete entry is usually replaced by a new (superseding) entry that receives a new PDB ID.
2 METHODS
For each amino acid μ, it is possible to define the difference between observed and predicted 13Cαchemical-shifts as:
| (1) |
where, 13Cμ,iα is the chemical shift of residue μ in conformation i out of Ω conformations. The average of the predicted chemical shifts over the Ω conformations is evaluated because proteins in solution exist as an ensemble of conformations.
The following procedure for mapping the Δμ values onto a 3D protein model was formulated. First, the Δμ value computed for each residue μ is smoothed by averaging it over the values of the two nearest-neighbor residues (see Supplementary Material for details). Second, the resulting averaged <Δμ> value is discretized according to the following rule:
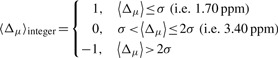 |
(2) |
The selection of the cut-off σ value of 1.7 ppm is explained in the Supplementary Material. Third, the <Δμ>integer values, 1, 0 and −1 are mapped onto a 3D protein model and associated with a color; blue, white and red, respectively. Implicit in this color-code assignment is the assumption that average differences per residue between observed and predicted 13Cα chemical shifts that are within ~1σ (blue) are considered small; within ~2σ (white) they are considered medium, i.e. being both blue and white considered as acceptable differences; and beyond 2σ (red), they are considered large differences and, hence, special attention should be attached to those residues. In addition, the color yellow was adopted to indicate the absence of the observed or computed 13Cα chemical shift value.
3 RESULTS
We found evidence (see Supplementary Material) indicating that the protonated form of histidine (His), rather than the neutral ones, namely, the Nδ1-H or Nε2-H tautomers form, respectively, leads to a better representation of the observed 13Cα chemical shifts. This observation, together with the well-documented effect of proline on the computed chemical shift of the preceding residue (Vila et al., 2010), are now taken into account in CheShift-2 predictions.
Figure 1A shows the color distribution obtained for three ensembles of conformations for the bovine cytochrome B5 protein. The first ensemble of conformations was obtained by NMR spectroscopy (PDB ID 1WDB); most of the flaws (red-colored residues) are located in the helices, which are very distorted. In the year 2003, 1WDB was superseded by 1HKO, also determined by NMR spectroscopy. According to CheShift-2, 1HKO is enriched in blue regions, indicating that it is indeed a better structural model than 1WDB. A third conformation (PDB ID 1CYO) is included for comparison with the previous two NMR-derived conformations. This third conformation was determined by X-ray diffraction at 1.5 Å resolution and is colored almost white/blue indicating that this X-ray model is essentially free of flaws.
Fig. 1.
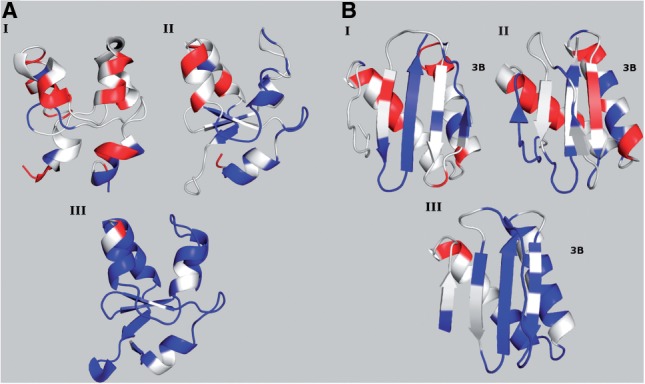
Conformations are colored according to CheShift-2. (A) Three conformations of the bovine cytochrome B5 protein are shown: 1WDB (I), rendered obsolete and replaced by 1HKO (II), both NMR-derived ensembles; and 1CYO (III) an X-ray-derived structure. (B) Three conformations of rabbit 8KDA dynein light chain protein are shown: 1BKQ (I), rendered obsolete and replaced by 1F3C (II), both NMR-derived ensembles; and 1CMI (III) an X-ray-derived structure.
As a second example, we analyzed the rabbit 8KDA dynein light chain protein (Fig. 1B). This protein was first, incorrectly, solved as a monomer (PDB ID 1BKQ), and then as a dimer (PDB ID 1F3C). 1BKQ was rendered obsolete and superseded by 1F3C. The most important difference between 1BKQ and 1F3C is the relative orientation of the third strand (highlighted as 3B in Fig. 1B) with respect to the rest of the protein. Specifically, in the 1BKQ conformation, strand 3B is part of a β-sheet of the monomer, while in 1F3C, strand 3B is part of the β-sheet of a monomer forming a dimer (not shown). Comparison of the color distribution obtained from 1BKQ and 1F3C could mislead the user to conclude that the sheet arrangement of Figure 1B (I) is better than that of Figure1B (II). It should be pointed out that CheShift-2 does not enable one to determine whether a given structure should be a monomer or dimer. The reason for this drawback of the method is due to the fact that 13Cα chemical shifts are a local property and, hence, our validation tool cannot be used to decide as to whether a topological arrangement is correct or not. Therefore, a correct interpretation of Figure 1B is that both structures (1BKQ and 1F3C) need further refinement. In contrast, 1CMI solved as a dimer by X-ray crystallography, at 2.5 Å resolution, is enriched in blue/white regions, confirming that the 1CMI protein is indeed a very good structure [see Fig. 1B (III)].
4 CONCLUSIONS
CheShift-2 constitutes a fast and accurate validation tool with which to determine the existence of local flaws in protein models. Examples analyzed in the present study show that, if the NMR-determined ensemble had not been solved at a high-quality level, a comparison with the corresponding structure determined by X-ray crystallography reveals that the X-ray structure is almost flawless and, hence, indicates that the detected flaws in the NMR-determined ensemble are not a bias of the method but a warning that the NMR-derived structure may benefit from further structural refinement.
This new physics-based validation tool, CheShift-2, should be used as a complementary one to other existing knowledge-based methods, such as WHAT IF (Vriend, 1990) and PROCHECK (Laskowski et al., 1993), or combined knowledge-based and physics-based methods, such as the PSVS package (Huang et al., 2005; Bhattacharya et al., 2007).
Supplementary Material
ACKNOWLEDGEMENTS
We thank P. Serrano at the Scripps Research Institute for valuable discussions regarding protein structure validation methods. The research was conducted by using the resources of Pople, a facility of the NSF Terascale Computing System at the Pittsburgh Supercomputer Center.
Funding: NIH (GM-14312) and NSF (MCB10-19767), USA; CONICET and UNSL (P-328402), Argentina.
Conflict of Interest: none declared.
REFERENCES
- Berman H.M., et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A., et al. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- Huang Y.J., et al. Protein NMR Recall, Precision and F-measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics. J. Am. Chem. Soc. 2005;127:1665–1674. doi: 10.1021/ja047109h. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., et al. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- Nabuurs S.B., et al. Traditional biomolecular structure determination by NMR spectroscopy allows for major errors. PLoS Comp. Biol. 2006;2:71–79. doi: 10.1371/journal.pcbi.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila J.A., Scheraga H.A. Assessing the accuracy of protein structures by quantum mechanical computations of13Cαchemical shifts. Acc. Chem. Res. 2009;42:1545–1553. doi: 10.1021/ar900068s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila J.A., et al. Quantum-mechanics-derived 13C chemical shift server (CheShift) for protein structure validation. Proc. Natl Acad. Sci. 2009;106:16972–16977. doi: 10.1073/pnas.0908833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila J.A., et al. Sequential nearest-neighbor effects on computed13Cαchemical shifts. J. Biomol. NMR. 2010;48:23–30. doi: 10.1007/s10858-010-9435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- Wishart D.S. Interpreting protein chemical shift data. Prog. Nucl. Magn. Reson. Spectrosc. 2011;58:62–67. doi: 10.1016/j.pnmrs.2010.07.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.