

Abstract
Genetic screens performed in worms identified major regulators of the epidermal growth factor receptor (EGFR) pathway, including the ubiquitin ligase Cbl/SLI-1. Here we focus on the less characterized Lst2 protein, and confirm suppression of MAPK signals. Unexpectedly, human Lst2, a monoubiquitinylated phosphoprotein, does not localize to endosomes, despite an intrinsic phosphoinositol-binding FYVE domain. By constructing an ubiquitinylation-defective mutant and an ubiquitin fusion, we conclude that endosomal localization of Lst2, along with an ability to divert incoming EGFR molecules to degradation in lysosomes, is regulated by ubiquitinylation/de-ubiquitinylation cycles. Consistent with bifurcating roles, Lst2 physically binds Trim3/BERP, which interacts with Hrs and a complex that biases cargo recycling. These results establish an ubiquitin-based endosomal switch of receptor sorting, functionally equivalent to the mechanism inactivating Hrs via monoubiquitinylation.
Introduction
To ensure fidelity of signaling outcomes, activated receptor tyrosine kinases (RTKs), such as the epidermal growth factor receptor (EGFR), are subjected to signal desensitizing mechanisms, primarily entailing accelerated endocytosis and degradation in lysosomes (Sorkin and Goh, 2008). Activated receptors concentrate over clathrin-coated pits at the plasma membrane, which invaginate to form coated vesicles. Vesicles then uncoat prior to fusing with tubulo-vesicular organelles, denoted early endosomes (EEs). Receptor cargos subsequently undergo sorting, either for recycling back to the plasma membrane or for lysosomal destruction, at late endosomal compartments, termed multi-vesicular bodies (MVBs). RTKs often undergo ubiquitinylation through recruitment of Cbl family ubiquitin ligases (Levkowitz et al., 1998; Miyake et al., 1998). Mono- and oligoubiquitins in the context of RTKs drive progression of cargos via endosomes, towards lysosomes (Haglund et al., 2003; Huang et al., 2006; Mosesson et al., 2003). Active sorting in endosomes is attributed to various ubiquitin-binding domains (UBDs) carried by multiple adaptors (Hurley et al., 2006).
Small GTPases of the Rab subfamily are also attributed pivotal functions: enrichment of specific Rabs in different endosomal compartments permits local activation of a particular complement of effectors, which execute requisite endocytic tasks (Zerial and McBride, 2001). Activated Rab5, which operates at the cell surface/EE interface, stimulates production of phosphatidylinositol 3-phosphate (PI3P) in EE membranes, thereby nucleating complexes of effector proteins like EEA1, which harbors a PI3P-binding domain called FYVE (Stenmark et al., 1996). In addition to EEA1, ~30 other proteins share the double zinc finger FYVE motif. Notably, PI3P binding by specific FYVE domain proteins is highly regulated both in cis and in trans. For example, a FYVE-containing amino-terminal fragment of Hrs failed to localize to EEs. This may be attributed to ancillary interactions involving the flanking coiled-coil domain (Raiborg et al., 2001), or dimerization of FYVE domains (Hayakawa et al., 2004). Similarly, the FYVE domain of EEA1 may not suffice for endosomal localization (Hayakawa et al., 2004). Instead, endosomal localization of EEA1 is thought to be complemented in living cells by p38-induced phosphorylation (Mace et al., 2005). Thus, FYVE-mediated localization of a variety of endocytic adaptors to EEs is a multi-focal regulatory node that impacts on both endocytosis and signaling. The present study extends the complexity to regulation by monoubiquitinylation.
Here, we describe for the first time a mammalian FYVE domain protein, termed hLst2, whose endosomal localization is regulated by monoubiquitinylation. Consistent with its primitive ortholog in C. elegans, which functions as a negative regulator of the worm’s EGFR (Yoo et al., 2004), cellular depletion of human Lst2 augments EGF-induced signaling. Like many endosomal adaptors, hLst2 undergoes constitutive monoubiquitinylation, but despite the intrinsic FYVE domain, it displays primarily non-endosomal localization. By identifying a specific lysine acceptor, we discover monoubiquitinylation as a means to prevent FYVE domain-dependent association of hLst2 with PI3P-enriched endosomes. In line with an unique ubiquitin/PI3P switch, a non-ubiquitinylated hLst2 localizes to EEs, promotes degradative sorting of activated EGFRs and reduces signaling.
Results
hLst2, an evolutionarily conserved FYVE domain protein
With a view to a potentially novel mechanism of EGFR restraint, we focused on Lst2, a molecule described solely in worms as a negative regulator of EGFR-dependent vulval morphogenesis (Yoo et al., 2004). To identify orthologs of Lst2, the C. elegans sequence was blasted against the NR-NCBI database. This yielded putative sequences deriving from higher eukaryotic species, all mapping to a single genomic locus, but no yeast ortholog was found. The human gene (locus: 4p16) encodes a polypeptide of 887 amino acids, which we denoted hLst2 (also termed ZFYVE28; Fig. 1A). hLst2 bears an archetypal FYVE domain, sharing several PI3P binding motifs with other FYVE domains (Fig. 1B). The majority of the hLst2 sequence, however, does not correspond to recognized motifs. Nevertheless, a multiple alignment of evolutionarily distant Lst2s revealed well-conserved regions (Supplementary Fig. 1), including the amino terminal portion (denoted ‘NDom’), and a segment upstream of the FYVE domain (denoted ‘CBox’). To study hLst2 expression and function, several monoclonal antibodies (mAbs) to hLst2 were generated, and employed to probe mouse tissues. As shown, murine Lst2 protein was highly enriched in brain (Fig. 1C, and RT-PCR data not shown). For a positive control, we used a lysate of HEK-293T cells transfected with a Flag-hLst2 plasmid. While this presented a specific band corresponding to the expected mass of hLst2 (96.5 kDa), the more prominent species corresponded to an apparent Mw of 130-140 kDa (Fig.1C). Extending our analysis to a panel of transformed human cell lines, the anomalous form of hLst2 again appeared as the major species (Fig. 1D), suggesting robust post-translational modifications.
Figure 1. hLst2, a conserved FYVE domain protein, restrains EGF signaling.
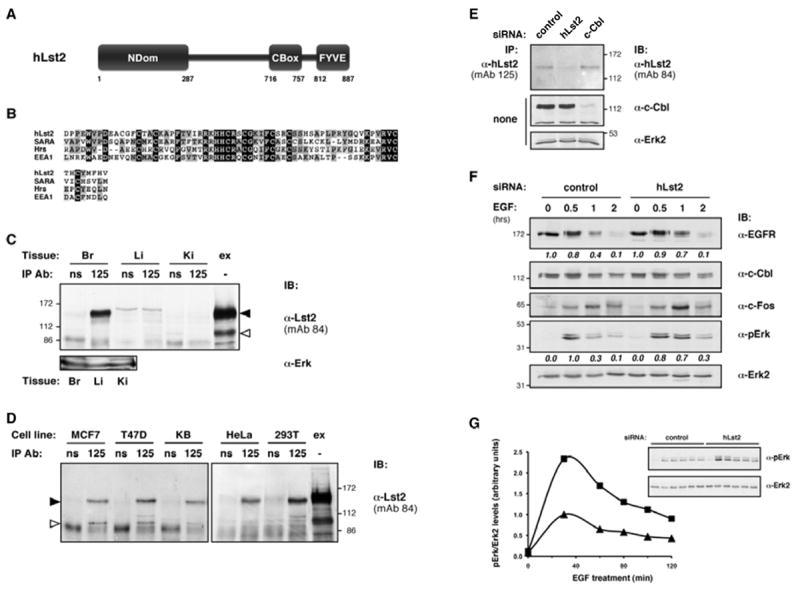
(A) Schematic depiction of hLst2; indicated are the carboxyl terminal FYVE domain, and evolutionary conserved regions we denoted ‘NDom’ and ‘CBox’.
(B) Alignment of the FYVE domains of hLst2, Smad Anchor for Receptor Activation (SARA), Hrs and EEA1.
(C and D) Lysates were prepared from several mouse tissues (panel C: Br, brain; Li, liver; Ki, kidney; ‘ex’ denotes cellular extract from HEK-293T cells ectopically expressing hLst2), or from the indicated panel of cell lines (panel D). Thereafter, lysates were subjected to immunoprecipitation (IP), either using normal mouse serum (ns) or mAb125 to hLst2, as indicated. Immunoprecipitates were analyzed by immunoblotting (IB) with the indicated antibodies, including mAb84 to hLst2. An open arrowhead marks a protein band consistent with the calculated Mw of hLst2 (96.5 kDa). The closed arrowhead indicates a major anti-hLst2 reactive species with slower than expected gel mobility.
(E) HeLa cells were transfected with siRNAs targeting either hLst2 or c-Cbl, or with control oligonucleotides. Cell lysates were subjected to IB with the indicated antibodies, either directly or following IP with an anti-Lst2 antibody.
(F) KB oral carcinoma cells transfected with siRNA duplexes, either control or hLst2-specific, were serum-starved for 12 hours, and then stimulated with EGF (100 ng/ml) at 37°C for the indicated time intervals, or else left untreated. Subsequently, cell lysates were analysed by IB with the antibodies indicated. Relative amounts of EGFR normalized to Cbl levels, and similarly, phosphorylated Erk normalized to Erk2 levels, were quantified using ImageJ (http://rsb.info.nih.gov/ij) and presented below the respective panels.
(G) Cells were transfected with control or hLst2-specific siRNA oligonucleotides, and analysed as in (F). Phosphorylated Erk levels relative to Erk2 (inset) were quantified and depicted graphically (closed triangles, control siRNA-treated cells; closed squares, hLst2 siRNA-treated cells).
Depletion of hLst2 in mammalian cells augments EGF signaling
Because Lst2 negatively regulates EGFR in worms, we studied effects of depleting hLst2 on EGFR signals. As shown, siRNA oligonucloetides targeting hLst2 mRNA markedly, and specifically, reduced expression of hLst2 (Fig. 1E). KB human cells transfected with hLst2-specific siRNA were then assayed for several downstream targets of EGFR. Interestingly, hLst2 depletion resulted in increased EGF signaling: specifically, we observed consistently higher Erk phosphorylation one hour after EGF stimulation. This coincided with delayed degradation of EGFR and enhanced expression of c-Fos (Fig. 1F). To corroborate these data, a similar experiment was performed that included better-resolved time intervals. As shown, cells transfected with siLst2 exhibited a clear and consistent increase in activated Erk (Fig. 1G). These results indicate that hLst2 negatively regulates EGF signaling to the Erk cascade, which is consistent with the purported activity of its ortholog in worms (Yoo et al., 2004).
Localization of hLst2 to endosomes is prevented by intramolecular determinants
Atypically, hLst2 ectopically expressed in HeLa cells distributed in a non-vesicular cytoplasmic pattern (Fig. 2A), distinct from the small puncta exhibited by EEA1 (Fig. 2B), a well-characterized resident of endosomes. As expected, an overexpressed Hrs manifested in aberrant structures (class E phenotype), but ectopic hLst2 did not coincide (Fig. 2B). These observations argue against an endosomal role for hLst2. Strikingly, an isolated FYVE domain coupled to a green fluorescent protein (GFP-hLst2FYVE) exhibited clear punctate distribution, which could be dissipated upon inhibition of PI3P production with wortmannin (Fig. 2C), and significantly overlapped with EEA1 (Fig. 2D). Hence, the FYVE domain in hLst2 bears autonomous avidity for putative PI3P-containing membranes. Assuming that the FYVE domain of hLst2 might be hindered by in cis interactions with the highly conserved NDom or CBox regions (Fig. 2E), we prepared a series of deletion mutants, individually lacking one of the three conserved regions. Remarkably, removal of either the NDom or the CBox redistributed hLst2 to punctate structures, while the FYVE domain deletion showed no detectable change (Fig. 2F). Further, GFP-Rab5, which serves as a marker for early endosomes, exhibited considerable overlap with either of the two mutants (Fig. 2G, and data not shown). Taken together, these results suggest that FYVE-dependent docking of hLst2 at PI3P-containing membranes may be hampered by mechanisms involving either NDom or CBox (see a summary of the results in Fig. 2G).
Figure 2. Endosomal localization of hLst2 is prevented in cis by either of the NDom or CBox regions.
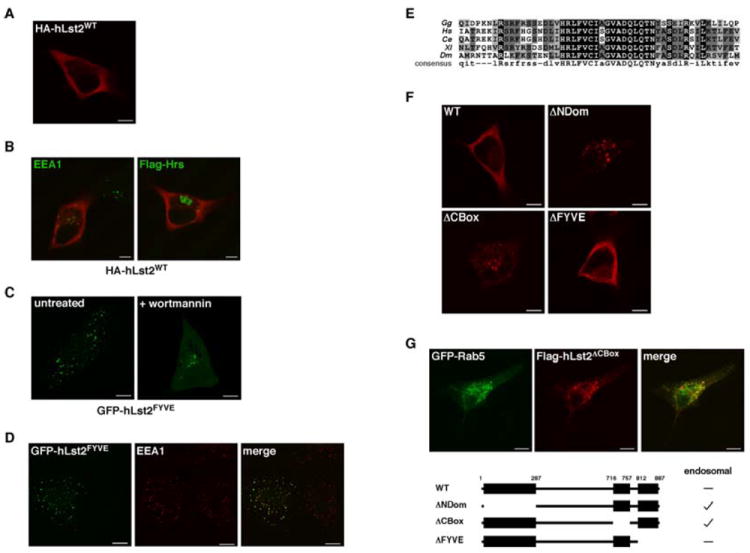
(A) HeLa cells transiently expressing HA-tagged hLst2 were fixed, permeabilized and incubated with an anti-HA antibody. Subsequently, cells were incubated with a Cy3-labeled secondary antibody, and examined by fluorescence microscopy. Shown is a confocal image of a representative cell. Scale bar, 10 μm.
(B) HeLa cells expressing HA-hLst2, either alone or together with Flag-Hrs, were processed for fluorescence microscopy using an anti-HA antibody, and either an anti-EEA1 or an anti-Flag antibody. Appropriate secondary antibodies were used for detection. Merged images indicating the distributions of EEA1 or Hrs (green) and hLst2 (red) are shown.
(C) HeLa cells transfected with the FYVE domain of hLst2 (aa 761-887) fused to GFP (GFP-hLst2FYVE) were treated with wortmannin (100 nM) for 30 min, or left untreated. Thereafter, cells were fixed and analyzed directly by fluorescence microscopy.
(D) HeLa cells expressing GFP-hLst2FYVE were fixed, permeabilized and incubated with an anti-EEA1 antibody, followed by a Cy3-conjugated secondary antibody. Confocal images showing the relative distributions of hLst2FYVE (green) and EEA1 (red) are presented.
(E) Alignment of the CBox derived from putative metazoan orthologs of hLst2. (Gg, G. gallus; Hs, H. sapiens; Ce, C. elegans; Xl, X. laevis; Dm, D. melanogaster).
(F) HeLa cells transfected with vectors encoding Flag-hLst2 or various deletion mutants thereof, as indicated, were analysed as in (A).
(G) HeLa cells co-expressing Flag-hLst2ΔCBox together with GFP-tagged Rab5 were analyzed as in (D). A scheme of the described deletion mutants of hLst2, individually lacking the NDom, CBox or FYVE domain (ΔNDom, ΔCBox or ΔFYVE, respectively), along with a summary of their endosomal status, is shown.
hLst2 is a phosphoprotein and undergoes CBox-dependent monoubiquitinylation at a conserved lysine acceptor
To address post-translational modifications, ectopic hLst2 was isolated and tryptic peptides subjected to LC-MS/MS analysis. Positive identifications included a subset of peptides derived from ubiquitin (data not shown). Further, after factoring in supplementary masses correlating with ubiquitinylation and phosphorylation, our analysis revealed the presence in hLst2 of a single lysine residue modified by ubiquitin (K87; Fig. 3A), as well as at least two phospho-acceptor sites (S586 and T870). Immunoblotting analyses corroborated these results: incubation of hLst2 with a broad reactivity phosphatase resulted in increased gel mobility (Fig. 3B), and anti-ubiquitin antibodies detected constitutive ubiquitinylation, which was enhanced upon transfection of ectopic ubiquitin (Fig. 3C). Based on the similar electrophoretic mobility of the endogenous hLst2 (Figs. 1C and 1D), we assume that rich post-translational modification is an attribute of this protein. Focusing on lysine-87, we noted that this residue comprises one of three conserved lysines within the NDom. Indeed, mutation of lysine-87 abolished ubiquitinylation of hLst2 concomitant with a reduction in apparent molecular weight, while the other candidates showed no effect (Fig. 3D). These results suggest modification of hLst2 by a single ubiquitin moiety, rather than polyubiquitin chains. Examining this further, an ubiquitin construct lacking major lysine residues that participate in chain elongation (denoted Ub4KR) was employed. As depicted, this mutant recapitulated the wild-type pattern in the context of hLst2, against a background of diminished polyubiquitinylation (Fig. 3E). In addition, using an inhibitor of the 26S proteasome (MG132), we detected no effect on hLst2 levels, in line with monoubiquitin serving a non-proteolytic role. Together, these observations strongly support predominant monoubiquitinylation of hLst2.
Figure 3. hLst2 undergoes constitutive phosphorylation and CBox-dependent monoubiquitinylation.
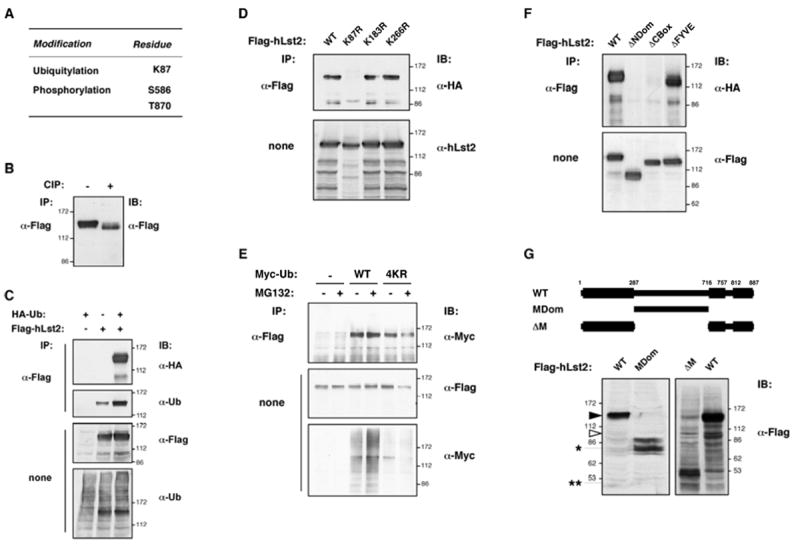
(A) Flag-hLst2 immunoprecipitates isolated from transfected HEK-293T cells were gel-resolved, stained, and the major specific band (apparent molecular weight p130-140) excised, and analysed by mass spectrometry. Results identifying a single ubiquitinylation site and two phosphorylation sites in hLst2 are depicted in the table.
(B) Lysates of HEK-293T cells expressing Flag-hLst2 were subjected to IP using an anti-Flag antibody. Immunoprecipitates were treated with calf intestinal phosphatase (CIP), or left untreated, as indicated, before immunoblotting (IB).
(C) Lysates of HEK-293T cells expressing Flag-hLst2 and HA-ubiquitin (HA-Ub) were subjected to IP and IB with the indicated antibodies, including an antibody to ubiquitin.
(D) Lysates of HeLa cells expressing HA-Ub together with wild-type Flag-hLst2WT or the indicated lysine-to-arginine point mutants thereof (Lst2K87R, hLst2K187R or hLst2K266R) were analyzed as in (C).
(E) HeLa cells transiently transfected with an expression vector for Flag-hLst2, together with plasmids encoding either Myc-ubiquitin (Myc-UbWT) or a multiple lysine mutant thereof (K11/29/48/63R; denoted Myc-Ub4KR), were treated with MG132 (12.5μM) for 4 hours at 37°C, or else left untreated, as indicated. Thereafter, cell lysates were analyzed as indicated. Note that the slightly lower ubiquitinylation of hLst2 in the presence of Ub4KR was not reproducible.
(F) HEK-293T cells expressing HA-ubiquitin (HA-Ub) together with Flag-hLst2, either wild-type (WT) or the deletion mutants indicated (Flag- hLst2MDom, -ΔCBox, or -ΔFYVE), were lysed, and analyzed as in (D).
(G), top, scheme showing the central portion of hLst2 in isolation (hLst2MDom) and a complementary deletion mutant lacking this region (hLst2-ΔM). Bottom, HEK-293T cells transiently expressing Flag-hLst2 (WT), or the indicated deletion mutants thereof, were lysed and analyzed using an anti-Flag antibody. Black and open arrowheads are as described in Figure 1(C); an asterisk and a pair of asterisks respectively indicate the observed and predicted gel mobility of hLst2MDom.
As a basic feature of hLst2 protein, we focused on the sequence determinants of monoubiquitylation. Interestingly, as well as deletion of the NDom (which harbors the acceptor lysine), the hLst2 mutant lacking the CBox was also unable to undergo ubiquitinylation, while the FYVE domain appeared dispensable (Fig. 3F). These results indicate that the CBox is critically involved in mediating hLst2 ubiquitinylation, and conversely, that a capacity to dock at PI3P-containing membranes is probably not required. It was also notable that all tested versions of hLst2 maintained a sizable mobility shift that could not be accounted for by ubiquitinylation and phosphorylation combined. Hence, a mutant lacking the relatively unconserved central portion of hLst2 (ΔM), and the complementary region in isolation (MDom) were examined. As shown, whereas MDom retained relatively slow gel mobility, ΔM appeared consistent with its molecular weight (~50 kDa; Fig. 3G). Employing mass spectrometry, however, we were unable to detect any further post-translational modification of p130-p140 (data not shown). In addition, the discrepancy between p130-140 and p97 remains unknown; the latter reacted with all antibodies to hLst2 (data not shown), underwent ubiquitinylation (Fig. 3F), and co-overexpressed with the longer form upon transfection. Notably, however, our results identify hLst2 as a robust target for phosphorylation, and CBox-mediated monoubiquitinylation.
Monoubiquitinylation of hLst2 regulates its endosomal localization
Because our results correlated endosomal localization of Lst2 with a lack of ubiquitinylation, we tested whether the largely non-ubiquitinylated mutant (hLst2K87R) coincides with endosomes. Addressing this, we employed live cell confocal microscopy to follow HeLa cells stably expressing GFP-tagged hLst2. Remarkably, GFP-hLst2K87R protein was indeed found to concentrate in clear puncta; GFP-hLst2WT, in contrast, displayed a dynamic, mainly reticular-like distribution (Fig. 4A, Supplementary Movies 1 and 2). Further, overexpression of hLst2K87R manifested aberrantly enlarged endosomes, while a FYVE-disabling cysteine mutation (hLst2K87R/C823A) abolished vesicular localization (Supplementary Fig. 2A), congruent with ubiquitinylation-mediated redistribution. On the other hand, the dynamic reticular pattern of hLst2WT was associated with microtubules, and it underwent redistribution upon treatment with a microtubule-severing agent, nocodazole (Supplementary Fig. 4 and Movies 3 and 4). To verify targeting of hLst2K87R to PI3P-enriched endosomes, we compared its distribution with that of two FYVE-containing endosomal proteins, EEA1 and Hrs. As shown, the results confirmed considerable co-localization of hLst2K87R with both proteins (Fig. 4B). Moreover, employing various fluorescently-tagged Rab GTPases, hLst2K87R exhibited significant overlap with three of the tested Rabs (5, 4 and 7), but was segregated from Rab11-containing structures (Supplementary Fig. 2B), in line with enrichment of PI3P in early and sorting endosomes (Vicinanza et al., 2008).
Figure 4. Monoubiquitinylation of hLst2 disables FYVE domain-dependent targeting to sorting endosomes.
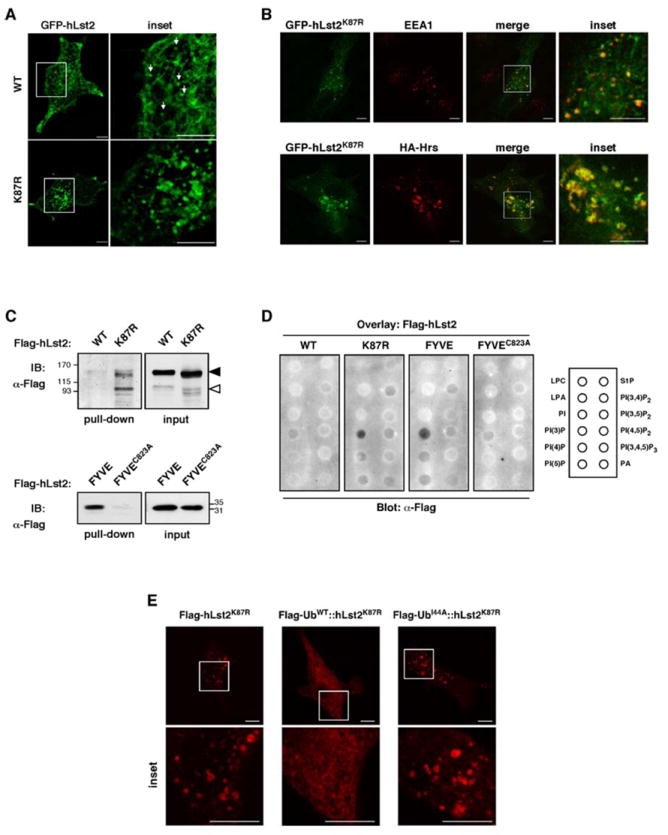
(A) Confocal sections of live HeLa cells stably expressing GFP-tagged hLst2WT or hLst2K87R, along with magnification of the squared area of each panel (inset). Note that hLst2WT exhibits largely reticular patterns, and possibly some small vesicles (arrows), but the mutant is largely vesicular. Scale bar, 10 μm.
(B) HeLa cells ectopically expressing GFP-hLst2K87R, either alone (upper row), or together with HA-tagged Hrs (lower row) were fixed, permeabilized, and incubated either with an anti-EEA1 antibody (upper row) or an anti-HA antibody (lower row), followed by an appropriate Cy3-labeled secondary antibody. Shown are confocal images of representative cells; a magnification the indicated area within the merge pane is also depicted (inset). Scale bar, 10μm.
(C) HEK-293T cells were transfected with vectors encoding His6-Flag-tagged hLst2, either wild-type (WT), the lysine point mutant (K87R), the isolated FYVE domain (hLst2FYVE) or a disabled point mutant thereof (hLst2FYVE/C823A). Cell lysates were incubated with a nickel resin, and immobilized proteins subsequently eluted in 250 mM imidazole. Equivalent amounts of the indicated hLst2 proteins were then incubated with PI3P-agarose beads, before washing and analysis by gel electrophoresis and IB with an anti-Flag antibody. Black and gray arrowheads are as in Figure 1(C).
(D) The indicated hLst2 proteins were isolated as in (B) and incubated with nitrocellulose membranes, which were pre-bound to the indicated panel of phospholipids (see rectangle). Thereafter, membranes were probed with an anti-Flag antibody. Abbreviations: LPC, lysophosphocholine; LPA, lysophosphatidic acid; PI, phosphoinositol; PI(3)P, phosphoinositol 3-phosphate; PI(4)P, phosphoinositol 4-phosphate; PI(5)P, phosphoinositol 5-phosphate; S1P, sphingosine 1-phosphate; PI(3,4)P2, phosphoinositol (3,4)-bisphosphate; PI(3,5)P2, phosphoinositol (3,5)-bisphosphate; PI(4,5)P2, phosphoinositol (4,5)-bisphosphate; PI(3,4,5)P2, phosphoinositol (3,4,5)-trisphosphate; PA, phosphatidic acid).
(E) HeLa cells were transiently transfected with constructs encoding a Flag-tagged ubiquitinylation-defective point mutant of hLst2 (Flag-hLst2K87R), a chimera comprised of hLst2K87R fused at the amino terminus to an ubiquitin moiety (Flag-UbWT∷hLst2K87R), or a derivative chimera that includes an isoleucine-44 mutation within the ubiquitin moiety (Flag-UbI44A∷hLst2K87R). Thereafter, cells were fixed, permeabilized and incubated with an anti-Flag antibody, followed by an appropriated Cy3-labeled secondary antibody. Confocal images of representative cells are shown; a magnification the boxed area is also depicted (inset). Scale bar, 10μm.
To directly test whether monoubiquitinylation prevents binding of the FYVE domain to PI3P-enriched membranes, we incubated hLst2 proteins with immobilized phosphoinositides. Specifically, hLst2WT or hLst2K87R were fused to a His6-Flag-tag and expressed in HEK-293T cells. Nickel-purified proteins, along with the isolated FYVE domain and a disabled point mutant thereof (FYVEC823A), were incubated with PI3P-conjugated beads, prior to immunoblotting. As expected, the isolated FYVE domain remained bound to the PI3P resin, while the mutant did not (Fig. 4C). Furthermore, whereas hLst2WT exhibited only minimal interaction with PI3P, hLst2K87R displayed enhanced binding (Fig. 4C). It is notable that this assay reflected preferential binding of the p97 variant to PI3P. In line with specificity, an overlay assay comparing various lipids corroborated the results and confirmed FYVE’s specificity to PI3P (Fig. 4D). To independently test ubiquitinylation-dependent sequestration of hLst2 away from PI3P-containing endosomes, an in-frame fusion of ubiquitin with hLst2K87R was generated (UbWT∷hLst2K87R); in parallel, we tested a similar construct incorporating a mutation within the ubiquitin moiety (isoleucine 44-to-alanine) at a locus commonly accessed by UBDs (UbI44A∷hLst2K87R; depicted in Supplementary Figure 3A). Expression of the chimeric proteins was confirmed in mammalian cells (Supplementary Figure 3B), and their localization analyzed by immunofluorescence microscopy. As shown, the UbWT∷hLst2K87R fusion assumed a diffuse pattern, in contrast to the punctate appearance of both hLst2K87R and the UbI44A∷hLst2K87R chimera (Fig. 4E). Predictably, the latter chimera significantly coincided with a co-expressed Hrs, but UbWT∷hLst2K87R did not (Supplementary Fig. 3C). Overall, these results ascribe to ubiquitin an in cis inhibitory effect on hLst2 binding to PI3P, probably by means of recruiting an ubiquitin-binding protein(s) to the isoleucine-44-centered hydrophobic patch.
Non-ubiquitinylated hLst2 co-localizes with EGFR in endosomes and promotes receptor degradation
Conceivably, Lst2 negatively regulates EGFR signaling, both in worms (Yoo et al., 2004) and in mammalian cells (Figs. 1F and 1G), by directly impinging on EGFR trafficking. To address this, internalization of fluorescently-labeled EGF in HeLa cells stably expressing hLst2WT or hLst2K87R was followed by live-cell confocal microscopy (Supplementary Movies 5 and 6). As shown, the non-ubiquitinylated mutant extensively colocalized with endocytosed EGF 30 min after EGF addition, however, hLst2WT displayed only limited overlap (Fig. 5A). Further, by transient transfection of hLst2, we noted that in cells overexpressing hLst2K87R, a fraction of EGFRs redistributed to large vesicles containing the mutant, in the absence of ligand, while no such basal effect was observed for hLst2WT (Supplementary Fig. 5). Of note, after adding EGF the receptors almost exclusively co-localized with hLst2K87R, but stimulation with EGF exerted no detectable redistribution of either ectopic form of hLst2. Receptor redistribution by endosome-anchored hLst2 may reflect an ability to promote EGFR degradation or inhibit recycling. To examine these options, we made use of an engineered HeLa cell-line in which endogenous receptor levels had been stably silenced by stable expression of an EGFR-specific siRNA (Zwang and Yarden, 2006). By transient transfection of hLst2 and siRNA-refractory EGFR, this set up allowed effective monitoring of co-expressing cell populations. As shown, receptor degradation became apparent after half an hour of treatment with EGF (Fig. 5B). Upon inclusion of hLst2, however, ligand-induced receptor degradation could be observed already after ten minutes, and the non-ubiquitinylated mutant manifested an even more pronounced destabilizing effect. These results suggest that increased hLst2 levels promote ligand-dependent EGFR degradation, and moreover, that the endosomal K87R mutant is more active in this capacity.
Figure 5. Endosomal hLst2 accelerates ligand-induced EGF receptor degradation.
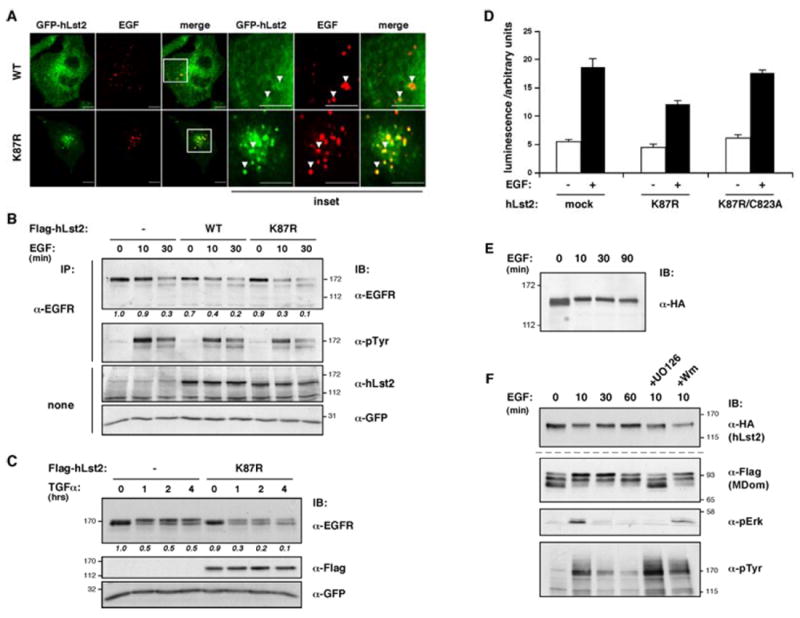
(A) Confocal sections were obtained 30 min after adding Alexa-fluor555-conjugated EGF (0.5 μg/ml) to HeLa cells expressing GFP-hLst2, either WT or K87R. Note the limited co-localization of hLst2WTand EGF (arrowheads in insets), as opposed to the extensive co-localization of hLst2K87R with EGF. Scale bar, 10 μm.
(B and C) HeLa cells stably expressing a siRNA expression construct targeting EGFR (HeLa-sEGFR) were transiently transfected with plasmids encoding siRNA-refractory EGFR, GFP, and as indicated, Flag-hLst2, either wild-type (WT) or the ubiquitinylation-defective mutant (K87R). Thirty-six hours post-transfection, cells were serum-starved for 12 hours, and stimulated with either EGF (10 ng/ml; panel B), or TGF-alpha (20 ng/ml; panel C) at 37°C for the indicated time periods. Thereafter, cell lysates were subjected to analysis with the indicated antibodies. Relative amounts of EGFR normalized to GFP levels are presented below the upper panel; quantification of bands was performed using ImageJ.
(D) HeLa cells were co-transfected with plasmids encoding a SRE-luciferase reporter, GFP and the indicated forms of hLst2. Following serum starvation, cells were treated with EGF (black columns) for 8 hrs, or else left untreated (white columns). Thereafter, cells were lysed, and the luminescence signal measured. Results were normalized to the GFP signal, and are presented as mean values (and S.D.; bars).
(E) HeLa cells expressing HA-tagged hLst2 were serum-starved for 12 hours before treatment with EGF (100 ng/ml) at 37°C for the indicated time periods. Thereafter, cells were lysed and analyzed by IB using an anti-HA antibody.
(F) Cells transiently expressing either wild-type HA-hLst2 (upper panel) or the Flag-tagged central portion of hLst2 (Flag-MDom; lower panels) were serum-starved for 12 hrs. Thereafter, cells were pre-treated with a MEK inhibitor (UO126; 5 μM) or with wortmannin (100 nM) for 30 min at 37°C, or else left untreated, as indicated, before stimulation with EGF (20 ng/ml) for the indicated time intervals, and analysis with the indicated antibodies.
In view of these results, we considered the possibility that non-ubiquitylated hLst2 is involved in sorting decisions; namely, whether to recycle or degrade EGFRs. To address this, we employed an alternative ligand for EGFR (TGFα), which is known to favor receptor recycling (Ebner and Derynck, 1991). Indeed, even after prolonged treatment with TGFα (up to 4 hours), we observed limited EGFR degradation, but in the presence of non-ubiquitinylated hLst2, ligand induction resulted in a marked receptor destabilization (Fig. 5C). To further test whether the enhanced rate of receptor degradation translates into reduced signaling outcome, we utilized a luciferase reporter gene fused to the serum response element (SRE), a transcriptional target of EGFR-activated Erk. Consistent with effects observed on EGFR degradation, expression of hLst2K87R reduced induction of the reporter protein, while a non-ubiquitinylated mutant with a disabled FYVE domain (K87R/C823A) restored similar levels of SRE activity (Fig. 5D). In sum, these results support a role for endosomal hLst2 in promoting EGFR downregulation in a manner dependent on its FYVE domain and state of ubiquitinylation.
Given a negative role in EGF signaling, and the critical functions played by EGFR in cancer (Citri and Yarden, 2006), the status of hLst2 in human tumors was of interest. For this purpose, a large cohort (272 cases) of invasive human breast carcinomas was employed, and sections probed with a mAb to hLst2. Our initial analyses revealed expression of hLst2 protein in a subset of Luminal A type tumors (Supplementary Table). hLst2-positive tumors were relatively differentiated and displayed limited metastasis to adjacent lymph nodes, in line with putative tumor suppression activity and our signaling data. Possible implications of low incidence of hLst2 expression in other types of cancers will entail further analyses.
EGF induces phosphorylation of hLst2
Tyrosine phosphorylation and ubiquitinylation of the Eps15 endocytic effector are regulated in an inducible manner by EGF. We detected no EGF-induced ubiquitinylation of hLst2 (data not shown). On the other hand, the analyses described below indicate that hLst2 is similarly phosphorylated, albeit on serine/threonine residues. As shown, EGF treatment caused a subtle, but reproducible, upshift of hLst2 (Fig. 5E), which is not due to tyrosine phosphorylation (data not shown). It is notable that the upshift of Lst2 was minor, and was not apparent in all our experiments. Nevertheless, using deletion mutants we enhanced the inducible modification and mapped it to the central MDom domain (Fig. 5F). Further, pharmacological inhibitors indicated that the ligand-stimulated effect is prevented by pre-treatment with a MEK inhibitor (UO126) but not with a PI3K inhibitor (wortmannin; Fig. 5F). Hence, in addition to constitutive phosphorylation, hLst2 is subjected to MEK-dependent serine/threonine phosphorylation.
hLst2 physically binds Trim3, a coordinator of endosomal trafficking
To shed light on mechanistic aspects of hLst2, we aimed at identifying interacting proteins. As bait, we expressed in bacteria the NDom of hLst2 flanked by a GST moiety and a His6-tag (denoted GNH). Nickel-immobilized GNH was mixed with a lysate of mouse brain, and associated proteins eluted, then re-immobilized on glutathione-conjugated beads. Samples were subsequently washed in increasing salt concentrations, resolved by electrophoresis and visualized with a protein stain (Fig. 6A). Of the three specific bands we analyzed by LC-MS/MS, only the ~85 kDa band yielded readable tryptic peptides, which identified the protein as the RBCC domain protein, Trim3 (also known as BERP or RNF22). ‘RBCC’ embodies an ordered union of a RING domain, one or two distinct zinc fingers, termed B-boxes, a coiled-coil region, and a divergent carboxyl terminus (Borden, 1998). In Trim3, additional domains complement the RBCC (Fig. 6B), including a filamin repeat, which forms rod-like structures in several actin cross-linking proteins, and a series of NHL repeats, resembling WD repeats in adopting a β-propeller configuration. The proposed association between Trim3 and hLst2 was confirmed in HEK-293T cells (Fig. 6C), but EGF exerted no effect on these physical interactions (Fig. 6D). Notably, hLst2 coincides almost entirely with more peripherally located Trim3-containing structures (Fig. 6E). Hence, hLst2 apparently encounters a sub-population of Trim3 molecules in peripheral cytoplasmic regions. The function of Trim3 is unknown, but interactions with two actin-associated proteins, myosin V and α-actinin-4, have been reported (El-Husseini et al., 2000; El-Husseini and Vincent, 1999), as well as an involvement in a vesicular recycling complex termed ‘CART’ (Yan et al., 2005). As we discuss below, these features provide clues into mechanisms underlying the ability of hLst2 to regulate vesicular trafficking, as well as signaling by tyrosine kinases like EGFR, both in worms and in mammals.
Figure 6. Identification of Trim3 as an interacting partner of hLst2.
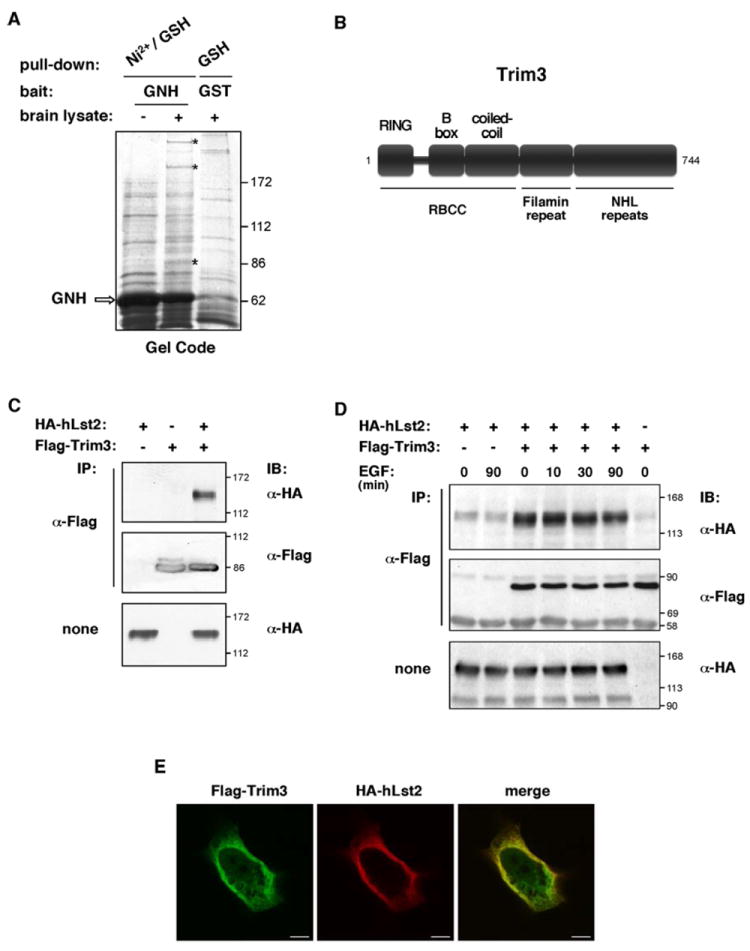
(A) Bacterially produced GNH (comprising the NDom of hLst2 flanked by an amino-terminal GST moiety and a carboxyl-terminal His6-tag) was purified on nickel-agarose beads (~50 μg protein) and incubated without or with lysate prepared from mouse brain (~30 mg protein). Immobilized proteins were then eluted in 250 mM imidazole, and rebound to GSH-agarose beads. For a parallel external control, we used GST alone coupled to GSH-agarose (~50 μg protein). After extensive washes, samples were subjected to gel electrophoresis and staining with Gel Code. Specific bands were dispatched for mass spectrometry analysis. A representative stained gel is shown. An arrow indicates the major band corresponding to GNH. Asterisks mark bands specific to the GNH sample exposed to brain lysate.
(B) Scheme depicting the domain structure of Trim3.
(C) HEK-293T cell monolayers were transfected with plasmids encoding Flag-Trim3 and HA-hLst2, as indicated. Forty-eight hours post-transfection, cell lysates were subjected to IP with an anti-Flag antibody, and IB with the indicated antibodies.
(D) HeLa cells transiently expressing HA-hLst2 and Flag-Trim3, as indicated, were serum-starved for twelve hours, and then treated with EGF (20ng/ml at 37°C) for the indicated time periods, or else left untreated. Thereafter, cells were lysed and analyzed with the indicated antibodies.
(E) HeLa cells co-expressing Flag-Trim3 and HA-hLst2 were fixed, permeabilized and incubated with mouse anti-Flag and rat anti-HA antibodies. For detection, appropriate Cy2- and Cy3-conjugated secondary antibodies were used. Shown are confocal images taken from the central plane of the cell. Scale bar, 10 μm.
Discussion
Internalization of cell-surface receptors, such as RTKs, remodels and restricts their signaling capacity; yet mechanisms that govern endocytosis are incompletely understood. In this study, we report a unique mode of regulation, in which removal of monoubiquitin from hLst2 activates its cargo sorting activity and negates growth factor signaling (Fig 7), consistent with the function of the invertebrate Lst2 uncovered through genetic analyses (Yoo et al., 2004).
Figure 7. Proposed model for Lst2 function in EGFR endocytosis.
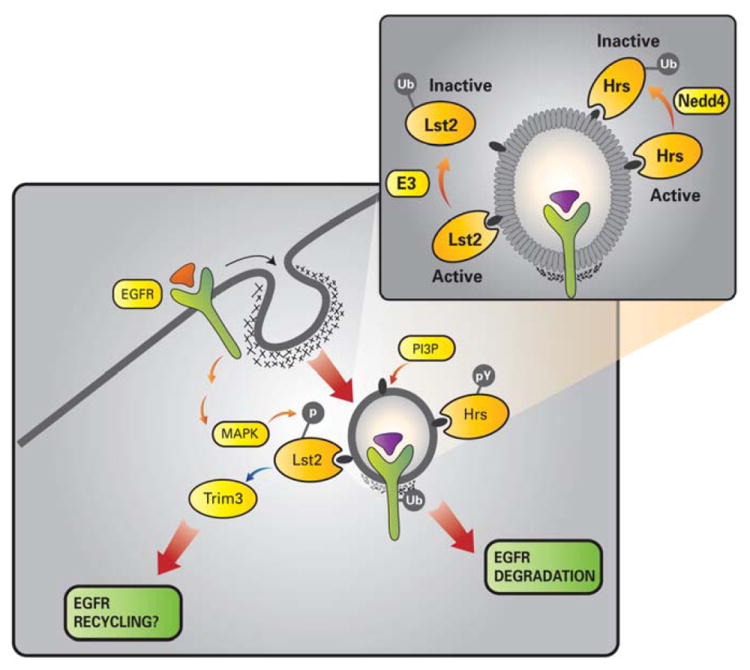
The scheme depicts localization of the newly described Lst2 protein to PI3P-enriched early endosomes, by virtue of its FYVE domain. Lst2 undergoes monoubiquitinylation at a conserved lysine residue (lysine 87), which results in impairment of its FYVE domain, and sequestration from endosomes (inset). Accordingly, we envisage an ubiquitinylation/deubiquitinylation cycle that allows intermittent recruitment of Lst2 to sorting endosomes during EGFR trafficking. In comparison, the extensively characterized FYVE domain protein, Hrs, remains membrane bound despite its ubiquitinylation by Nedd4 family E3 ubiquitin ligases. Note that EGF-induced and MAPK-mediated serine/threonine phosphorylation of Lst2 contrasts with tyrosine phosphorylation of Hrs. We find that Lst2 associates with the RBCC protein, Trim3, which in turn couples to the CART cargo recycling complex, comprising α-actinin-4 and myosin V.
An ubiquitinylation-dependent switch for PI3P binding
Proteins that bear FYVE domains are most often targeted to PI3P-enriched membranes, yet the steady-state distribution of hLst2 did not coincide with patterns exhibited by other family members (Fig. 2B). FYVE domains are, nevertheless, not homogeneous and sequence deviation around consensus residues may impinge on qualities of binding (Kutateladze, 2006). Mutational analysis of the specific ubiquitin acceptor site in hLst2 subsequently resolved this discrepancy: hLst2 monoubiquitinylation inhibits its PI3P binding capability (Fig 4). Our in vitro experiments excluded the possibility that hLst2 harbors an intrinsic ubiquitin binding function (data not shown), which would explain intramolecular steric hindrance of the FYVE domain. Still, several other scenarios may explain impairment of FYVE domain activity. First, constitutive phosphorylation of hLst2 may be involved; curiously, one of the target acceptor sites we identified by mass spectrometry (threonine-870) resides within the FYVE domain (Fig. 3A), and phosphorylation of EEA1 and Rabenosyn-5 within their FYVE domains promotes endosome targeting (Mace et al., 2005). According to an alternative model, which is strongly supported by our ubiquitin-hLst2 fusion chimera (Fig. 4E), ubiquitin may serve as a docking site for an UBD-containing protein, which could hamper the FYVE domain. The putative UBD proteins involved in sequestration of hLst2 may play pivotal roles in receptor sorting for degradation, but their identity remains unknown.
hLst2 regulation by E3 ligases and de-ubiquitinylating enzymes
The described monoubiquitin switch anticipates the existence of an E3 ligase and deubiquitinylating enzyme pairing that acts upon hLst2. Regarding their relative contribution, constitutive ubiquitinylation indicates that hLst2 is robustly coupled to its cognate ligase. To pursue possible E3 ligases, we employed an isolated CBox as bait, but were unable to identify any recognized participants of the ubiquitinylation machinery (data not shown). A growing list of ligases is associated with endocytic processes: Cbl and Nedd4 family proteins, as well as Trim3, however, did not influence hLst2 ubiquitinylation (data not shown). Other feasible possibilities include multiple ligases implicated in trafficking within the Notch pathway, such as Mindbomb and Neuralised, Parkin, which targets Eps15, and Mdm2, which ubiquitinylates β-arrestin. On the opposing side, it will be critical to identify the deubiquitylating factor in hLst2 regulation. As with ligases, several DUBs have emerged as potential regulators of endocytosis. For instance, genetic and biochemical studies implicate the Drosophila protein ‘fat facets’ and its mammalian ortholog, FAM, as cognate DUBs for Epsin proteins (Chen et al., 2002). AMSH and UBPY are DUBs that both converge on the STAM adaptor protein. Interestingly, these enzymes appear to exert opposing effects on lysosomal degradation of activated RTKs, though their precise involvement remains unclear (Alwan and van Leeuwen, 2007; McCullough et al., 2006).
Ubiquitinylation-dependent sequestration from endosomes: an emerging motif to regulate trafficking and signaling
Multiple endosomal adaptor proteins undergo ubiquitin conjugation, which is often not associated with their proteasomal degradation (Mukhopadhyay and Riezman, 2007), but consequences of adaptor ubiquitinylation remain unclear. Several reports point to a role in regulation of subcellular localization: for instance, monoubiquitinylation of endocytic proteins, such as Eps15 and Sts2, may result in intramolecular interactions between self-ubiquitin and their UBDs, which may prevent adaptor association with other ubiquitinylated targets (Hoeller et al., 2006). Likewise, a recent study into regulation of Rabex-5 reports that while endosomal localization of Rabex-5 is dependent on its ubiquitin-binding capability, the monoubiquinylated protein appears enriched in the cytosol (Mattera and Bonifacino, 2008). Hence, as with Lst2, ubiquitinylation-mediated sequestration of endosomal adaptors from target membranes may prove to be a widespread regulatory mechanism. In this scenario, an ubiquitinylation/deubiquitinylation cycle may operate in temporal and spatial coordination with receptor trafficking, thereby enabling timely engagement of Lst2, and other ubiquitin-conjugated endocytic effector proteins, such as Hrs (see Fig. 7).
An emergent sorting machinery involving Trim3
By a yeast two-hybrid screen, the carboxyl terminal β-propeller region of rat Trim3 was found to interact with unconventional class V myosins (El-Husseini and Vincent, 1999). Myosin V proteins are highly processive actin-based molecular motors that function in transport of diverse intracellular cargoes (Reck-Peterson et al., 2000). In a further screen using the RBCC domain of Trim3, an interaction was reported with the non-muscle actin-bundling protein, α-actinin-4 (El-Husseini et al., 2000). The so-called ‘CART’ complex (comprising Hrs, α-actinin-4, myosin V and Trim3) is proposed to mediate transferrin receptor recycling, though its disruption did not appear to affect EGFR trafficking (Yan et al., 2005). Another study has suggested that Hrs may actively recycle specific cargoes, such as certain GPCRs, independently of its well-known sorting role at the MVB (Hanyaloglu et al., 2005). Class V myosins have been widely implicated in recycling and in transport of endosomes to the cell surface. Hence, hLst2 may participate in a novel sorting machinery involving Trim3, in line with the evolutionarily conserved role of Lst2 in trafficking of RTKs. Future studies will address potential bifurcating roles of Lst2, and associated protein complexes, in sorting of RTKs to degradation or recycling.
Experimental procedures
Materials
All cell lines were from American Type Culture Collection (Manassas, VA). Control (siGENOME Non-Targeting siRNA pool) and hLst2-specific siRNA oligonucleotides were purchased from Dharmacon Research (Lafayette, CO). Antibodies were from Alexis Biochemicals (San Diego, CA), Roche Molecular Biochemicals (Mannheim, Germany; anti-HA), Covance (Princeton, NJ; anti-ubiquitin, clone P4G7), Sigma (anti phospho-Erk and anti-Flag) and Cell Signaling (Beverly, MA; anti-EEA1). Alexa Fluor 555-labelled EGF was from Molecular Probes (Eugene, OR). To generate monoclonal anti-hLst2 antibodies, mice were immunized with recombinant full-length GST-hLst2. Splenocytes from a selected mouse were fused with NSO myeloma cells, and hybridomas selected and cloned by limiting dilution. For a polyclonal anti-hLst2 antibody, rabbits were immunized with a recombinant amino-terminal portion (NDom) of hLst2.
Protein-lipid overlay and lipid bead-protein pull-down assays
The overlay assay was performed using purified His-Flag tagged hLst2 proteins and nitrocellulose membranes spotted with specific lipids (Echelon Biosciences, Salt Lake City, UT). Albumin-blocked membranes were incubated overnight at 4°C with purified hLst2 proteins (1 μg) in TBST. Membranes were then washed and processed for detection, using an anti-Flag antibody. For a lipid-protein pull-down assay, PI3P-agarose beads (Echelon Biosciences, Salt Lake City, UT) were incubated with purified hLst2 protein (1 μg) in binding buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 0.25% Nonidet P-40) for 3 hrs at 4°C. Tightly bound proteins were eluted by boiling in gel sample buffer.
DNA constructs and bacterial expression
cDNA for human hLst2 was provided by the Kazusa Human cDNA Project and subcloned downstream of a peptide tag by PCR into the pCDNA3 vector. The Flag-hLst2-MDom construct was produced by PCR amplification of the central region of hLst2 (aa 288-715). GFP- hLst2WT, GFP-hLst2K87R and GFP-Flag-FYVE expression vectors were generated by PCR and cloning into pEGFP-C1 (Clontech, Mountain View, CA). Plasmids encoding His-Flag-hLst2 and its derivatives were made by subcloning into the pcDNA3HisB vector (Invitrogen, Carlsbad, CA). Chimeric constructs encoding in-frame fusions of ubiquitin (aa 1-75; either wild-type or an isoleucine 44-to-alanine point mutant thereof) with hLst2K87R (denoted UbWT∷hLst2K87R and UbI44A∷hLst2K87R, respectively) were generated by overlap extension PCR, using Flag-ubiquitin- and hLst2K87R-encoding plasmids as templates. To generate the ‘GNH’ construct, an oligo-deoxynucleotide duplex coding for a hexa-histidine tag was inserted in frame carboxyl terminal to the GST-NDom-expressing sequence.
Cell lines, transfection, lysate preparation and immunoblotting analyses
HeLa cells were transfected using the jetPEI reagent (PolyPlus, Illkirch, France). A derivative cell-line, stably expressesing EGFR-specific siRNA, along with immunoblotting procedures have been described (Zwang and Yarden, 2006). Transfection of siRNAs was performed using HiPerFect reagent (Qiagen, Valencia, CA).
SRE reporter assay
Cells were transfected with vectors encoding green fluorescent protein (GFP), various forms of hLst2, and a plasmid containing the serum response element (SRE) fused to a luciferase reporter gene. Thereafter, cells were split into 24-well plates and serum-starved overnight, before treatment with EGF (20 ng/ml) for 8 hrs at 37°C. Light intensity measured using a luminometer, and normalized to GFP expression levels.
Immunofluorescence microscopy
Paraformaldehyde-fixed cells were permeabilized for 10 min at 22°C with 0.2% Triton X-100. For labeling, cover slips were incubated for 1 hour at room temperature with a primary antibody, followed by an additional hour with Cy2- or Cy3-conjugated secondary antibodies. Confocal microscopy was performed using a Zeiss Axiovert 100 TV microscope (Oberkochen, Germany) with a 63X/1.4 plan-Apochromat objective. Live cell imaging is described under Supplementary Procedures.
Mass spectrometry
GelCode-stained, excised gel bands were reduced (10 mM DTT), alkylated (10mM iodoacetamide), and digested overnight with trypsin (Promega, Madison, WI) at a 1:100 enzyme to substrate ratio. The tryptic peptides were resolved by reverse-phase chromatography using a linear gradient of 5 to 95% acetonitrile with 0.1% formic acid. Mass spectrometry was performed by an ion-trap mass spectrometer (LCQ-DecaXP, Thermo, San Jose, CA) in a positive ion mode. Repetitive full MS scan was used followed by collision-induced dissociation of the three most dominant ions selected from the first MS scan, or from a calculated mass list.
Supplementary Material
Acknowledgments
We thank Drs. S. Lev and S. Vincent (University of British Columbia) for plasmids. This work was supported in part by grants from the National Cancer Institute (grant CA72981), the German-Israel Foundation, Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and the Israel Science Foundation funded by the Israel Academy of Sciences. Y.Y. is the Harold and Zelda Goldenberg Professorial Chair of Molecular Cell Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alwan HA, van Leeuwen JE. UBPY-mediated epidermal growth factor receptor (EGFR) de-ubiquitination promotes EGFR degradation. J Biol Chem. 2007;282:1658–1669. doi: 10.1074/jbc.M604711200. [DOI] [PubMed] [Google Scholar]
- Borden KL. RING fingers and B-boxes: zinc-binding protein-protein interaction domains. Biochem Cell Biol. 1998;76:351–358. doi: 10.1139/bcb-76-2-3-351. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang B, Fischer JA. A specific protein substrate for a deubiquitinating enzyme: Liquid facets is the substrate of Fat facets. Genes Dev. 2002;16:289–294. doi: 10.1101/gad.961502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- Ebner R, Derynck R. Epidermal growth factor and transforming growth factor-alpha: differential intracellular routing and processing of ligand-receptor complexes. Cell Regul. 1991;2:599–612. doi: 10.1091/mbc.2.8.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Kwasnicka D, Yamada T, Hirohashi S, Vincent SR. BERP, a novel ring finger protein, binds to alpha-actinin-4. Biochem Biophys Res Commun. 2000;267:906–911. doi: 10.1006/bbrc.1999.2045. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Vincent SR. Cloning and characterization of a novel RING finger protein that interacts with class V myosins. J Biol Chem. 1999;274:19771–19777. doi: 10.1074/jbc.274.28.19771. [DOI] [PubMed] [Google Scholar]
- Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003;5:461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, McCullagh E, von Zastrow M. Essential role of Hrs in a recycling mechanism mediating functional resensitization of cell signaling. Embo J. 2005;24:2265–2283. doi: 10.1038/sj.emboj.7600688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa A, Hayes SJ, Lawe DC, Sudharshan E, Tuft R, Fogarty K, Lambright D, Corvera S. Structural basis for endosomal targeting by FYVE domains. J Biol Chem. 2004;279:5958–5966. doi: 10.1074/jbc.M310503200. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Crosetto N, Blagoev B, Raiborg C, Tikkanen R, Wagner S, Kowanetz K, Breitling R, Mann M, Stenmark H, et al. Regulation of ubiquitin-binding proteins by monoubiquitination. Nat Cell Biol. 2006;8:163–169. doi: 10.1038/ncb1354. [DOI] [PubMed] [Google Scholar]
- Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell. 2006;21:737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem J. 2006;399:361–372. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutateladze T. Phosphatidylinositol 3-phosphate recognition and membrane docking by the FYVE domain. Biochim Biophys Acta. 2006;1761:868–877. doi: 10.1016/j.bbalip.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998;12:3663–3674. doi: 10.1101/gad.12.23.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace G, Miaczynska M, Zerial M, Nebreda AR. Phosphorylation of EEA1 by p38 MAP kinase regulates mu opioid receptor endocytosis. Embo J. 2005;24:3235–3246. doi: 10.1038/sj.emboj.7600799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Bonifacino JS. Ubiquitin binding and conjugation regulate the recruitment of Rabex-5 to early endosomes. Embo J. 2008 doi: 10.1038/emboj.2008.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Row PE, Lorenzo O, Doherty M, Beynon R, Clague MJ, Urbe S. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr Biol. 2006;16:160–165. doi: 10.1016/j.cub.2005.11.073. [DOI] [PubMed] [Google Scholar]
- Miyake S, Lupher ML, Jr, Druker B, Band H. The tyrosine kinase regulator Cbl enhances the ubiquitination and degradation of the platelet-derived growth factor receptor alpha. Proc Natl Acad Sci U S A. 1998;95:7927–7932. doi: 10.1073/pnas.95.14.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, Szollosi J, Yarden Y. Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J Biol Chem. 2003;278:21323–21326. doi: 10.1074/jbc.C300096200. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Bremnes B, Mehlum A, Gillooly DJ, D’Arrigo A, Stang E, Stenmark H. FYVE and coiled-coil domains determine the specific localisation of Hrs to early endosomes. J Cell Sci. 2001;114:2255–2263. doi: 10.1242/jcs.114.12.2255. [DOI] [PubMed] [Google Scholar]
- Reck-Peterson SL, Provance DW, Jr, Mooseker MS, Mercer JA. Class V myosins. Biochim Biophys Acta. 2000;1496:36–51. doi: 10.1016/s0167-4889(00)00007-0. [DOI] [PubMed] [Google Scholar]
- Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2008;314:3093–3106. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Aasland R, Toh BH, D’Arrigo A. Endosomal localization of the autoantigen EEA1 is mediated by a zinc-binding FYVE finger. J Biol Chem. 1996;271:24048–24054. doi: 10.1074/jbc.271.39.24048. [DOI] [PubMed] [Google Scholar]
- Vicinanza M, D’Angelo G, Di Campli A, De Matteis MA. Function and dysfunction of the PI system in membrane trafficking. Embo J. 2008 doi: 10.1038/emboj.2008.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Sun W, Kujala P, Lotfi Y, Vida T, Bean A. CART: an Hrs/actinin-4/BERP/myosin V protein complex required for efficient receptor recycling. Virchows Arch. 2005;16:2470–2482. doi: 10.1091/mbc.E04-11-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Bais C, Greenwald I. Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulval development. Science. 2004;303:663–666. doi: 10.1126/science.1091639. [DOI] [PubMed] [Google Scholar]
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- Zwang Y, Yarden Y. p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. Embo J. 2006;25:4195–4206. doi: 10.1038/sj.emboj.7601297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.