

Abstract
The introduction of DNA microarrays and DNA sequencing technologies in medical genetics and diagnostics has been a challenge that has significantly transformed medical practice and patient management. Because of the great advancements in molecular genetics and the development of simple laboratory technology to identify the mutations in the causative genes, also the diagnostic approach to epilepsy has significantly changed. However, the clinical use of molecular cytogenetics and high-throughput DNA sequencing technologies, which are able to test an entire genome for genetic variants that are associated with the disease, is preparing a further revolution in the near future. Molecular Karyotype and Next-Generation Sequencing have the potential to identify causative genes or loci also in sporadic or non-familial epilepsy cases and may well represent the transition from a genetic to a genomic approach to epilepsy.
1. Introduction
In the last decades a large number of gene discoveries have changed our views of idiopathic and symptomatic epilepsy [1]. Indeed, idiopathic epilepsy has the considerable genetic advantage to be found very often in informative autosomal dominant families that have been of great relevance to map and to positional clone the causative gene, opening insight into the biology and molecular pathology of this condition [2, 3].
The search of epilepsy genes has allowed the identification of several genes in idiopathic generalized epilepsy (Table 1), the vast majority of which are channelopathies [4, 5] or affect the activity of excitatory or inhibitory neurotransmitters in central nervous system [6]. It is possible that the dominant nature of these genes due to the multisubunit composition of the molecules have greatly overestimated the role of their mutations in the disease.
Table 1.
Disease genes identified in generalized myoclonic epilepsy, febrile seizures, absences (37 genes).
| Gene Symbol | Gene name and description |
|---|---|
| ALDH7A1 | Aldehyde dehydrogenase 7 family, member A1 |
| BRD2 | Bromodomain containing 2 |
| CACNA1A | Calcium channel, voltage-dependent, P/Q type, alpha 1A subunit |
| CACNA1H | Calcium channel, voltage-dependent, T type, alpha 1H subunit |
| CACNB4 | Calcium channel, voltage-dependent, beta 4 subunit |
| CASR | Calcium-sensing receptor |
| CHRNA2 | Cholinergic receptor, nicotinic, alpha 2 (neuronal) |
| CHRNA4 | Cholinergic receptor, nicotinic, alpha 4 |
| CHRNB2 | Cholinergic receptor, nicotinic, beta 2 (neuronal) |
| CLCN2 | Chloride channel 2 |
| CSTB | Cystatin B (stefin B) |
| EFHC1 | EF-hand domain (C-terminal) containing 1 |
| EPM2A | Epilepsy, progressive myoclonus type 2A, Lafora disease (laforin) |
| GABRA1 | Gamma-aminobutyric acid (GABA) A receptor, alpha 1 |
| GABRB3 | Gamma-aminobutyric acid (GABA) A receptor, beta 3 |
| GABRD | Gamma-aminobutyric acid (GABA) A receptor, delta |
| GABRG2 | Gamma-aminobutyric acid (GABA) A receptor, gamma 2 |
| GPR98 | G protein-coupled receptor 98 |
| GRIN2A | Glutamate receptor, ionotropic, N-methyl D-aspartate 2A |
| GRIN2B | Glutamate receptor, ionotropic, N-methyl D-aspartate 2B |
| KCNMA1 | Potassium large conductance calcium-activated channel, subfamily M, alpha member 1 |
| KCNQ2 | Potassium voltage-gated channel, KQT-like subfamily, member 2 |
| KCNQ3 | Potassium voltage-gated channel, KQT-like subfamily, member 3 |
| KCTD7 | Potassium channel tetramerisation domain containing 7 |
| MBD5 | Methyl-CpG-binding domain protein 5 |
| ME2 | Malic enzyme 2, NAD(+)-dependent, mitochondrial |
| NHLRC1 | NHL repeat containing 1 |
| PCDH19 | Protocadherin 19 |
| PRICKLE1 | Prickle homolog 1 (Drosophila) |
| PRICKLE2 | Prickle homolog 2 (Drosophila) |
| SCARB2 | Scavenger receptor class B, member 2 |
| SCN1A | Sodium channel, voltage-gated, type I, alpha subunit |
| SCN1B | Sodium channel, voltage-gated, type I, beta subunit |
| SCN2A | Sodium channel, voltage-gated, type II, alpha subunit |
| SCN9A | Sodium channel, voltage-gated, type IX, alpha subunit |
| SLC2A1 | Solute carrier family 2 (facilitated glucose transporter), member 1 |
| TBC1D24 | TBC1 domain family, member 24 |
Other important insights came from the discoveries of causative genes of syndromic epilepsy (Table 2) [7] and other disorders where epilepsy is associated with encephalopathies (Table 3) [8], mental retardation with brain malformation (Table 4) [9, 10], other neurologic conditions including neuronal migration disorders (Table 5) [11], and inborn errors of metabolism (Tables 6 and 7) [12, 13]. Without any doubt, these discoveries have been great advances in the field; however, their impact on the management of epileptic patients was limited because of the failure to collect significant genetic information from each patient to distinguish the large number of genetic defects that can lead to the disease. Therefore, genetic testing was possible only for few or selected family cases.
Table 2.
Disease genes identified in syndromic epilepsy (47 genes).
| Gene symbol | Gene name and description | Syndrome |
|---|---|---|
| ARFGEF2 | ADP-ribosylation factor GEF2 | Periventricular heterotopia |
| ARHGEF9 | Cdc42 GEF 9 | Hyperekplexia with epilepsy |
| A2BP1 | Ataxin 2-binding protein 1 (RNA binding protein fox-1 homolog 1) | Mental retardation and epilepsy |
| ASPA | Aspartoacylase | Canavan syndrome |
| ATP1A2 | ATPase, Na/K transporting, alpha 2 polypeptide | Familial hemiplegic migraine |
| ATP2A2 | ATPase, Ca transporting, cardiac muscle, slow twitch 2 | Darier-White syndrome |
| ATP6V0A2 | ATPase, H+ transporting, lysosomal V0 subunit a2 | Cutis laxa with epilepsy and mental retardation |
| CACNA1A | Calcium channel, voltage-dependent, P/Q type, alpha 1A subunit | Familial hemiplegic migraine |
| CCDC88C | Coiled-coil domain containing 88C | Hydrocephalus with medial diverticulum |
| CLCNKA | Chloride channel Ka | Bartter syndrome |
| CLCNKB | Chloride channel Kb | Bartter syndrome |
| COH1 | Cohen syndrome protein 1—vacuolar protein sorting 13 homolog B | Cohen syndrome |
| DLGAP2 | Discs, large (Drosophila) homolog-associated protein 2 | Progressive epilepsy with mental retardation |
| GFAP | Glial fibrillary acidic protein | Alexander disease |
| GLI3 | GLI family zinc finger 3 | Pallister-hall syndrome |
| GLRA1 | Glycine receptor, alpha 1 | Hyperekplexia |
| GLRB | Glycine receptor, beta | Hyperekplexia |
| GPHN | Gephyrin | Hyperekplexia |
| KCNA1 | Potassium voltage-gated channel, shaker-related | Episodic ataxia |
| KCNJ1 | Potassium inwardly rectifying channel, subfamily J, member 1 | Bartter syndrome |
| KCNJ10 | Potassium inwardly rectifying channel, subfamily J, member 10 | Seizures, deafness, ataxia, mental retardation |
| KIAA1279 | Kinesin family member 1 binding protein | Goldberg-Shprintzen |
| LAMA2 | Laminin, alpha 2 | Merosin deficiency |
| LBR | Lamin B receptor | Pelger-Huet syndrome |
| LGI1 | Leucine-rich, glioma inactivated 1 | Autosomal dominant lateral temporal lobe epilepsy |
| MLC1 | Megalencephalic leukoencephalopathy with subcortical cysts 1 | Megalencephalic leukoencephalopathy with cysts |
| MLL2 | Myeloid/lymphoid or mixed-lineage leukemia 2 | Kabuki syndrome |
| NF1 | Neurofibromin 1 | Neurofibromatosis |
| NIPBL | Nipped-B homolog (Drosophila) | Cornelia de Lange syndrome |
| PANK2 | Pantothenate kinase 2 | Neurodegeneration with brain iron accumulation |
| PI12 | Serpin peptidase inhibitor, clade I (neuroserpin), member 1 | Encephalopathy with neuroserpin inclusion bodies |
| PIGV | Phosphatidylinositol glycan anchor biosynthesis, class V | Hyperphosphatasia with mental retardation |
| PLA2G6 | Phospholipase A2, group VI (cytosolic, calcium independent) | Infantile neuroaxonal dystrophy |
| RAI1 | Retinoic acid induced 1 | Smith Magenis syndrome |
| SCN8A | Sodium channel, voltage gated, type VIII, alpha subunit | Cerebellar atrophy, ataxia, and mental retardation |
| SETBP1 | SET binding protein 1 | Schinzel-Giedion midface retraction syndrome |
| SHH | Sonic hedgehog | Holoprosencephaly |
| SLC4A10 | Solute carrier family 4, sodium bicarbonate transporter, member 10 | Epilepsy with mental retardation |
| SLC6A5 | Solute carrier family 6 (neurotransmitter transporter, glycine), member 5 | Hyperekplexia |
| SMC1A | Structural maintenance of chromosomes 1A | Cornelia de lange syndrome |
| SMC3 | Structural maintenance of chromosomes 3 | Cornelia de lange syndrome |
| SYNGAP1 | Synaptic Ras GTPase activating protein 1 | Epilepsy and mental retardation |
| TBX1 | T-box 1 | Di George syndrome |
| TSC1 | Tuberous sclerosis 1 | Tuberous sclerosis |
| TSC2 | Tuberous sclerosis 2 | Tuberous sclerosis |
| VPS13A | Vacuolar protein sorting 13 homolog A | Neuroacanthocytosis |
| ZEB2 | Zinc finger E-box binding homeobox 2 | Mowat-Wilson syndrome |
Table 3.
Disease genes identified in epileptic encephalopathies (30 genes).
| Gene symbol | Gene Name and Description | Diseases |
|---|---|---|
| ARHGEF9 | Cdc42 guanine nucleotide exchange factor (GEF) 9 | Early infantile epileptic encephalopathy |
| ARX | Aristaless related homeobox | Early infantile epileptic encephalopathy |
| CDKL5 | Cyclin-dependent kinase-like 5 | Early infantile epileptic encephalopathy |
| CNTNAP2 | Contactin associated protein-like 2 | Pitt Hopkins syndrome |
| FOXG1 | Forkhead box G1 | Rett syndrome |
| GABRG2 | Gamma-aminobutyric acid (GABA) A receptor, gamma 2 | Early infantile epileptic encephalopathy |
| GRIN2A | Glutamate receptor, ionotropic, N-methyl D-aspartate 2A | Early infantile epileptic encephalopathy |
| GRIN2B | Glutamate receptor, ionotropic, N-methyl D-aspartate 2B | Early infantile epileptic encephalopathy |
| MAPK10 | Mitogen-activated protein kinase 10 | Lennox Gastaut syndrome |
| MECP2 | Methyl CpG binding protein 2 | Rett syndrome |
| NRXN1 | Neurexin 1 | Pitt Hopkins Syndrome |
| PCDH19 | Protocadherin 19 | Early infantile epileptic encephalopathy |
| PNKP | Polynucleotide kinase 3'-phosphatase | Early infantile epileptic encephalopathy |
| RNASEH2A | Ribonuclease H2, subunit A | Aicardi-Goutieres syndrome |
| RNASEH2B | Ribonuclease H2, subunit B | Aicardi-Goutieres syndrome |
| RNASEH2C | Ribonuclease H2, subunit C | Aicardi-Goutieres syndrome |
| SAMHD1 | SAM domain and HD domain 1 | Aicardi-Goutieres syndrome |
| SCN1A | Sodium channel, voltage-gated, type I, alpha subunit | Early infantile epileptic encephalopathy |
| SCN1B | Sodium channel, voltage-gated, type I, beta subunit | Early Infantile epileptic encephalopathy |
| SCN2A | Sodium channel, voltage-gated, type II, alpha subunit | Early infantile epileptic Encephalopathy |
| SCN9A | Sodium channel, voltage-gated, type IX, alpha subunit | Early infantile epileptic encephalopathy |
| SLC2A1 | Solute carrier family 2 (facilitated glucose transporter), member 1 | GLUT1 deficiency syndrome |
| SLC25A22 | Solute carrier family 25 (mitochondrial carrier: glutamate), member 22 | Early infantile epileptic encephalopathy |
| SLC9A6 | Solute carrier family 9 (sodium/hydrogen exchanger), member 6 | Angelman syndrome |
| SPTAN1 | Spectrin, alpha, non-erythrocytic 1 (alpha-fodrin) | Early infantile epileptic encephalopathy |
| STXBP1 | Syntaxin binding protein 1 | Early infantile epileptic encephalopathy |
| TCF4 | Transcription factor 4 | Pitt Hopkins syndrome |
| TREX1 | Three prime repair exonuclease 1 | Aicardi-Goutieres syndrome |
| UBE3A | Ubiquitin protein ligase E3A | Angelman syndrome |
| ZEB2 | Zinc finger E-box binding homeobox 2 | Mowat-Wilson syndrome |
Table 4.
Epilepsy with mental retardation and brain malformations.
| Gene symbol | Name | Disease |
|---|---|---|
| (a) Mental retardation (25 genes) | ||
|
| ||
| ARHGEF9 | Cdc42 guanine nucleotide exchange factor (GEF) 9 | Early infantile epileptic encephalopathy |
| ARX | Aristaless related homeobox | Early infantile epileptic encephalopathy |
| ATP6AP2 | ATPase, H+ transporting, lysosomal accessory protein 2 | Epilepsy with XLMR* |
| ATRX | Alpha thalassemia/mental retardation syndrome X-linked | Epilepsy with XLMR* |
| CASK | Calcium/calmodulin-dependent serine protein kinase (MAGUK family) | Mental retardation and microcephaly |
| CDKL5 | Cyclin-dependent kinase-like 5 | Early infantile epileptic encephalopathy |
| CUL4B | Cullin 4B | Epilepsy with XLMR* |
| CXORF5 | Oral-facial-digital syndrome 1 | Simpson-Golabi-Behmel syndrome |
| DCX | Doublecortin | Lissencephaly |
| FGD1 | FYVE, RhoGEF and PH domain containing 1 | Aarskog-Scott syndrome |
| GPC3 | Glypican 3 | Simpson-Golabi-Behmel syndrome |
| GRIA3 | Glutamate receptor, ionotrophic, AMPA 3 | Epilepsy with XLMR* |
| HSD17B10 | Hydroxysteroid (17-beta) dehydrogenase 10 | Epilepsy with XLMR* |
| JARID1C | Lysine (K)-specific demethylase 5C | Epilepsy with XLMR* |
| OPHN1 | Oligophrenin 1 | Epilepsy with XLMR* |
| PAK3 | P21 protein (Cdc42/Rac)-activated kinase 3 | Epilepsy with XLMR* |
| PHF6 | PHD finger protein 6 | Borjeson Forssmann Lehmann syndrome |
| PLP1 | Proteolipid protein 1 | Pelizaeus-Merzbacher disease |
| PQBP1 | Polyglutamine binding protein 1 | Epilepsy with XLMR* |
| RAB39B | RAB39B, member RAS oncogene family | Epilepsy with XLMR* |
| SLC9A6 | Solute carrier family 9 (sodium/hydrogen exchanger), member 6 | Angelman-Like syndrome |
| SMC1A | Structural maintenance of chromosomes 1A | Cornelia De Lange syndrome |
| SMS | Spermine synthase | Epilepsy with XLMR* |
| SRPX2 | Sushi-repeat containing protein, X-linked 2 | Rolandic epilepsy |
| SYP | Synaptophysin | Epilepsy with XLMR* |
| *XLMR: X-linked mental retardation | ||
|
| ||
| (b) Joubert syndrome (10 genes) | ||
|
| ||
| AHI1 | Abelson helper integration site 1 | Joubert syndrome |
| ARL13B | ADP-ribosylation factor-like 13B | Joubert syndrome |
| CC2D2A | Coiled-coil and C2 domain containing 2A | Joubert syndrome |
| CEP290 | Centrosomal protein 290 kDa | Joubert syndrome |
| CXORF5 | Oral-facial-digital syndrome 1 | Joubert syndrome |
| INPP5E | Inositol polyphosphate-5-phosphatase, 72 kDa | Joubert syndrome |
| NPHP1 | Nephronophthisis 1 (juvenile) | Joubert syndrome |
| RPGRIP1L | Retinitis pigmentosa GTPase regulator interacting protein 1 like | Joubert syndrome |
| TMEM67 | Transmembrane protein 67 | Joubert syndrome |
| TMEM216 | Transmembrane protein 216 | Joubert syndrome |
|
| ||
| (c) Lissencephaly and polymicrogyria (18 genes) | ||
|
| ||
| COL18A1 | Collagen, type XVIII, alpha 1 | Polymicrogyria |
| CPT2 | Carnitine palmitoyltransferase 2 | Polymicrogyria |
| DCX | Doublecortin | Lissencephaly |
| EOMES | Eomesodermin | Polymicrogyria |
| FGFR3 | Fibroblast growth factor receptor 3 | Polymicrogyria |
| FLNA | Filamin A, alpha | Periventricular heterotopia |
| GPR56 | G protein-coupled receptor 56 | Polymicrogyria |
| PAFAH1B1 | Platelet-activating factor acetylhydrolase 1b, regulatory subunit 1 (45kDa) | Lissencephaly |
| PAX6 | Paired box 6 | Polymicrogyria |
| PEX7 | Peroxisomal biogenesis factor 7 | Polymicrogyria |
| RAB3GAP1 | RAB3 GTPase activating protein subunit 1 (catalytic) | Warburg microsyndrome |
| RELN | Reelin | Lissencephaly |
| SNAP29 | Synaptosomal-associated protein, 29 kDa | Cerebral dysgenesis |
| SRPX2 | Sushi-repeat containing protein, X-linked 2 | Rolandic epilepsy |
| TUBA1A | Tubulin, alpha 1a | Lissencephaly |
| TUBA8 | Tubulin, alpha 8 | Polymicrogyria |
| TUBB2B | Tubulin, beta 2B | Polymicrogyria |
| VDAC1 | Voltage-dependent anion channel 1 | Polymicrogyria |
|
| ||
| (d) Severe microcephaly and pontocerebellar hypoplasia (22 genes) | ||
|
| ||
| ASPM | Asp (abnormal spindle) homolog, microcephaly associated (Drosophila) | Microcephaly |
| ATR | Ataxia telangiectasia and Rad3 related | Microcephaly |
| BUB1B | Budding uninhibited by benzimidazoles 1 homolog beta (yeast) | Microcephaly |
| CASK | Calcium/calmodulin-dependent serine protein kinase (MAGUK family) | Microcephaly |
| CDK5RAP2 [Microcephaly] | CDK5 regulatory subunit associated protein 2 | Microcephaly |
| CENPJ | Centromere protein J | Microcephaly |
| CEP152 | Centrosomal protein 152 kDa | Microcephaly |
| LIG4 | Ligase IV, DNA, ATP-dependent | Microcephaly |
| MCPH1 | Microcephalin 1 | Microcephaly |
| MED17 | Mediator complex subunit 17 | Microcephaly |
| NHEJ1 | Nonhomologous end-joining factor 1 | Microcephaly |
| PCNT | Pericentrin | Microcephalic osteodysplastic Dwarfism |
| PNKP | Polynucleotide kinase 3'-phosphatase | Microcephaly |
| PQBP1 | Polyglutamine binding protein 1 | X-linked mental retardation |
| RARS2 | Arginyl-tRNA synthetase 2, mitochondrial | Pontocerebellar hypoplasia |
| SLC25A19 | Solute carrier family 25 (mitochondrial thiamine pyrophosphate carrier), member 19 | Microcephaly |
| STIL | SCL/TAL1 interrupting locus | Microcephaly |
| TSEN2 | tRNA splicing endonuclease 2 homolog (S. cerevisiae) | Pontocerebellar hypoplasia |
| TSEN34 [Pontocerebellar Hypoplasia] | tRNA splicing endonuclease 34 homolog (S. cerevisiae) | Pontocerebellar hypoplasia |
| TSEN54 [Pontocerebellar Hypoplasia] | tRNA splicing endonuclease 54 homolog (S. cerevisiae) | Pontocerebellar hypoplasia |
| VRK1 | Vaccinia related kinase 1 | Pontocerebellar hypoplasia |
| WDR62 | WD repeat domain 62 | Microcephaly, cortical malformations and mental retardation |
|
| ||
| (e) Walker-Warburg syndrome (WWS) or muscle, eye and brain disease (6 genes) anomalies type A2 (MDDGA2) | ||
|
| ||
| FKRP | Fukutin-related protein | Walker-Warburg syndrome |
| FKTN | Fukutin | Walker-Warburg syndrome |
| LARGE | Like-glycosyltransferase | Walker-Warburg syndrome |
| POMGNT1 | Protein O-linked mannose beta1,2-N-acetylglucosaminyltransferase | Walker-Warburg syndrome |
| POMT1 | Protein-O-mannosyltransferase 1 | Walker-Warburg syndrome |
| POMT2 | Protein-O-mannosyltransferase 2 | Walker-Warburg Syndrome |
|
| ||
| (f) Holoprosencephaly (HPE) (8 genes) | ||
|
| ||
| FGF8 | Fibroblast growth factor 8 (androgen-induced) | Holoprosencephaly |
| GLI2 | GLI family zinc finger 2 | Holoprosencephaly 9 |
| GLI3 | GLI family zinc finger 3 | Greig cephalopolysyndactyly syndrome |
| PTCH1 | patched 1 | Holoprosencephaly 7 |
| SHH | Sonic Hedgehog | Holoprosencephaly 3 |
| SIX3 | SIX homeobox 3 | Holoprosencephaly 2 |
| TGIF1 | TGFB-induced factor homeobox 1 | Holoprosencephaly 4 |
| ZIC2 | Zic family member 2 | Holoprosencephaly 5 |
Table 5.
Epilepsy with other neurological problems.
| Gene symbol | Name | Disease |
|---|---|---|
| (a) Leukodystrophies (20 genes) | ||
|
| ||
| ARSA | Arylsulfatase A | Leukodystrophy metachromatic (MLD) |
| ASPA | Aspartoacylase | Canavan disease |
| EIF2B1 | Eukaryotic translation initiation factor 2B, subunit 1 alpha, 26 kDa | Leukodystrophy |
| EIF2B2 | Eukaryotic translation initiation factor 2B, subunit 2 beta, 39 kDa | Leukodystrophy |
| EIF2B3 | Eukaryotic translation initiation factor 2B, subunit 3 gamma, 58 kDa | Leukodystrophy |
| EIF2B4 | Eukaryotic translation initiation factor 2B, subunit 4 delta, 67 kDa | Leukodystrophy |
| EIF2B5 | Eukaryotic translation initiation factor 2B, subunit 5 epsilon, 82 kDa | Leukodystrophy |
| GALC | Galactosylceramidase | Leukodystrophy globoid cell (GLD) |
| GFAP | Glial fibrillary acidic protein | Alexander disease |
| MLC1 | Megalencephalic leukoencephalopathy with subcortical cysts 1 | Megalencephalic leukoencephalopathy |
| NOTCH3 | Notch3 | CADASIL |
| PLP1 | Proteolipid protein 1 | Leukodystrophy hypomyelinating type 1 (HLD1) |
| PSAP | Prosaposin | Leukodystrophy metachromatic |
| RNASEH2A | Ribonuclease H2, subunit A | Aicardi-Goutieres syndrome type 4 (AGS4) |
| RNASEH2B | Ribonuclease H2, subunit B | Aicardi-Goutieres syndrome type 2 (AGS2) |
| RNASEH2C | Ribonuclease H2, subunit C | Aicardi-Goutieres syndrome type 3 (AGS3 |
| SAMHD1 | SAM domain and HD domain 1 | Aicardi-Goutieres syndrome type 5 (AGS5) |
| SDHA | Succinate dehydrogenase complex, subunit A, flavoprotein (Fp) | Leigh syndrome |
| SUMF1 | Sulfatase modifying factor 1 | Multiple sulfatase deficiency (MSD) |
| TREX1 | Three prime repair exonuclease 1 | Aicardi-Goutieres syndrome type 1 (AGS1) |
|
| ||
| (b) Migraine (6 genes) | ||
|
| ||
| ATP1A2 | ATPase, Na+/K+ transporting, alpha 2 polypeptide | Migraine familial hemiplegic type 2 (FHM2) |
| CACNA1A | Calcium channel, voltage-dependent, P/Q type, alpha 1A subunit | Spinocerebellar ataxia type 6 (SCA6) |
| NOTCH3 | Notch 3 | CADASIL |
| POLG | Polymerase (DNA directed), gamma | Progressive external ophthalmoplegia |
| SCN1A | Sodium channel, voltage-gated, type I, alpha subunit | Migraine familial hemiplegic type 3 (FHM3) |
| SLC2A1 | Solute carrier family 2 (facilitated glucose transporter), member 1 | GLUT1 deficiency type 1 (GLUT1DS1) syndrome |
|
| ||
| (c) Disorders of Ras-MAPK pathway with epilepsy (13 genes) | ||
|
| ||
| BRAF | V-raf murine sarcoma viral oncogene homolog B1 | Cardiofaciocutaneous (CFC ) syndrome |
| CBL | Cas-Br-M (murine) ecotropic retroviral transforming sequence | Noonan syndrome-like disorder (NSL) |
| HRAS | V-Ha-ras Harvey rat sarcoma viral oncogene homolog | Faciocutaneoskeletal (FCSS) syndrome |
| KRAS | V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog | Noonan type 3 (NS3) syndrome |
| MAP2K1 | Mitogen-activated protein kinase kinase 1 | cardiofaciocutaneous (CFC) syndrome |
| MAP2K2 | Mitogen-activated protein kinase kinase 2 | cardiofaciocutaneous (CFC) syndrome |
| NF1 | Neurofibromin 1 | Neurofibromatosis type 1 |
| NRAS | Neuroblastoma RAS viral (v-ras) oncogene homolog | Noonan type 6 (NS6) syndrome |
| PTPN11 | Protein tyrosine phosphatase, non-receptor type 11 | LEOPARD type 1 (LEOPARD1) syndrome |
| RAF1 | V-raf-1 murine leukemia viral oncogene homolog 1 | Noonan type 5 (NS5) syndrome |
| SHOC2 | Soc-2 suppressor of clear homolog (C. elegans) | Noonan syndrome-like with loose anagen hair |
| SOS1 | Son of sevenless homolog 1 (Drosophila) | Noonan type 4 (NS4) syndrome |
| SPRED1 | Sprouty-related, EVH1 domain containing 1 | Neurofibromatosis type 1-like syndrome |
|
| ||
| (d) Hyperekplexia (5 genes) | ||
|
| ||
| ARHGEF9 | Cdc42 guanine nucleotide exchange factor (GEF) 9 | Hyperekplexia with epilepsy |
| GLRA1 | Glycine receptor, alpha 1 | Hyperekplexia with epilepsy |
| GLRB | Glycine receptor, beta | Hyperekplexia with epilepsy |
| GPHN | Gephyrin | Hyperekplexia with epilepsy |
| SLC6A5 | solute carrier family 6 (neurotransmitter, transporter, glycine), member 5 | Hyperekplexia with epilepsy |
|
| ||
| (e) Neuronal migration disorders (31 genes) | ||
|
| ||
| ARFGEF2 | ADP-ribosylation factor guanine nucleotide-exchange factor 2 (brefeldin A-inhibited) | Microcephaly |
| ARX | Aristaless-related homeobox | Early infantile epileptic encephalopathy |
| COL18A1 | Collagen, type XVIII, alpha 1 | Polymicrogyria |
| COL4A1 | Collagen, type IV, alpha 1 | Porencephaly |
| CPT2 | Carnitine palmitoyltransferase 2 | Polymicrogyria |
| DCX | Doublecortin | Lissencephaly |
| EMX2 | Empty spiracles homeobox 2 | Schizencephaly |
| EOMES | Eomesodermin | Polymicrogyria |
| FGFR3 | Fibroblast growth factor receptor 3 | Polymicrogyria |
| FKRP | Fukutin related protein | Walker-Warburg syndrome |
| FKTN | Fukutin | Walker-Warburg syndrome |
| FLNA | Filamin A, alpha | Periventricular heterotopia |
| GPR56 | G protein-coupled receptor 56 | Polymicrogyria |
| LAMA2 | Laminin, alpha2 | Merosin deficiency |
| LARGE | Like-glycosyltransferase | Walker-Warburg syndrome |
| PAFAH1B1 | Platelet-activating factor acetylhydrolase 1b, regulatory subunit 1 (45 kDa) | Lissencephaly |
| PAX6 | Paired box 6 | Polymicrogyria |
| PEX7 | Peroxisomal biogenesis factor 7 | Polymicrogyria |
| POMGNT1 | Protein O-linked mannose beta1,2-N-acetylglucosaminyltransferase | Walker-Warburg syndrome |
| POMT1 | Protein O-mannosyltransferase 1 | Walker-Warburg syndrome |
| POMT2 | Protein O-mannosyltransferase 2 | Walker-Warburg syndrome |
| PQBP1 | Polyglutamine binding protein 1 | X-linked mental retardation |
| RAB3GAP | RAB3 GTPase activating protein subunit 1 (catalytic) | Warburg microsyndrome |
| RELN | Reelin | Lissencephaly |
| SNAP29 | Synaptosomal-associated protein, 29 kDa | Cerebral dysgenesis |
| SRPX2 | Sushi-repeat containing protein, X-linked 2 | Rolandic epilepsy |
| TUBA1A | Tubulin, alpha 1a | Lissencephaly |
| TUBA8 | Tubulin, alpha 8 | Polymicrogyria |
| TUBB2B | Voltage-dependent anion channel 1 | Polymicrogyria |
| VDAC1 | Voltage-dependent anion channel 1 | Polymicrogyria |
| WDR62 | WD repeat domain 62 | Microcephaly, cortical malfor, mental retardatation |
Table 6.
Inherited errors of metabolism with epilepsy (49 genes).
| Gene symbol | Defective enzyme name | Disease |
|---|---|---|
| ABCC8 | ATP-binding cassette, subfamily C (CFTR/MRP), member 8 | Hypoglcemia |
| ACY1 | Aminoacylase1 | Aminoacylase1 deficiency |
| ADSL | Adenylosuccinate lyase | Adenylosuccinase deficiency |
| AGA | Aspartylglucosaminidase | Aspartylglucosaminuria |
| ALDH4A1 | Aldehyde dehydrogenase 4 family, member A1 | Hyperprolinemia |
| ALDH5A1 | Aldehyde dehydrogenase 5 family, member A1 | Succinic Semialdehyde dehydrogenase deficiency |
| ALDH7A1 | Aldehyde dehydrogenase 7 family, member A1 | Pyridoxine deficiency |
| ARG1 | Liver arginase | Argininemia |
| ARSA | Arylsulfatase A | Metachromatic leukoodystrophy |
| ASPA | Aspartoacylase | Canavan disease |
| ATIC | 5-aminoimidazole-4-carboxamide ribonucleotide (AICAr) formyltransferase/IMP cyclohydrolase | AICAr transformylase/IMP cyclohydrolase deficiency (ATIC Deficiency) |
| BTD | Biotinidase | Biotinidase deficiency |
| CPT2 | Carnitine palmitoyltransferase 2 | Carnitine palmitoyltransferase II deficiency |
| CTSA | Cathepsin A | Galactosialidosis |
| DPYD | Dihydropyrimidine dehydrogenase | Dihydropyrimidine dehydrogenase deficiency |
| ETFA | Electron-transfer-flavoprotein, alpha polypeptide | Glutaraciduria |
| ETFB | Electron-transfer-flavoprotein, beta polypeptide | Glutaraciduria |
| ETFDH | Electron-transferring-flavoprotein dehydrogenase | Glutaraciduria |
| FH | Fumarate hydratase | Fumarase deficiency |
| FOLR1 | Folate receptor 1 (adult) | Cerebral folate transport deficiency |
| FUCA1 | Fucosidase, alpha-L- 1, tissue | Fucosidosis |
| GALC | Galactosylceramidase | Krabbe disease |
| GAMT | Guanidinoacetate N-methyltransferase | Guanidinoacetate N-methyltransferase deficiency |
| GCDH | Glutaryl-CoA dehydrogenase | Glutaraciduria |
| GCSH | Glycine cleavage system protein H (aminomethyl carrier) | Glycine encephalopathy |
| GCST | Glycine cleavage system protein T (aminomethyltransferase) | Glycine encephalopathy |
| GLB1 | Galactosidase, beta 1 | Gangliosidosis |
| GLDC | Glycine dehydrogenase (decarboxylating) | Glycine encephalopathy |
| GNE | Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase | Sialuria |
| HEXA | Hexosaminidase A (alpha polypeptide) | Gangliosidosis |
| HEXB | Hexosaminidase B (beta polypeptide) | Gangliosidosis |
| HPD | 4-Hydroxyphenylpyruvate dioxygenase | Tyrosinemia |
| L2HGDH | L-2-Hydroxyglutarate dehydrogenase | L-2-Hydroxyglutaric aciduria |
| LAMA2 | Laminin, alpha 2 | Muscular dystrophy |
| MOCS1 | Molybdenum cofactor synthesis 1 | Molybdene cofactor deficiency |
| MOCS2 | Molybdenum cofactor synthesis 2 | Molybdene cofactor deficiency |
| NEU1 | Sialidase 1 (lysosomal sialidase) | Neuraminidase deficiency |
| NPC1 | Niemann-Pick disease, type C1 | Niemann-Pick disease |
| NPC2 | Niemann-Pick disease, type C2 | Niemann-Pick disease |
| PGK1 | Phosphoglycerate kinase 1 | GAMT deficiency |
| PRODH | Proline dehydrogenase (oxidase) 1 | Hyperprolinemia |
| PSAP | Prosaposin | Krabbe disease |
| QDPR | Quinoid dihydropteridine reductase | Hyperphenylalaninemia |
| SLC17A5 | Solute carrier family 17 (anion/sugar transporter), member 5 | Sialuria |
| SLC25A15 | Solute carrier family 25 (mitochondrial carrier; ornithine transporter) member 15 | Ornithine translocase deficiency |
| SLC46A1 | Solute carrier family 46 (folate transporter), member 1 | Folate malabsorption |
| SMPD1 | Sphingomyelin phosphodiesterase 1, acid lysosomal | Niemann pick disease |
| SUMF1 | Sulfatase modifying factor 1 | Sulfatidosis |
| SUOX | Sulfite oxidase | Sulfitoxidasis |
Table 7.
Other inherited errors of metabolism with epilepsy.
| Gene symbol | Defective enzyme name | Disease |
|---|---|---|
| (a) Congenital Disorder of Glycosylation (CDG) (23 genes) | ||
|
| ||
| ALG1 | N-linked glycosylation 1, beta-1,4-mannosyltransferase homolog | CDG |
| ALG2 | N-linked glycosylation 2, alpha-1,3-mannosyltransferase homolog | CDG |
| ALG3 | N-linked glycosylation 3, alpha-1,3-mannosyltransferase homolog | CDG |
| ALG6 | N-linked glycosylation 6, alpha-1,3-glucosyltransferase homolog | CDG |
| ALG8 | N-linked glycosylation 8, alpha-1,3-glucosyltransferase homolog | CDG |
| ALG9 | N-linked glycosylation 9, alpha-1,3-glucosyltransferase homolog | CDG |
| ALG12 | N-linked glycosylation 12, alpha-1,3-glucosyltransferase homolog | CDG |
| B4GALT1 | UDP-Gal: betaGlcNAc beta 1,4-galactosyltransferase, polypeptide 1 | CDG |
| COG1 | Component of oligomeric golgi complex 1 | CDG |
| COG7 | Component of oligomeric golgi complex 7 | CDG |
| COG8 | Component of oligomeric golgi complex 8 | CDG |
| DOLK | Dolichol kinase | CDG |
| DPAGT1 | Dolichyl-phosphate (UDP-N-acetylglucosamine) N-acetyl glucosamine phosphotransferase 1 (GlcNAc-1-P transferase) |
CDG |
| DPM1 | Dolichyl-phosphate mannosyltransferase polypeptide 1, catalytic subunit | CDG |
| DPM3 | Dolichyl-phosphate mannosyltransferase polypeptide 3 | CDG |
| MOGS | Mannosyl-oligosaccharide glucosidase | CDG |
| MGAT2 | Mannosyl (alpha-1,6-)-glycoprotein beta-1,2-N-acetylglucosaminyltransferase | CDG |
| MPDU1 | Mannose-P-dolichol utilization defect 1 | CDG |
| MPI | Mannose phosphate isomerase | CDG |
| PMM2 | Phosphomannomutase 2 | CDG |
| RFT1 | Requiring fifty three 1 homolog | CDG |
| SLC35A1 | Solute carrier family 35 (CMP-sialic acid transporter), member A1 | CDG |
| SLC35C1 | Solute carrier family 35, member C1 | CDG |
|
| ||
| (b) Neuronal ceroid lipofuscinosis (NCL) (8 genes) | ||
|
| ||
| CLN3 | Ceroid-lipofuscinosis, neuronal 3 | NLC |
| CLN5 | Ceroid-lipofuscinosis, neuronal 5 | NLC |
| CLN6 | Ceroid-lipofuscinosis, neuronal 6 | NLC |
| CLN8 | Ceroid-lipofuscinosis, neuronal 8 | NLC |
| CTSD | Cathepsin D | NLC |
| MFSD8 | Major facilitator superfamily domain containing 8 | NLC |
| PPT1 | Palmitoyl-protein thioesterase 1 | NLC |
| TPP1 | Tripeptidyl peptidase I | NLC |
|
| ||
| (c) Defects of mitochondrial metabolism including coenzyme Q deficiency (35 genes) | ||
|
| ||
| APTX | Aprataxin | Coenzyme Q10 Deficiency |
| ATPAF2 | ATP synthase mitochondrial F1 complex assembly factor 2 | ATPase deficiency |
| BCS1L | BCS1-like | Leigh syndrome |
| C12ORF65 | Chromosome 12 open reading frame 65 | Leigh syndrome |
| C8ORF38 | Chromosome 8 open reading frame 38 | Leigh syndrome |
| CABC1 | Chaperone activity of bc1 complex-like, mitochondria | Coenzyme Q10 deficiency |
| COQ2 | Coenzyme Q2 homolog, prenyltransferase (yeast) | Coenzyme Q10 deficiency |
| COQ9 | Coenzyme Q9 homolog (S. cerevisiae) | Coenzyme Q10 deficiency |
| COX10 | COX10 homolog, cytochrome c oxidase assembly protein, heme A: farnesyltransferase (yeast) | Leigh syndromeCOX10 |
| COX15 | COX15 homolog, cytochrome c oxidase assembly protein (yeast) | Leigh syndrome |
| DLD | Dihydrolipoamide dehydrogenase | Leigh syndrome |
| GCSH | Glycine cleavage system protein H (aminomethyl carrier) | Glycine encephalopathy |
| GCST | Aminomethyltransferase (glycine cleavage system protein T) | Glycine encephalopathy |
| GLDC | Glycine dehydrogenase (decarboxylating) | Glycine encephalopathy |
| HSD17B10 | Hydroxysteroid (17-beta) dehydrogenase 10 | HSD17B10 deficiency |
| LRPPRC | Leucine-rich PPR-motif containing | Leigh syndrome |
| NDUFA2 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 2,8 kDa | Leigh syndrome |
| NDUFS1 | NADH dehydrogenase (ubiquinone) Fe-S protein 1, 75 kDa | Leigh syndrome |
| NDUFS3 | NADH dehydrogenase (ubiquinone) Fe-S protein 3, 30 kDa | Leigh syndrome |
| NDUFS4 | NADH dehydrogenase (ubiquinone) Fe-S protein 4, 18 kDa | Leigh syndrome |
| NDUFS7 | NADH dehydrogenase (ubiquinone) Fe-S protein 7, 20 kDa | Leigh syndrome |
| NDUFS8 | NADH dehydrogenase (ubiquinone) Fe-S protein 8, 23 kDa | Leigh syndrome |
| NDUFV1 | NADH dehydrogenase (ubiquinone) flavoprotein 1, 51 kDa | Leigh syndrome |
| PC | Pyruvate carboxylase | Leigh syndrome |
| PDHA1 | Pyruvate dehydrogenase (lipoamide) alpha 1 | Leigh syndrome |
| PDSS1 | Prenyl (decaprenyl) diphosphate synthase, subunit 1 | Coenzyme Q10 deficiency |
| PDSS2 | Prenyl (decaprenyl) diphosphate synthase, subunit 2 | Coenzyme Q10 deficiency] |
| POLG | Polymerase (DNA directed), gamma | Mitochondrial DNA depletion Syndrome |
| RARS2 | Arginyl-tRNA synthetase 2, mitochondrial | Pontocerebellar hypoplasia |
| SCO2 | SCO cytochrome oxidase deficient homolog 2 (yeast) | Leigh syndrome |
| SDHA | Succinate dehydrogenase complex, subunit A, flavoprotein (Fp) | Leigh syndrome |
| SURF1 | Surfeit 1 | Leigh syndrome |
| TACO1 | Translational activator of mitochondrially encoded cytochrome c oxidase I | Leigh syndrome |
| TMEM70 | Transmembrane protein 70 | Encephalocardiomyopathy |
| VDAC1 | voltage-dependent anion channel 1 | VDAC deficiency |
|
| ||
| (d) Mucopolysaccharidosis (MPS) and mucolipidosis (MLP) (15 genes) | ||
|
| ||
| ARSB | Arylsulfatase B | MPS 6 (Maroteaux-Lamy syndrome) |
| GALNS | Galactosamine (N-acetyl)-6-sulfate sulfatase | MPS 4A (Morquio syndrome) |
| GLB1 | Galactosidase, beta 1 | GM1-gangliosidosis |
| GNPTAB | N-acetylglucosamine-1-phosphate transferase, alpha and beta subunits | Mucolipidosi 2 (I cell disease) and 3A |
| GNPTG | N-acetylglucosamine-1-phosphate transferase, gamma subunit | Mucolipidosi 3C |
| GNS | Glucosamine (N-acetyl)-6-sulfatase | MPS 3D (Sanfilippo D syndrome) |
| GUSB | Glucuronidase, beta | MPS 7 (Sly syndrome) |
| HGSNAT | Heparan-alpha-glucosaminide N-acetyltransferase | MPS 3C (Sanfilippo C syndrome) |
| HYAL1 | Hyaluronoglucosaminidase 1 | MPS 9 |
| IDS | Iduronate 2-sulfatase | MPS 2 (Hunter syndrome) |
| IDUA | Iduronidase, alpha-L- | MPS 1H (Hurler syndrome) |
| MCOLN1 | Nucolipin 1 | Mucolipidosi 4 |
| NAGLU | N-acetylglucosaminidase, alpha | MPS 3B (Sanfilippo B syndrome) |
| SGSH | N-sulfoglucosamine sulfohydrolase | MPS 3A (Sanfilippo A syndrome) |
| SUMF1 | Sulfatase modifying factor 1 | Multiple sulfatase deficiency |
|
| ||
| (e) Peroxisome biogenesis disorders (PBD) (9 genes): Zellweger syndrome (ZWS): neonatal adrenoleukodystrophy (NALD): infantile refsum disease (IRD): rhizomelic chondrodysplasia punctata type 1 (RCDP1) | ||
|
| ||
| PEX1 | Peroxisomal biogenesis factor 1 | ZWS-NADL-IRD |
| PEX2 | Peroxisomal biogenesis factor 2 | ZWS-IRD |
| PEX3 | Peroxisomal biogenesis factor 3 | ZWS |
| PEX5 | Peroxisomal biogenesis factor 5 | ZWS-NADL |
| PEX6 | Peroxisomal biogenesis factor 6 | ZWS |
| PEX7 | Peroxisomal biogenesis factor 7 | RCDP1 |
| PEX12 | Peroxisomal biogenesis factor 12 | ZWS |
| PEX14 | Peroxisomal biogenesis factor 14 | ZWS |
| PEX26 | Peroxisomal biogenesis factor 26 | ZWS-NADL-IRD |
Technical improvements in human chromosomes recognition and better definition of chromosome regions realized by increasing the number of detectable chromosome bands have provided higher resolution of normal and pathological karyotype. It is today well established an association between epileptic seizures and chromosome abnormalities recognized by high-resolution chromosome banding [14, 15]. However, the type and the size of the chromosome defects are not always easy to detect even by the highest-resolution cytogenetic techniques available for light microscopes.
The identification of the specific genetic defect in a patient with epilepsy may clarify the diagnosis (diagnostic testing), suggest the prognosis, assist with treatment and management (e.g., the use of a ketogenic diet in glucose transporter type 1 deficiency syndrome or the avoidance of lamotrigine, phenytoin, and carbamazepine in Dravet syndrome), elucidate the risk of a disease in family members and future children, and save the patient from further diagnostic evaluation and potentially invasive testing.
In asymptomatic subjects with increased risk of seizures because of a family history, genetic test may predict onset of epilepsy (predictive testing) [16, 17]. Despite such potential benefits, genetic testing has also potential harms, such as its ethical, legal, and social implications, and the potential for stigma, distress, adverse labeling, and nonconfidentiality that exists in the setting of inadequate safeguards against discrimination [18]. Considering that our understanding of the epidemiology and clinical utility of genetic testing in the epilepsies is incomplete, the assessment of these potential benefits and harms is particularly complex and is closely linked to the clinical scenario.
The International League Against Epilepsy (ILAE) Genetic Commission presented a tool in the approach to specific tests for epilepsy [16]. According to ILAE report, the diagnostic genetic testing is “very useful” in individual affected by early-onset spasms, X-linked infantile spasms, Dravet and related syndromes, Ohtahara syndrome, epilepsy and mental retardation limited to females, early-onset absence epilepsy, autosomal dominant nocturnal frontal lobe epilepsy, and epilepsy with paroxysmal exercise-induced dyskinesia; the predictive testing is “very useful” in unaffected relatives of individuals affected by Dravet syndrome and epilepsy and mental retardation limited to females [16]. Considering the potential harms, genetic testing should always be performed with the patient's consent or parental consent in the case of minors. A team approach, including a genetic counselor, a psychologist, and a social worker, is recommended throughout the process of evaluation.
In the last years a number of new molecular genetic technologies became available and they promise to change genetic testing for epilepsy, allowing to extend genetic analysis also to sporadic or nonfamilial cases. Two are the major new technologies that can affect the management of epileptic patients: Oligonucleotide Arrays Comparative Genomic Hybridization (Array-CGH) and Next-Generation Sequencing (NGS).
2. Molecular Karyotype
During the last 50 years cytogenetics has evolved from simple chromosome counting or banded chromosome morphological identification under light microscope to a molecular approach where chromosomes are analyzed through sophisticated computer system for their ability to hybridize to specific oligonucleotides spanning the entire genome [19]. Array-CGH is nowadays a basic diagnostic tool for clinical diagnosis of several types of developmental delays [20], intellectual disabilities [21, 22], and congenital abnormalities [23]. Epilepsy is also enjoying several advantages from the use of this technology that significantly improves diagnostic resolution of classic cytogenetics [24, 25].
Chromosomes did not become individually identifiable before the discovery that several procedures could create reproducible, permanent, and specific banding patterns [26, 27]. This was fundamental for gene mapping and positional cloning of disease genes and also revealed a large number of rare and subtle pathological conditions that disturbed the normal band patterning of chromosomes. The improvements of high-resolution banding techniques allowed the identification of several subtle chromosomal abnormalities associated with epilepsy [14, 15]. The possibility to study these chromosomes regions with specific hybridization DNA probes through fluorescence in situ hybridization (FISH) greatly improved sensitivity to detect small chromosomal aberrations in specific regions [28].
Today molecular karyotyping is rapidly replacing conventional cytogenetics and FISH. This name refers to the analysis of all chromosomes using hybridization to standard DNA sequences arranged on a “chip” rather than microscope observation. The technological development of this approach allows now clinicians to evaluate the entire genome for copy number variants (CNVs, duplications, deletions) in a single test. The high resolution of this approach is however limited by the difficulty to identify balanced chromosome translocations or inversions, even if this powerful technique recognizes in many of them microdeletions or cryptic anomalies at the chromosomal breakpoints. Detection of deletions or duplications is based on the comparison of two genomes (Figure 1). Labelled patient DNA is cohybridized with control DNA to an array spotted with oligonucleotide DNA probes spanning the entire genome at critical intervals. The distance between these oligonucleotide sequences in the genome marks the resolution of the technique and can be as low as 1000 bp. The intensity of the signal from patient and control are then read and normalized by an electronic scanning device coupled with a software that generates a graphic plot of intensities for each probe.
Figure 1.
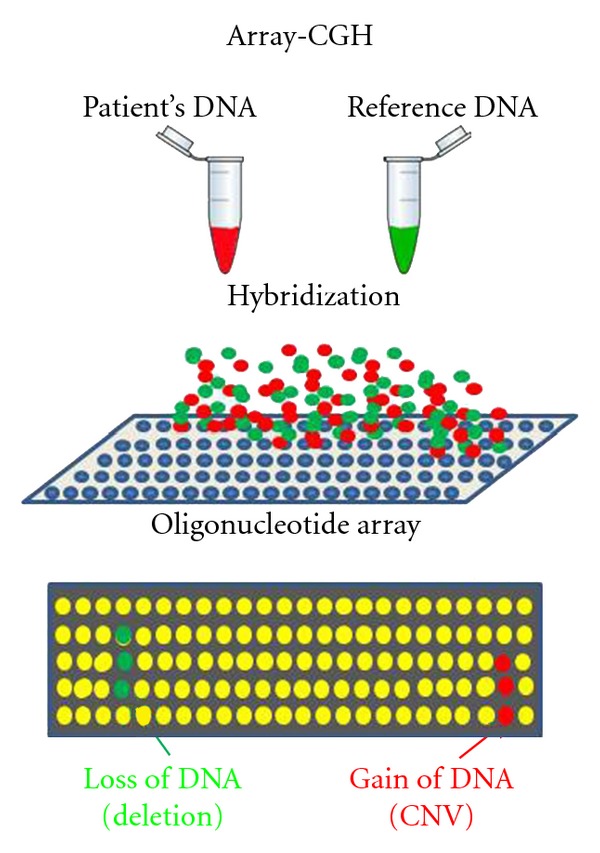
A search of Online Mendelian Inheritance in Man (OMIM) clinical synopsis with the term “seizure” reveals that there are at least 754 mendelian disorders in which epilepsy is or can be part of the clinical condition, but not the main feature. Many of these disorders can be associated with DNA sequence mutations or subtle chromosomal anomalies that can be conveniently detected by array-CGH. Some will be private or sporadic cases and others will be familial. With the widespread use of CGH in both circumstances, many more genetic events will be reported in patients and the genetic aetiology will be recognized making possible over the time to saturate the genome with all possible loci and events that have an epileptogenic role.
3. Next-Generation Sequencing
From the publication of the draft of human genome sequence in Nature, on 15th February 2001 issue, our view and knowledge of human genome has considerably changed [29] and the technologies to sequence DNA are today of common use in diagnostic practice and much cheaper. Chain termination or Sanger's method [30] was largely used for the Human Genome Project and has dominated the past decades. The logic of this technology was to create by synthesis a population of DNA fragments of different size each one terminated at all possible positions by one of the four labelled dideoxynucleotides (ddNTPs) terminators. Separation of these fragments by polyacrylamide gel or capillary electrophoresis allowed the reading of the sequence through the first developed sequencing machines that could distinguish the fluorescence emitted by the blocking ddNTP [31]. Therefore, these are considered the “first generation” of DNA sequencing technologies.
The need to reduce the cost of large sequencing projects has stimulated the development of a variety of cheaper sequencing technologies that are generally called “Next-Generation Sequencing” (NGS) [32, 33]. The final goal of this new field is to reduce the cost of human genome sequencing till or lower than $1,000 per genome to make it available for common medical practice and diagnostic use [34]. The development of further third-generation sequencing technologies should make possible to sequence single DNA molecules in real time with a cost that it is projected to be very close to the goal [35].
The development of NGS platforms was a major progress in the technology because, differently from Sanger method, rather than producing about one thousand nucleotides for run, they are able to produce orders of magnitude more sequence data using massive parallel process, resulting in substantial increase of data at a lower cost per nucleotide [36, 37].
Several commercial platforms are today available, including Roche/454 [38], Illumina/Solexa and Life Technologies/SOLiD (Table 8(a)). In very general terms these platforms follow similar process that includes: (a) template preparation by breaking large DNA macromolecule to generate short fragment libraries with platform-specific synthetic DNA adapters at the fragment ends, (b) massive and parallel clonal amplification of individual DNA fragment molecules on glass slide or microbeads by PCR [39] to generate a sufficient copy number of the labelled fragment to be detected by the machine optical system, and (c) sequencing by several cycles of extensions that are repeated and detected automatically to create short reads [40]. The data of these reads are then collected by the device, and the alignment of the short reads with specific software allows to rebuild the initial template sequence. Helicos and Pacific Biosystem platforms (Table 8(b)) are substantially different because they use a more advanced laser-based detection system that does not require massive parallel amplification with the considerable advantages to simplify preparation process, to eliminate PCR-induced bias and errors, and to make easy data collection. Ion Torrent developed an entirely new approach to sequencing based on hydrogen ion release when a nucleotide is incorporated into a DNA strand by polymerase (Table 8) [41]. An ion sensor can detect hydrogen ions and convert this ion chemical signal to digital sequence information eliminating the need of optical reading at each dNTP incorporation.
Table 8.
Comparison of commercially available sequencing platforms.
| (a) Massive parallel clonal amplification with optical detection | |||
|---|---|---|---|
| Roche 454 | Life Technologies SOLiD | Illumina | |
|
| |||
| Library amplification | emPCR* | emPCR* | On glass |
| Sequencing | Incorporation of unlabeled dNTPs | Ligase-mediated addition of fluorescent oligoNTPs (2bp) | Incorporation of end-blocked fluorescent dNTPs |
| Detection | Light emission from release of PPi | Fluorescence emission from ligated dye-labeled oligoNTPs | Fluorescence emission from incorporated labeled oligoNTPs |
| Progression | Unlabeled dNTPS added in base-specific fashion | Chemical cleavage removes dye and oligoNTP | Chemical cleavage fluorescent dye and blocking group |
| Errors | Insertion/deletion | End of read | End of read |
| Length | 400 bp | 75 bp | 150 bp |
| Overall yield/run | 500 Mbp | >100 Gbp | 200 Gbp |
|
| |||
| (b) Fluorescent and semiconductor single molecule sequencing | |||
|
| |||
| Helicos | Pacific biosystem | Iontorrent | |
|
| |||
| Library amplification | N/A-tSMS** | N/A-SMRT*** sequencing | Optional PCR |
| Sequencing | Incorporation of fluorescent labeled dNTPs | Polymerase incorporation terminal phosphate labeled dNTPs | Polymerase incorporation of dNTPs releases H+ |
| Detection | Laser-induced emission from incorporated dNTP | Real time detection of fluorescent dye in polymerase active site | Semiconductor ion sensor detects H+ released during dNTPs incorporation |
| Progression | Chemical cleavage of dNTP fluorescent group | N/A fluorescent dyes are removed as PPi with dNTPs incorporation | H+ signal during each dNTP incorporation is converted in voltage signal |
| Errors | Insertion/deletion | Insertion/deletion | Insertion/deletion |
| Length | 35 bp | 1000 bp | 200–400 bp |
| Overall yield/run | 21–37 Gbp | >100 Gbp | 1 Gbp |
*emPCR (emulsion PCR) is an amplification method where DNA library fragments are mixed with beads and PCR reagents in an oil emulsion that allows massive amplification of bead-DNA in a single reaction.
**tSMS: true Single Molecule Sequencing.
***SMRT (Single Molecule Real Time)
bp: base pair, Mbp: Mega base pair (106 bp), Gbp: Giga base pair (109 bp), dNTP: deoxynucleotide-tri-phosphate, PPi: pyrophosphate.
Other third-generation platforms under development make use of nanophotonic visualization chamber, ion semiconductor, electron microscopy, a variety of nanotechnologies like nanopores (Oxford Nanopore Technologies), nanochannels (BioNanomatrix/nanoAnalyzer), nanoparticles (GE Global Research), nanoballs (Complete Genomics), nanowells (CrackerBio), nanoknifes (Renveo), and specially engineered sensor DNA polymerase (VisiGen Biotechnologies) [42]. They promise even larger and faster data production although they are still under development and a few years away from commercial use. In principle also DNA microarrays could allow sequencing by hybridization using ultrafast nonenzymatic methods (Genizon BioSciences) and somebody even suggests that mass spectrometry might be used to determine mass differences between DNA fragments produced by chain termination [43].
The beginning of several individual genome projects has gradually decreased the cost of sequencing an individual genome, and it is likely that the $1,000 cost per person will be reached in few years. In medicine, the “personal genome” age made possible by NGS will be an important milestone for the entire genomic field and will mark a transition from single gene testing to whole genome evaluation [44].
It is impossible to predict today which NGS will eventually dominate genomic research, but it is sure that cost reductions, sequencing speed, and better accuracy will make NGS an essential molecular tool in several areas of biology and medicine.
Although the cost of whole genome sequencing has dropped significantly, it remains a major obstacle since it can reach $100,000 for a single individual. However, targeting sequencing of specific regions of interest can decrease the overall cost and improve efficiency of NGS making this technology ready for diagnostic use [45].
Also the field of epilepsy is potentially affected by NGS. Indeed too many genes and genetic conditions can be associated with epilepsy to make impossible for the clinicians a general use of specific monogene test for the vast majority of nonsyndromic or idiopathic epileptic patients. NGS is changing this situation by targeting several genome regions where known epilepsy genes are located and using enrichment techniques to significantly reduce the cost and improve efficiency. Targeted sequencing usually tests all protein-coding exons (functional exosome) which only requires roughly 5% as much sequencing than whole genome. This strategy will reduces the cost to about $3,000 or even less per single individual. Targeted selection technologies have been marketed and successfully used in different NGS projects and are becoming the tool of choice in several conditions, including epilepsy [46].
4. Targeting Sequencing and Epilepsy Gene Panels
A diagnostic panel is the contemporaneous targeted sequencing of a number of known genes that have already been identified as cause of a particular disease. A diagnostic panel is very different from whole genome or exome sequencing. Only genes clearly associated with a disease are examined. The genes included in the panel can be decided by the prescribing physician or by ad hoc committees of experts that can reach a consensus on the number and type of genes to test making commercially available diagnostic panel kits for specific diseases. This strategy should make easier to detect genetic variants that after validation by Sanger sequencing can be interpreted as the cause of the disease. Of course diagnostic panels and targeted sequencing make sense only if the condition is caused by several or very large number of genes. Many genetic disorders fall in this condition and are excellent candidate for the development of diagnostic panels. Epilepsy is an excellent example of such situation since it is a relative frequent disease affecting 1% of the population in a variety of forms, at different ages, with different progression. A genetic cause of epilepsy can be reasonably supposed in sporadic cases if trauma, tumor, or infection can be ruled out. In such circumstance all genetic information about epilepsy genes identified over the years in familial cases can be used to identify the causative gene through an epilepsy diagnostic panel. Indeed in the case of epilepsy the identified genes are so many that they can be classified in subpanels of genes that underline a common clinical entity (Table 9). Clinical considerations may suggest the clinician to include in a diagnostic panel other genes or genes from different sub-panels. At the present the first diagnostic panels for epilepsy that can analyze up to 400 different genes (CeGaT GmbH) are commercially available [47].
Table 9.
Epilepsy diagnostic panels.
| Subpanels With Homogeneous Clinical Entities | Table | Number of genes |
|---|---|---|
| Myoclonic epilepsy, febrile seizures, absences | 1 | 37 |
| Encephalopathies | 3 | 30 |
| X-linked mental retardation (XLMR) | 4(a) | 25 |
| Joubert syndrome | 4(b) | 10 |
| Lissencephaly and polymicrogyria | 4(c) | 18 |
| Severe Microcephaly and pontocerebellar hypoplasia | 4(d) | 22 |
| Walker-Warburg syndrome | 4(e) | 6 |
| Holoprosencephaly | 4(f) | 8 |
| Leukodystrophies | 5(a) | 20 |
| Migraine | 5(b) | 6 |
| Disorders of the Ras-MAPK pathway | 5(c) | 13 |
| Hyperekplexia for defective glycine neurotransmission | 5(d) | 5 |
| Neuronal migration disorders | 5(e) | 31 |
| Inherited errors of metabolism | 6 | 49 |
| Congenital disorder of glycosylation (CDG) | 7(a) | 23 |
| Neuronal ceroid lipofuscinosis (NCL) | 7(b) | 8 |
| Defects of mitochondrial metabolism including coenzyme Q deficiency | 7(c) | 35 |
| Mucopolisaccaridosis and mucolipidosis | 7(d) | 15 |
| Peroxisome biogenesis disorders (PBD) | 7(e) | 9 |
| Syndromic epilepsy | 2 | 47 |
5. Future Perspectives and Conclusions
The genetics of epilepsy has evolved from ion channel and neurotransmitter receptor subunits to newly discovered genes highlighting the importance of different pathways in the epileptogenesis. Furthermore, it has been demonstrated that copy number variations collectively explain a larger portion of idiopathic epilepsy than any single gene. These studies have identified structural genomic variations associated with idiopathic epilepsy, representing a change from the conventional knowledge that chromosome microarray analysis is useful only for patients with intellectual disability or dysmorphism [48]. Genetic testing techniques are rapidly evolving and whole exome or whole genome sequencing, performing at increasingly cheaper costs, will allow rapid discovery of other pathogenic mutations, variants in noncoding DNA, and copy number variations encompassing several genes. This rapidly accumulating genetic information will expand our understanding of epilepsy, and will allow more rational and effective treatment. However, along with the ability to identify genetic variants potentially associated with epilepsy it is imperative to validate genetic associations and analyze their clinical significance.
Is it worth? The main objective of a diagnostic panel for epilepsy is to discover the molecular defect in all possible cases to create a specific and personalized treatment of the disease than can be pharmacologically different for different types of molecular defects. Personalized therapy will be possible only within a genomic medicine. But genomic medicine at the same time will raise other questions: what to do if more than one genetic variant is identified in the same epileptic patient? Can we understand how genetic interactions will modulate the disease severity and prognosis? Can interaction of specific genetic variants and environmental factors modulate the clinical spectrum of the disorder? What to do if the diagnostic panel is inconclusive? Are the costs affordable? These are only few questions that the genomics of epilepsy will raise. The answers will require time, a lot of sequencing, and probably the development of new and cheaper sequencing technologies.
Acknowledgments
This work was supported by grants from Telethon, MIUR, Università del Molise (DISPeS), Università di Genova (DOBIG), Regione Molise, and Assomab to S. Garofalo. The authors thank Dr. Saskia Biskup for kindly sharing information before publication and for inspiring with her work the authors.
References
- 1.Rees MI. The genetics of epilepsy—the past, the present and future. Seizure. 2010;19(10):680–683. doi: 10.1016/j.seizure.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Jefferys JGR. Advances in understanding basic mechanisms of epilepsy and seizures. Seizure. 2010;19(10):638–646. doi: 10.1016/j.seizure.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 3.Scharfman HE. The neurobiology of epilepsy. Current Neurology and Neuroscience Reports. 2007;7(4):348–354. doi: 10.1007/s11910-007-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cossette P. Channelopathies and juvenile myoclonic epilepsy. Epilepsia. 2010;51(1):30–32. doi: 10.1111/j.1528-1167.2009.02439.x. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann-Horn F, Jurkat-Rott K, Lerche H, Weber Y. Hereditary channelopathies in neurology. Advances in Experimental Medicine and Biology. 2010;686:305–334. doi: 10.1007/978-90-481-9485-8_18. [DOI] [PubMed] [Google Scholar]
- 6.Werner F-M, Covenas R. Classical neurotransmitters and neuropeptides involved in generalized epilepsy: a focus on antiepileptic drugs. Current Medicinal Chemistry. 2011;18(32):4933–4948. doi: 10.2174/092986711797535191. [DOI] [PubMed] [Google Scholar]
- 7.Beghi E. The concept of the epilepsy syndrome: how useful is it in clinical practice? Epilepsia. 2009;50(5):4–10. doi: 10.1111/j.1528-1167.2009.02112.x. [DOI] [PubMed] [Google Scholar]
- 8.Nicita F, De Liso P, Danti FR, et al. The genetics of monogenic idiopathic epilepsies and epileptic encephalopathies. Seizure. 2012;21(1):3–11. doi: 10.1016/j.seizure.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Prince E, Ring H. Causes of learning disability and epilepsy: a review. Current Opinion in Neurology. 2011;24(2):154–158. doi: 10.1097/WCO.0b013e3283444c70. [DOI] [PubMed] [Google Scholar]
- 10.Bell B, Lin JJ, Seidenberg M, Hermann B. The neurobiology of cognitive disorders in temporal lobe epilepsy. Nature Reviews Neurology. 2011;7(3):154–164. doi: 10.1038/nrneurol.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu JS. Molecular genetics of neuronal migration disorders. Current Neurology and Neuroscience Reports. 2011;11(2):171–178. doi: 10.1007/s11910-010-0176-5. [DOI] [PubMed] [Google Scholar]
- 12.Cook P, Walker V. Investigation of the child with an acute metabolic disorder. Journal of Clinical Pathology. 2011;64(3):181–191. doi: 10.1136/jcp.2009.067884. [DOI] [PubMed] [Google Scholar]
- 13.Prasad AN, Hoffmann GF. Early onset epilepsy and inherited metabolic disorders: diagnosis and management. Canadian Journal of Neurological Sciences. 2010;37(3):350–358. doi: 10.1017/s0317167100010246. [DOI] [PubMed] [Google Scholar]
- 14.Battaglia A, Guerrini R. Chromosomal disorders associated with epilepsy. Epileptic Disorders. 2005;7(3):181–192. [PubMed] [Google Scholar]
- 15.Singh R, Gardner RJM, Crossland KM, Scheffer IE, Berkovic SF. Chromosomal abnormalities and epilepsy: a review for clinicians and gene hunters. Epilepsia. 2002;43(2):127–140. doi: 10.1046/j.1528-1157.2002.19498.x. [DOI] [PubMed] [Google Scholar]
- 16.Ottman R, Hirose S, Jain S, et al. Genetic testing in the epilepsies—report of the ILAE Genetics Commission. Epilepsia. 2010;51(4):655–670. doi: 10.1111/j.1528-1167.2009.02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pong AW, Pal DK, Chung WK. Developments in molecular genetic diagnostics: an update for the pediatric epilepsy specialist. Pediatric Neurology. 2011;44(5):317–327. doi: 10.1016/j.pediatrneurol.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Shostak S, Ottman R. Ethical, legal, and social dimensions of epilepsy genetics. Epilepsia. 2006;47(10):1595–1602. doi: 10.1111/j.1528-1167.2006.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furey TS, Haussler D. Integration of the cytogenetic map with the draft human genome sequence. Human Molecular Genetics. 2003;12(9):1037–1044. doi: 10.1093/hmg/ddg113. [DOI] [PubMed] [Google Scholar]
- 20.Yu S, Bittel DC, Kibiryeva N, Zwick DL, Cooley LD. Validation of the Agilent 244K oligonucleotide array-based comparative genomic hybridization platform for clinical cytogenetic diagnosis. American Journal of Clinical Pathology. 2009;132(3):349–360. doi: 10.1309/AJCP1BOUTWF6ERYS. [DOI] [PubMed] [Google Scholar]
- 21.Regier DA, Friedman JM, Marra CA. Value for money? array genomic hybridization for diagnostic testing for genetic causes of intellectual disability. American Journal of Human Genetics. 2010;86(5):765–772. doi: 10.1016/j.ajhg.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang B, Zhu H, Shen Y, et al. Genome-wide oligonucleotide array comparative genomic hybridization for etiological diagnosis of mental retardation: a multicenter experience of 1499 clinical cases. Journal of Molecular Diagnostics. 2010;12(2):204–212. doi: 10.2353/jmoldx.2010.090115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulley JC, Mefford HC. Epilepsy and the new cytogenetics. Epilepsia. 2011;52(3):423–432. doi: 10.1111/j.1528-1167.2010.02932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mefford HC, Muhle H, Ostertag P, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genetics. 2010;6(5, article e1000962) doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paris Conference (1971), supplement (1975): standardization in human cytogenetics. Cytogenetics Cell Genetics. 1975;15(4):203–238. [PubMed] [Google Scholar]
- 27.Shaffer LG, Tommerup N. An International System for Cytogenetic Nomenclature. Basel, Switzerland: S. Karger; 2005. [Google Scholar]
- 28.Speicher MR, Ballard SG, Ward DC. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nature Genetics. 1996;12(4):368–375. doi: 10.1038/ng0496-368. [DOI] [PubMed] [Google Scholar]
- 29.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470(7333):187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 30.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith LM, Sanders JZ, Kaiser RJ. Fluorescence detection in automated DNA sequence analysis. Nature. 1986;321(6071):674–679. doi: 10.1038/321674a0. [DOI] [PubMed] [Google Scholar]
- 32.Shendure J, Ji H. Next-generation DNA sequencing. Nature Biotechnology. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 33.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends in Genetics. 2008;24(3):133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 34.Mardis ER. Anticipating the 1,000 dollar genome. Genome biology. 2007;7(7):p. 112. doi: 10.1186/gb-2006-7-7-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mardis ER. A decade's perspective on DNA sequencing technology. Nature. 2011;470(7333):198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- 36.Mardis ER. New strategies and emerging technologies for massively parallel sequencing: applications in medical research. Genome Medicine. 2009;1(4) doi: 10.1186/gm40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venter JC, Levy S, Stockwell T, Remington K, Halpern A. Massive parallelism, randomness and genomic advances. Nature Genetics. 2003;33:219–227. doi: 10.1038/ng1114. [DOI] [PubMed] [Google Scholar]
- 38.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452(7189):872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 39.Shendure J, Porreca GJ, Reppas NB, et al. Molecular biology: accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309(5741):1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 40.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Chiodini R, Badr A, Zhang G. The impact of next-generation sequencing on genomics. Journal of Genetics and Genomics. 2011;38(3):95–109. doi: 10.1016/j.jgg.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mardis ER. Next-generation DNA sequencing methods. Annual Review of Genomics and Human Genetics. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 44.Schuster SC. Next-generation sequencing transforms today’s biology. Nature Methods. 2008;5(1):16–18. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- 45.Senapathy P, Bhasi A, Mattox J, Dhandapany PS, Sadayappan S. Targeted genome-wide enrichment of functional regions. PLoS ONE. 2010;5(6) doi: 10.1371/journal.pone.0011138. Article ID e11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biskup S. Next-generation sequencing in genetic diagnostics. LaboratoriumsMedizin. 2010;34(6):305–309. [Google Scholar]
- 47.Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing in diagnostics of seizure disorders. doi: 10.1111/j.1528-1167.2012.03516.x. Epilepsia. In press. [DOI] [PubMed] [Google Scholar]
- 48.Poduri A, Lowenstein D. Epilepsy genetics-past, present, and future. Current Opinion in Genetics and Development. 2011;21(3):325–332. doi: 10.1016/j.gde.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]